


Data Structures and Algorithms for k-th Nearest Neighbours Conformational Entropy Estimation



Abstract
1. Introduction
2. Materials and Methods
2.1. Entropy
2.2. Entropy Estimation Using the kNN Method
2.3. Data Structures and Algorithms for k-th Nearest Neighbours Computation
- ;
- ;
- .
- Given a list of n numbers, the sorting problem can be optimally solved in time.
- Given a list of n numbers and an index i, the selection problem (i.e., the problem of finding the i-th smallest number) can be solved in time (for example, using the Medians-of-medians algorithm [30]). It follows that the median of a list can also be calculated in linear time.
2.3.1. Naive Algorithms
One-Dimensional Approach
- Perform an ordering in time.
- For each point select the k-th neighbours in time.
Multidimensional Approach
- Calculate all the distances between p and all the other points in time.
- Find , a point that has the k-th smallest distance from p using a linear time selection algorithm.
- Select all the j points whose distance from p is strictly less than and then arbitrary select points whose distance is exactly equal to , if there are more than one such points.
2.3.2. Space Partitioning Algorithms
| Algorithm 1 General all-k-nn algorithm using a tree structure |
functionALL-KNN() BUILD-TREE() ▹ Tree building NEW-ARRAY(n) ▹ Space allocation for solution for each do QUERY() ▹ Tree traversal end for return end function |
| Algorithm 2 General tree construction |
functionBUILD-TREE() if then return MAKE-LEAF() end if MAKE-INTERNAL-NODE BUILD-TREE BUILD-TREE return u end function |
2.3.3. K-D Tree
The Data Structure
The Search Algorithm
Analysis
2.3.4. VP Tree
The Data Structure
The Search Algorithm
- The input is the same as for K-D Tree.
- We maintain a max-priority queue of length k with the same meaning as for K-D Tree.
- In each call, we visit the node u, and if u is a leaf node the steps performed are the same as for K-D Trees.
Analysis
2.4. Software Availability
3. Results
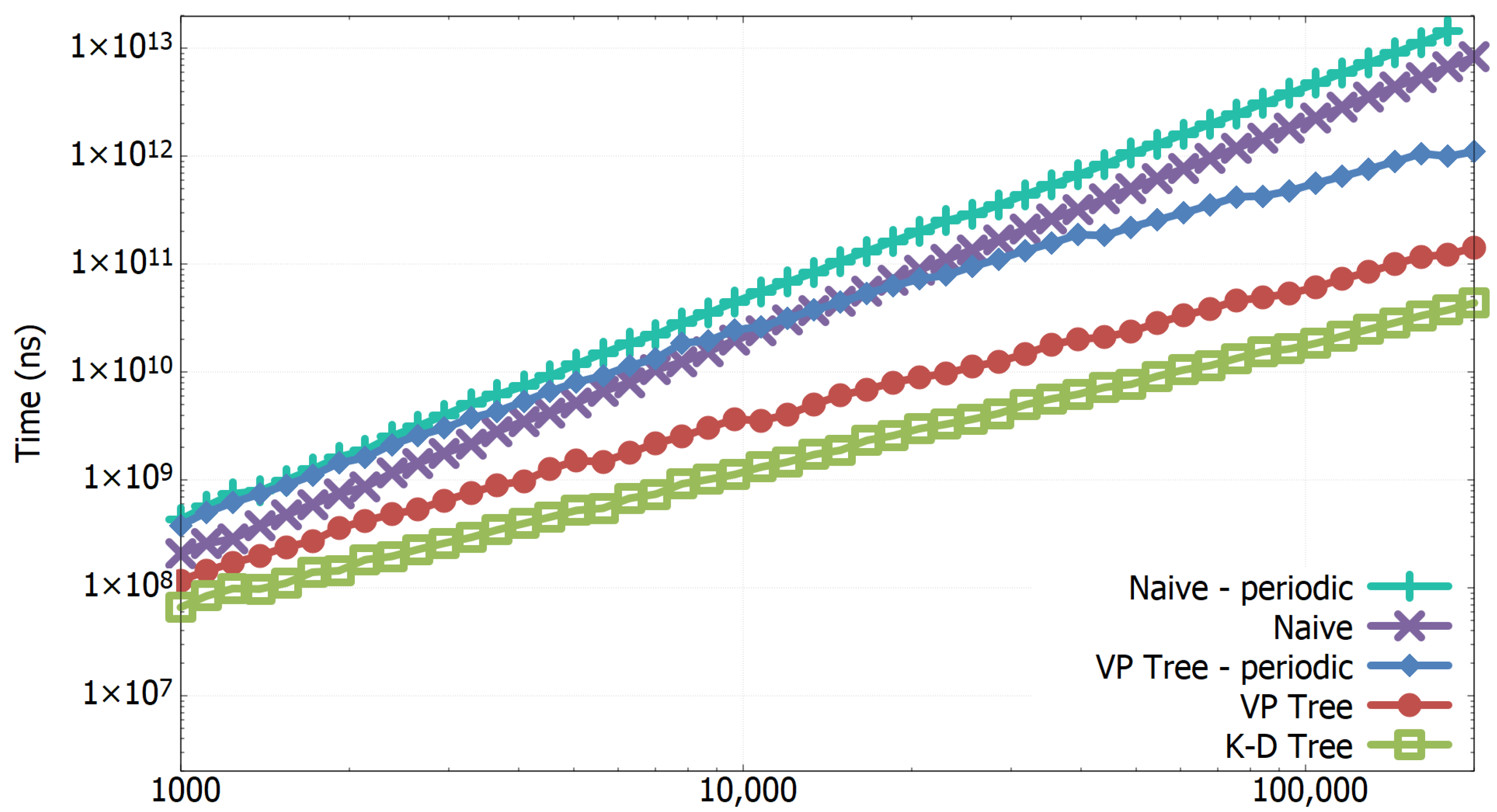
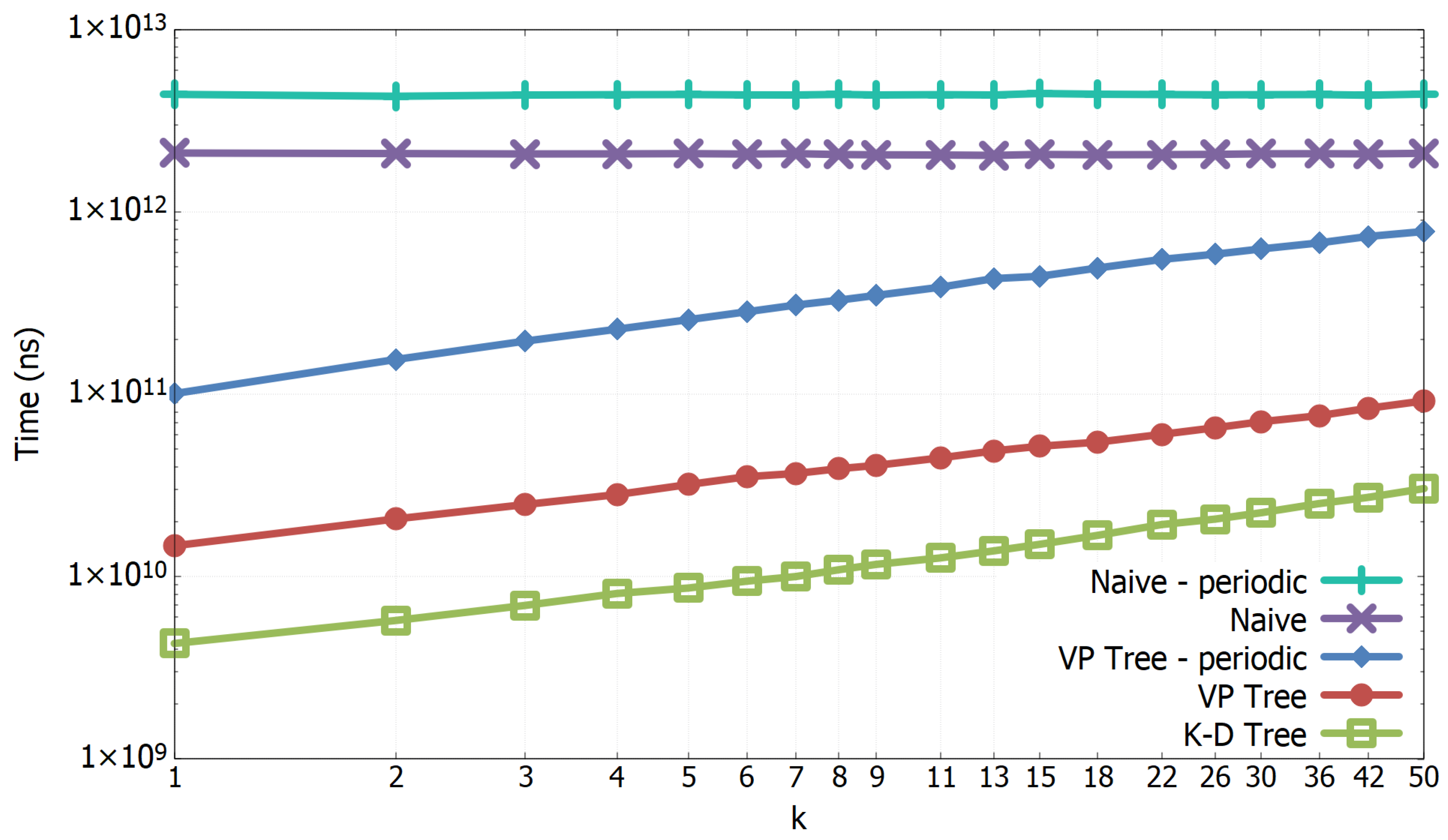
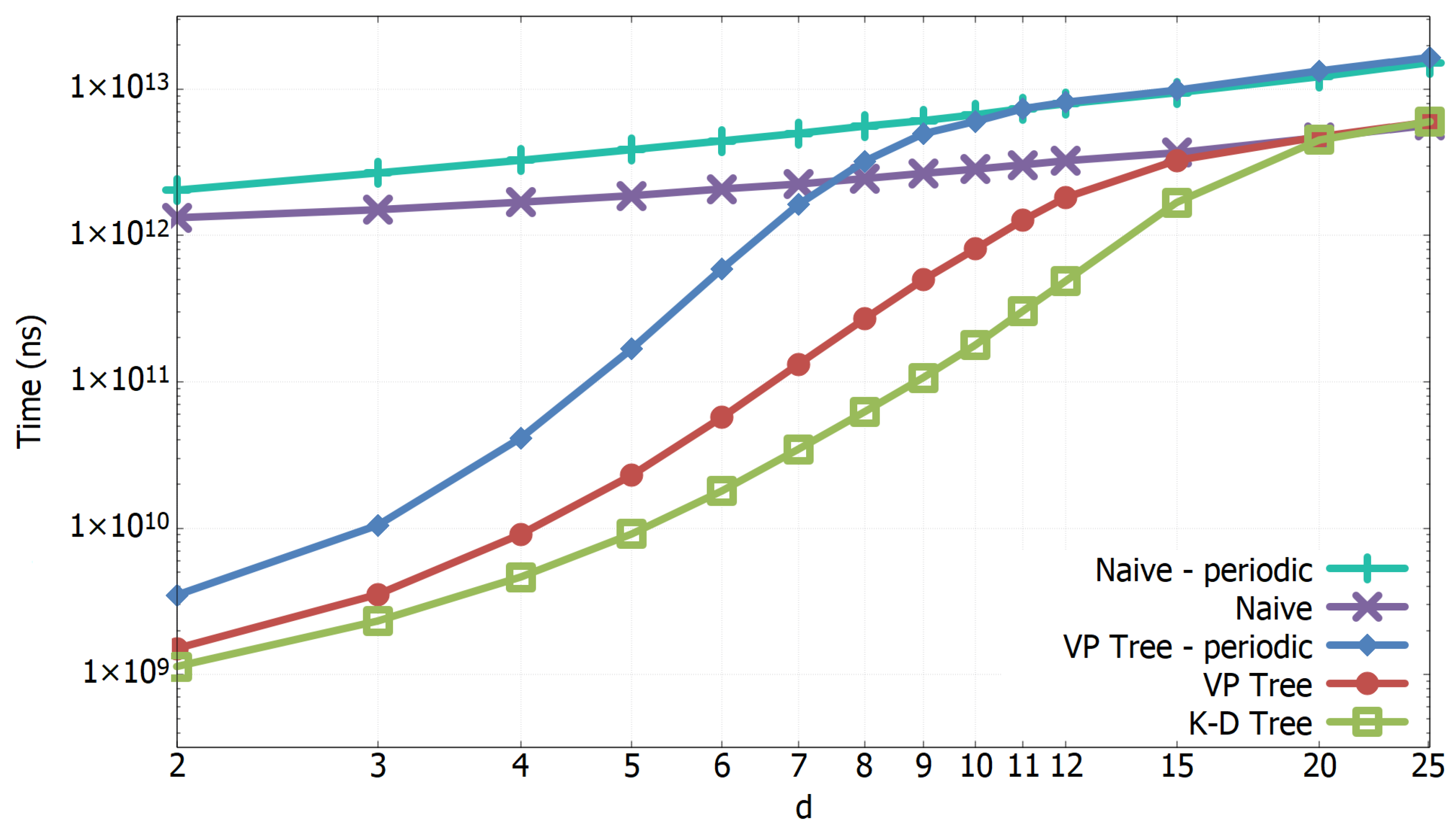
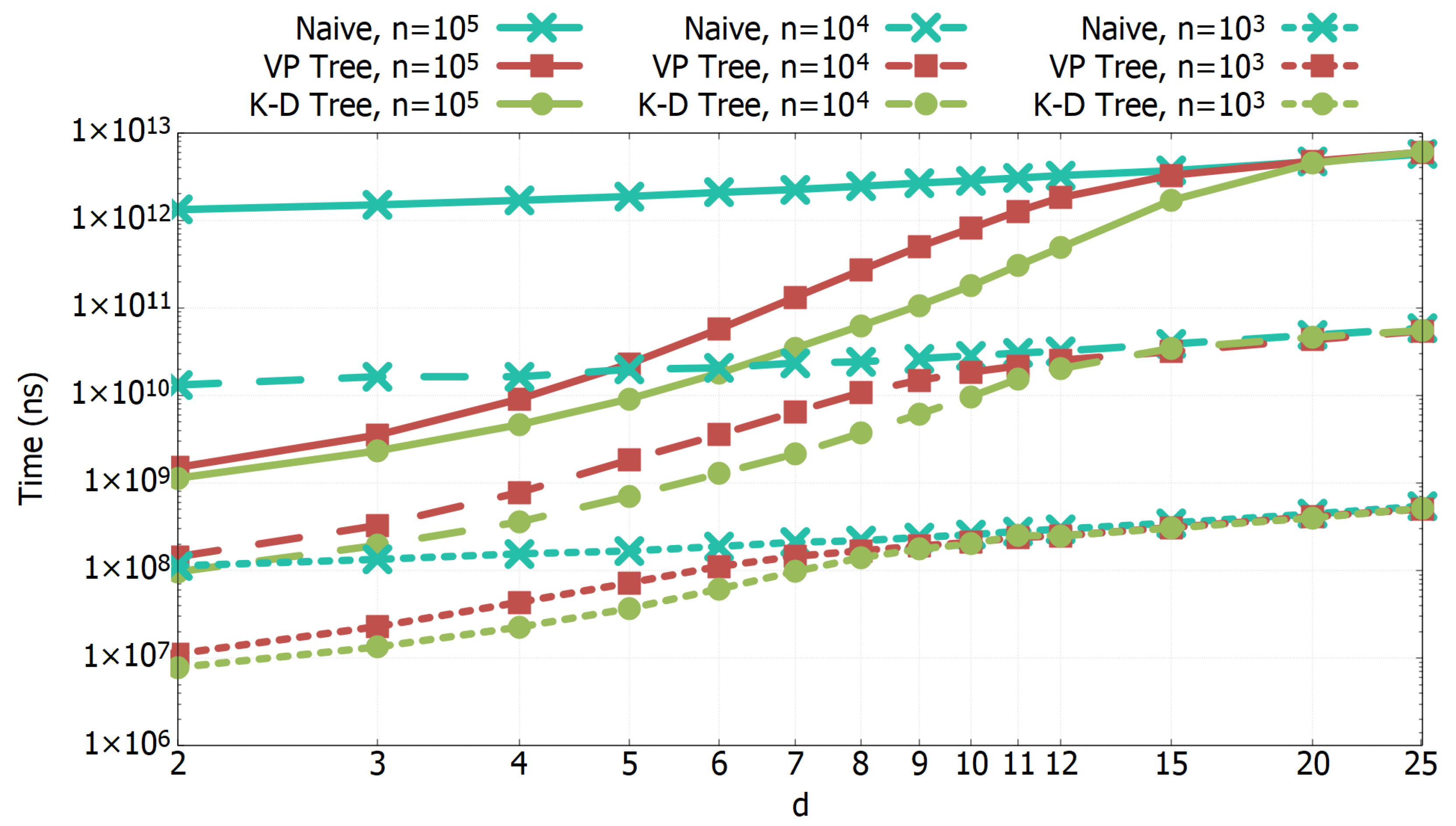
3.1. Experimental Results of All-k-nn Algorithms
3.2. Entropy of Amino Acids in Random Conformations
3.3. Entropy of a Small Folded Protein
4. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| kNN | k-th nearest neighbour |
| K-D | K dimensional |
| VP | Vantage Point |
References
- Wereszczynski, J.; McCammon, J.A. Statistical mechanics and molecular dynamics in evaluating thermodynamic properties of biomolecular recognition. Q. Rev. Biophys. 2012, 45, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Torrie, G.M.; Valleau, J.P. Nonphyisical sampling distributions in Monte Carlo free-energy estimation: Umbrella sampling. J. Comput. Phys. 1977, 23, 187–199. [Google Scholar] [CrossRef]
- Straatsma, T.P.; McCammon, J.A. Multiconfiguration thermodynamic integration. J. Chem. Phys. 1991, 95, 1175–1188. [Google Scholar] [CrossRef]
- Laio, A.; Parrinello, M. Escaping free energy minima. Proc. Natl. Acad. Sci. USA 2002, 99, 12562–12566. [Google Scholar] [CrossRef] [PubMed]
- Fogolari, F.; Corazza, A.; Esposito, G. Free Energy, Enthalpy and Entropy from Implicit Solvent End-Point Simulations. Front. Mol. Biosci. 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Gilson, M.K.; Given, J.A.; Bush, B.L.; McCammon, J.A. The statistical-thermodynamic basis for computation of binding affinities: A critical review. Biophys. J. 1997, 72, 1047–1069. [Google Scholar] [CrossRef]
- Roux, B.; Simonson, T. Implicit solvent models. Biophys. Chem. 1999, 78, 1–20. [Google Scholar] [CrossRef]
- Hnizdo, V.; Darian, E.; Fedorowicz, A.; Demchuk, E.; Li, S.; Singh, H. Nearest-neighbor nonparametric method for estimating the configurational entropy of complex molecules. J. Comput. Chem. 2007, 28, 655–668. [Google Scholar] [CrossRef] [PubMed]
- Fogolari, F.; Maloku, O.; Dongmo Foumthuim, C.J.; Corazza, A.; Esposito, G. PDB2ENTROPY and PDB2TRENT: Conformational and Translational–Rotational Entropy from Molecular Ensembles. J. Chem. Inf. Model. 2018, 58, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Killian, B.J.; Yundenfreund Kravitz, J.; Somani, S.; Dasgupta, P.; Pang, Y.P.; Gilson, M.K. Configurational entropy in protein-peptide binding: Computational study of Tsg101 ubiquitin E2 variant domain with an HIV-derived PTAP nonapeptide. J. Mol. Biol. 2009, 389, 315–335. [Google Scholar] [CrossRef]
- Killian, B.J.; Yundenfreund Kravitz, J.; Gilson, M.K. Extraction of configurational entropy from molecular simulations via an expansion approximation. J. Chem. Phys. 2007, 127, 024107. [Google Scholar] [CrossRef] [PubMed]
- King, B.M.; Tidor, B. MIST: Maximum Information Spanning Trees for dimension reduction of biological data sets. Bioinformatics 2009, 25, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- King, B.M.; Silver, N.W.; Tidor, B. Efficient calculation of molecular configurational entropies using an information theoretic approximation. J. Phys. Chem. B 2012, 116, 2891–2904. [Google Scholar] [CrossRef]
- Beirlant, J.; Dudewicz, E.; Gyorfi, L.; der Meulen EC, V. Nonparametric entropy estimation: An overview. Int. J. Math. Stat. Sci. 1997, 4, 17–39. [Google Scholar]
- Paninski, L. Estimation of Entropy and Mutual Information. Neural Comput. 2003, 15, 1191–1253. [Google Scholar] [CrossRef]
- le Brigant, A.; Puechmorel, S. Approximation of Densities on Riemannian Manifolds. Entropy 2019, 21, 43. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Scott, D.W. Nonparametric density estimation for high-dimensional data—Algorithms and applications. Wiley Interdiscip. Rev. Comput. Stat. 2019, 11, e1461. [Google Scholar] [CrossRef]
- Kozachenko, L.F.; Leonenko, N.N. Sample estimates of entropy of a random vector. Probl. Inform. Transm. 1987, 23, 95–101. [Google Scholar]
- Singh, H.; Misra, N.; Hnizdo, V.; Fedorowicz, A.; Demchuk, E. Nearest neighbor estimate of entropy. Am. J. Math. Manag. Sci. 2003, 23, 301–321. [Google Scholar] [CrossRef]
- Darian, E.; Hnizdo, V.; Fedorowicz, A.; Singh, H.; Demchuk, E. Estimation of the absolute internal-rotation entropy of molecules with two torsional degrees of freedom from stochastic simulations. J. Comput. Chem. 2005, 26, 651–660. [Google Scholar] [CrossRef]
- Hnizdo, V.; Tan, J.; Killian, B.J.; Gilson, M.K. Efficient calculation of configurational entropy from molecular simulations by combining the mutual-information expansion and nearest-neighbor methods. J. Comput. Chem. 2008, 29, 1605–1614. [Google Scholar] [CrossRef] [PubMed]
- Fogolari, F.; Corazza, A.; Fortuna, S.; Soler, M.A.; VanSchouwen, B.; Brancolini, G.; Corni, S.; Melacini, G.; Esposito, G. Distance-based configurational entropy of proteins from molecular dynamics simulations. PLoS ONE 2015, 10, e0132356. [Google Scholar] [CrossRef] [PubMed]
- Huggins, D.J. Estimating translational and orientational entropies using the k-nearest neighbors algorithm. J. Chem. Theory Comput. 2014, 10, 3617–3625. [Google Scholar] [CrossRef] [PubMed]
- Fogolari, F.; Dongmo Foumthuim, C.J.; Fortuna, S.r.; Soler, M.A.; Corazza, A.; Esposito, G. Accurate Estimation of the Entropy of Rotation–Translation Probability Distributions. J. Chem. Theory Comput. 2016, 12, 1–8. [Google Scholar] [CrossRef]
- Fogolari, F.; Esposito, G.; Tidor, B. Entropy of two-molecule correlated translational-rotational motions using the kth nearest neighbour method. J. Chem. Theory Comput. 2021, 17, 3039–3051. [Google Scholar] [CrossRef] [PubMed]
- Heinz, L.P.; Grubmüller, H. Computing spatially resolved rotational hydration entropies from atomistic simulations. J. Chem. Theory Comput. 2020, 16, 108–118. [Google Scholar] [CrossRef]
- Heinz, L.P.; Grubmüller, H. Per|Mut: Spatially resolved hydration entropies from atomistic simulati ons. J. Chem. Theory Comput. 2021, 17, 2090–2091. [Google Scholar] [CrossRef]
- Fogolari, F.; Esposito, G. Optimal Relabeling of Water Molecules and Single-Molecule Entropy Estimation. Biophysica 2021, 1, 279–296. [Google Scholar] [CrossRef]
- Brown, J.; Bossomaier, T.; Barnett, L. Review of Data Structures for Computationally Efficient Nearest-Neighbour Entropy Estimators for Large Systems with Periodic Boundary Conditions. J. Comput. Sci. 2017, 23, 109–117. [Google Scholar] [CrossRef]
- Cormen, T.H.; Leiserson, C.E.; Rivest, R.L.; Stein, C. Introduction to Algorithms, 3rd ed.; MIT Press: Cambridge, MA, USA, 2009. [Google Scholar]
- Vaidya, P.M. An O(n log n) Algorithm for the All-nearest. Neighbors Problem. Discret. Comput. Geom. 1989, 4, 101–115. [Google Scholar] [CrossRef]
- Bentley, J.L. Multidimensional Binary Search Trees Used for Associative Searching. Commun. ACM 1975, 18, 509–517. [Google Scholar] [CrossRef]
- Friedman, J.H.; Bentley, J.L.; Finkel, R.A. An Algorithm for Finding Best Matches in Logarithmic Expected Time. ACM Trans. Math. Softw. 1977, 3, 209–226. [Google Scholar] [CrossRef]
- Yianilos, P. Data Structures and Algorithms for Nearest Neighbor Search in General Metric Spaces. In Proceedings of the Fourth Annual ACM-SIAM Symposium on Discrete Algorithms, Austin, TX, USA, 25–27 January 1993; Volume 93, pp. 311–321. [Google Scholar] [CrossRef]
- Frenkel, D.; Smit, B. Understanding Molecular Simulation: From Algorithms to Applications, 2nd ed.; Academic Press: San Diego, CA, USA, 2002. [Google Scholar]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Touw, W.G.; Baakman, C.; Black, J.; te Beek, T.A.; Krieger, E.; Joosten, R.P.; Vriend, G. A series of PDB-related databanks for everyday needs. Nucleic Acids Res. 2014, 43, D364–D368. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Dunbrack, R.L. PISCES: A protein sequence culling server. Bioinformatics 2003, 19, 1589–1591. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| aa. | n | d | Time (s) |
|---|---|---|---|
| ALA | 87,225 | 2 | 1.7 |
| ARG | 60,855 | 6 | 24.8 |
| ASN | 78,167 | 4 | 10.5 |
| ASP | 104,071 | 4 | 13.3 |
| CYS | 11,596 | 4 | 0.8 |
| GLN | 45,820 | 5 | 9.6 |
| GLU | 82,224 | 5 | 21.6 |
| GLY | 165,129 | 2 | 4.3 |
| HIS | 35,293 | 4 | 3.6 |
| ILE | 50,498 | 4 | 5.7 |
| LEU | 87,053 | 4 | 11.5 |
| LYS | 74,522 | 6 | 37.2 |
| MET | 17,022 | 5 | 2.8 |
| PHE | 45,531 | 4 | 4.8 |
| PRO | 136,006 | 2 | 3.1 |
| SER | 97,054 | 4 | 10.5 |
| THR | 78,923 | 4 | 9.0 |
| TRP | 15,222 | 4 | 1.3 |
| TYR | 39,659 | 5 | 8.0 |
| VAL | 64,047 | 3 | 3.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borelli, R.; Dovier, A.; Fogolari, F. Data Structures and Algorithms for k-th Nearest Neighbours Conformational Entropy Estimation. Biophysica 2022, 2, 340-352. https://doi.org/10.3390/biophysica2040031
Borelli R, Dovier A, Fogolari F. Data Structures and Algorithms for k-th Nearest Neighbours Conformational Entropy Estimation. Biophysica. 2022; 2(4):340-352. https://doi.org/10.3390/biophysica2040031
Chicago/Turabian StyleBorelli, Roberto, Agostino Dovier, and Federico Fogolari. 2022. "Data Structures and Algorithms for k-th Nearest Neighbours Conformational Entropy Estimation" Biophysica 2, no. 4: 340-352. https://doi.org/10.3390/biophysica2040031
APA StyleBorelli, R., Dovier, A., & Fogolari, F. (2022). Data Structures and Algorithms for k-th Nearest Neighbours Conformational Entropy Estimation. Biophysica, 2(4), 340-352. https://doi.org/10.3390/biophysica2040031