
Exploring the Dynamics of Shikimate Kinase through Molecular Mechanics


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
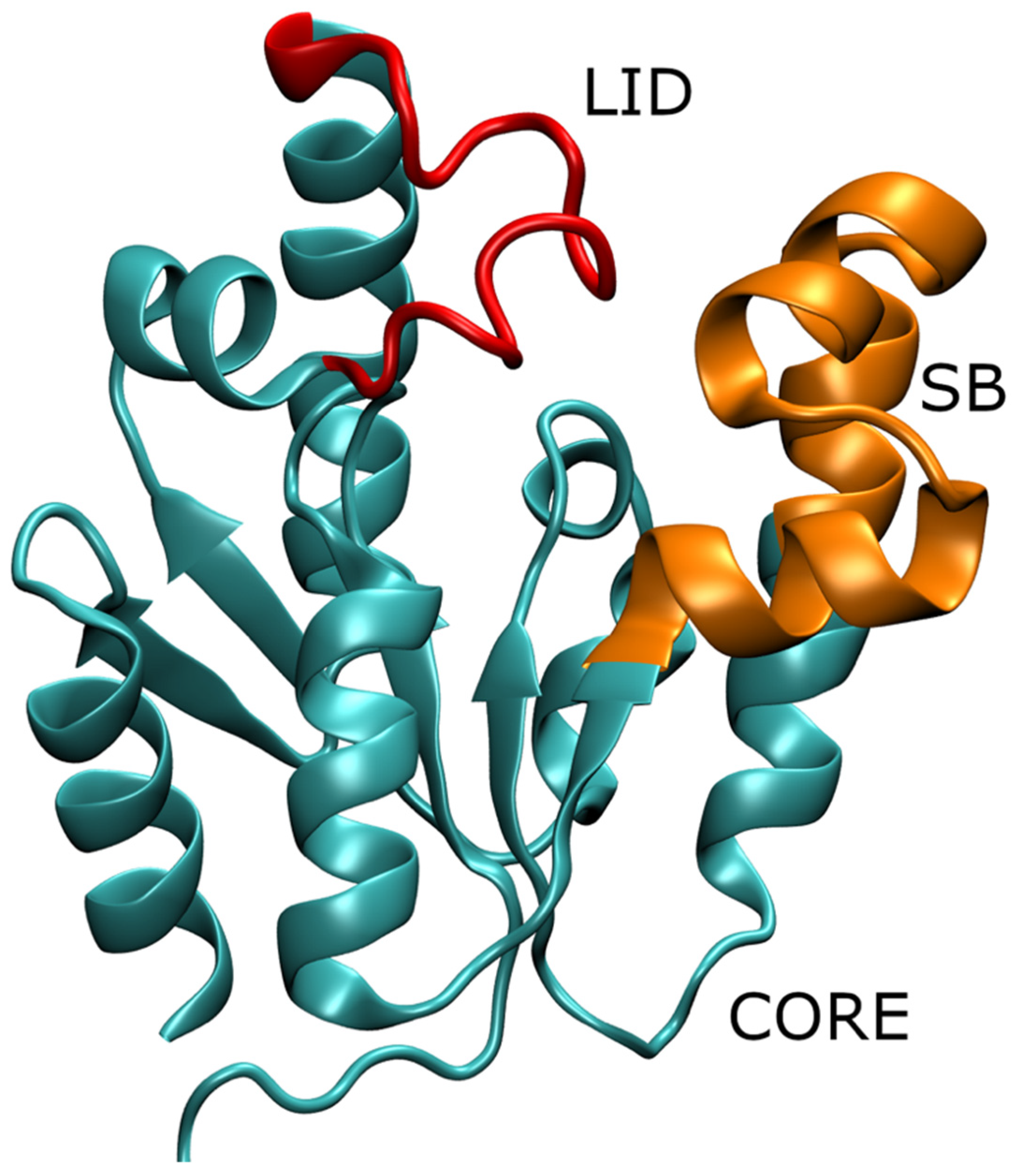
:1. Introduction
2. Materials and Methods
2.1. Simulation Setup
2.2. Analysis
3. Results and Discussion
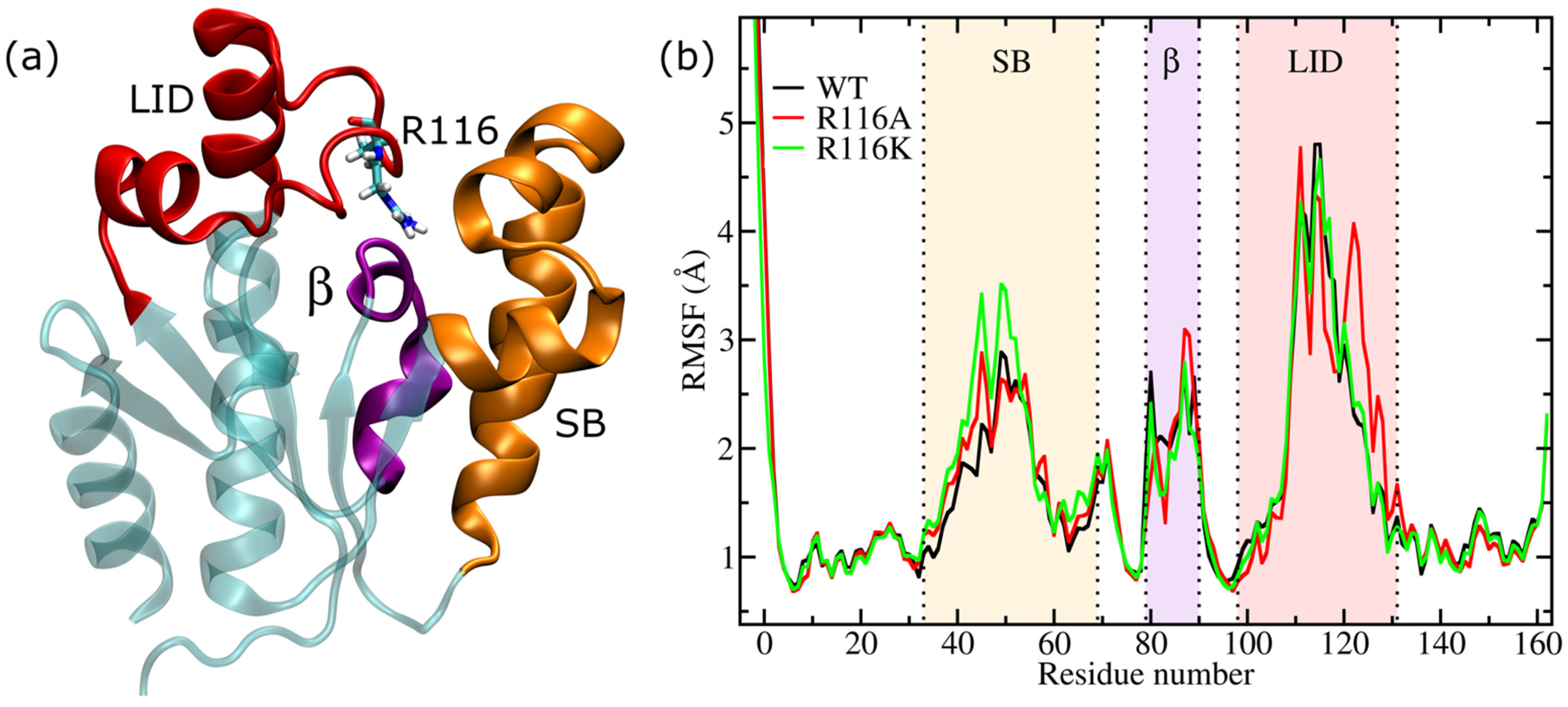
3.1. Root Mean Square Fluctuation (RMSF)
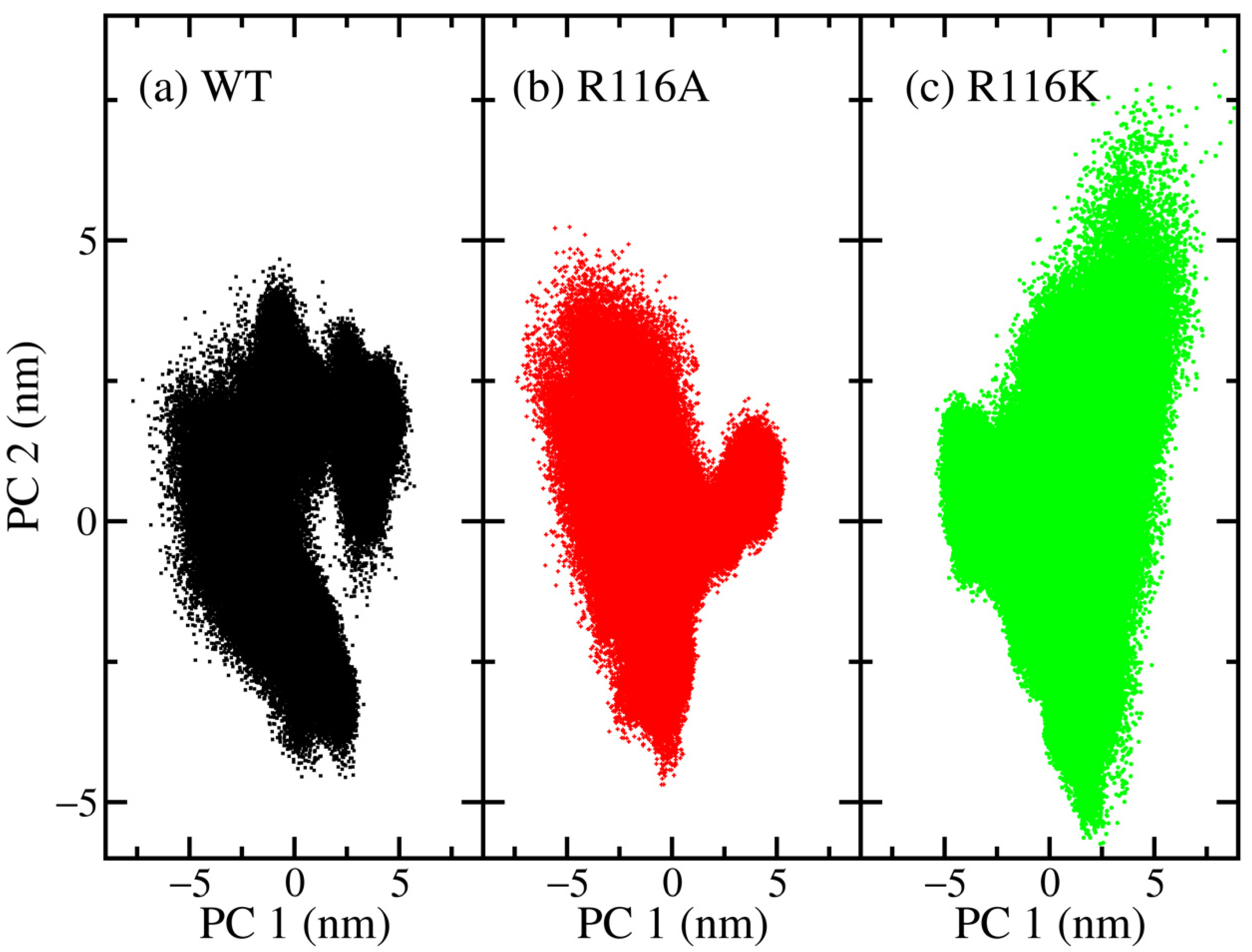
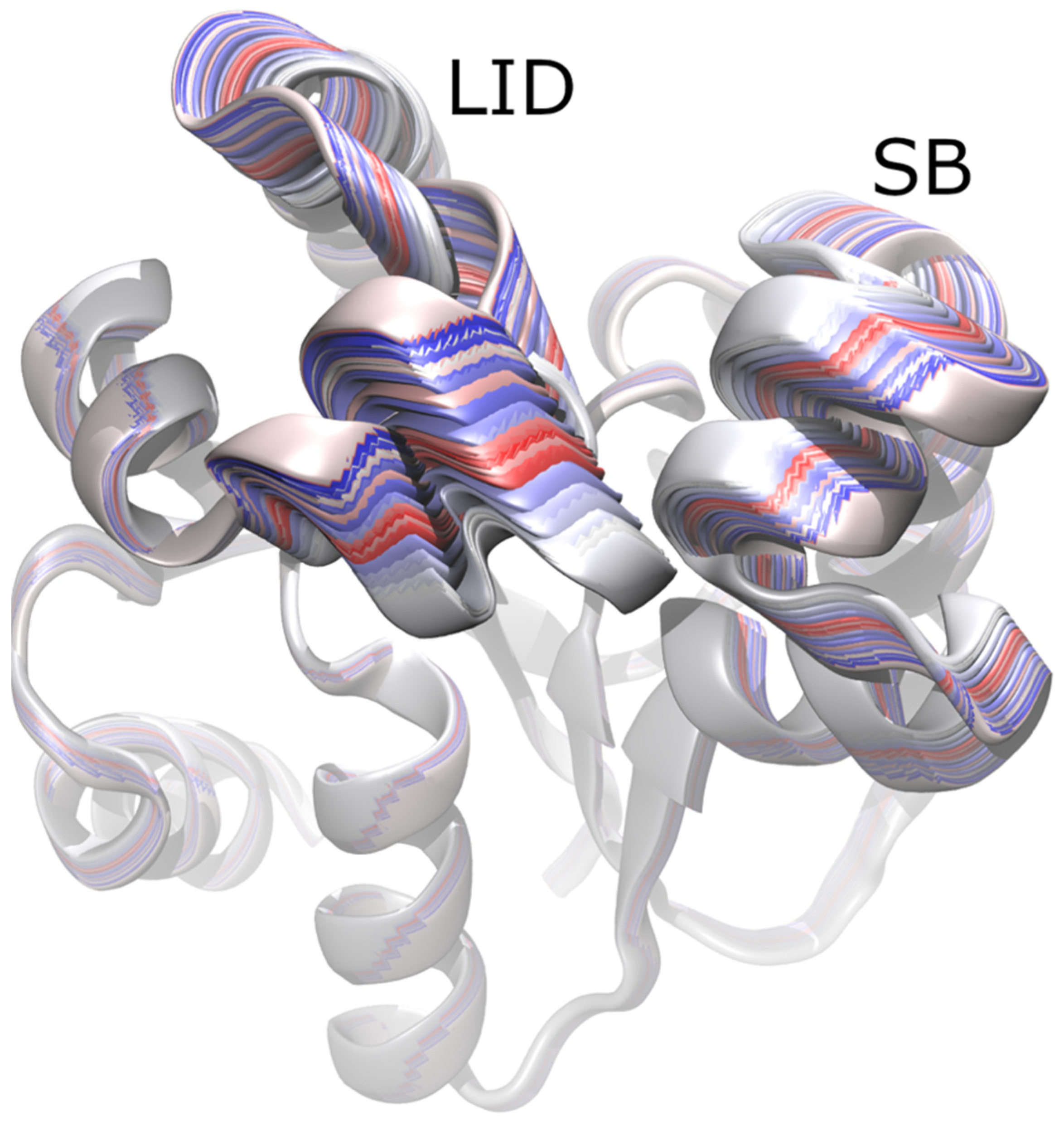
3.2. Principal Component Analysis (PCA)
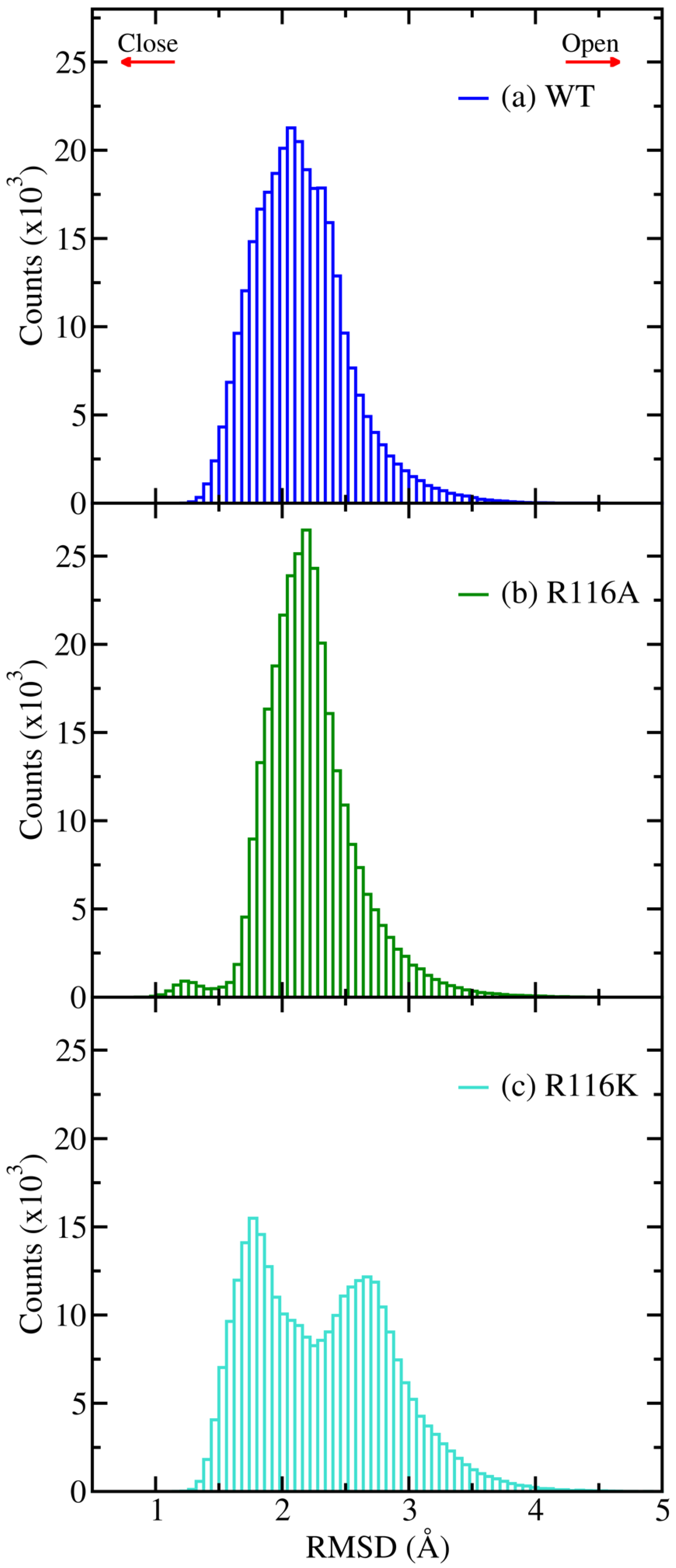
3.3. Root Mean Square Deviation (RMSD)
4. Conclusions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grillo, I.B.; Bachega, J.F.R.; Timmers, L.F.S.M.; Caceres, R.A.; de Souza, O.N.; Field, M.J.; Rocha, G.B. Theoretical Characterization of the Shikimate 5-Dehydrogenase Reaction from Mycobacterium tuberculosis by Hybrid QC/MM Simulations and Quantum Chemical Descriptors. J. Mol. Model. 2020, 26, 297. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.E.S.; Duque, M.A.; de Freitas, T.F.; Galina, L.; Timmers, L.F.S.M.; Bizarro, C.V.; Machado, P.; Basso, L.A.; Ducati, R.G. Mycobacterium tuberculosis Shikimate Pathway Enzymes as Targets for the Rational Design of Anti-Tuberculosis Drugs. Molecules 2020, 25, 1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coracini, J.D.; Azevedo, W.F. de Shikimate Kinase, a Protein Target for Drug Design. Curr. Med. Chem. 2014, 21, 592–604. [Google Scholar] [CrossRef]
- Cheng, W.-C.; Chen, Y.-F.; Wang, H.-J.; Hsu, K.-C.; Lin, S.-C.; Chen, T.-J.; Yang, J.-M.; Wang, W.-C. Structures of Helicobacter pylori Shikimate Kinase Reveal a Selective Inhibitor-Induced-Fit Mechanism. PLoS ONE 2012, 7, e33481. [Google Scholar] [CrossRef] [Green Version]
- Wolf-Watz, M.; Thai, V.; Henzler-Wildman, K.; Hadjipavlou, G.; Eisenmesser, E.Z.; Kern, D. Linkage between Dynamics and Catalysis in a Thermophilic-Mesophilic Enzyme Pair. Nat. Struct. Mol. Biol. 2004, 11, 945–949. [Google Scholar] [CrossRef] [PubMed]
- Villali, J.; Kern, D. Choreographing an Enzyme’s Dance. Curr. Opin. Chem. Biol. 2010, 14, 636–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, J.; Wang, X.; Luo, H.; Gu, P. Understanding the Catalytic Mechanism and the Nature of the Transition State of an Attractive Drug-Target Enzyme (Shikimate Kinase) by Quantum Mechanical/Molecular Mechanical (QM/MM) Studies. Chem.—Eur. J. 2017, 23, 16380–16387. [Google Scholar] [CrossRef]
- Ojeda-May, P. Exploring the Mechanism of Shikimate Kinase through Quantum Mechanical and Molecular Mechanical (QM/MM) Methods. Biophysica 2021, 1, 25. [Google Scholar] [CrossRef]
- Gu, Y.; Reshetnikova, L.; Li, Y.; Wu, Y.; Yan, H.; Singh, S.; Ji, X. Crystal Structure of Shikimate Kinase from Mycobacterium tuberculosis Reveals the Dynamic Role of the LID Domain in Catalysis. J. Mol. Biol. 2002, 319, 779–789. [Google Scholar] [CrossRef]
- Hénin, J.; Lelièvre, T.; Shirts, M.R.; Valsson, O.; Delemotte, L. Enhanced Sampling Methods for Molecular Dynamics Simulations. arXiv 2022, arXiv:220204164. [Google Scholar]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the Additive CHARMM All-Atom Protein Force Field Targeting Improved Sampling of the Backbone ϕ, ψ and Side-Chain Χ1 and Χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackerell, A.D.; Feig, M.; Brooks, C.L. Extending the Treatment of Backbone Energetics in Protein Force Fields: Limitations of Gas-Phase Quantum Mechanics in Reproducing Protein Conformational Distributions in Molecular Dynamics Simulations. J. Comput. Chem. 2004, 25, 1400–1415. [Google Scholar] [CrossRef] [PubMed]
- Foloppe, N.; MacKerell, A.D., Jr. All-Atom Empirical Force Field for Nucleic Acids: I. Parameter Optimization Based on Small Molecule and Condensed Phase Macromolecular Target Data. J. Comput. Chem. 2000, 21, 86–104. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Spoel, D.v.d.; et al. GROMACS 4.5: A High-Throughput and Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Páll, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS. In Proceedings of the Solving Software Challenges for Exascale, Stockholm, Sweden, 2–3 April 2014; Springer: Cham, Germany, 2014; pp. 3–27. [Google Scholar]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; Spoel, D.v.d. GROMACS 3.0: A Package for Molecular Simulation and Trajectory Analysis. Mol. Model. Annu. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N⋅log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef]
- Nosé, S.; Klein, M.L. Constant Pressure Molecular Dynamics for Molecular Systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Pearson, K.L., III. On Lines and Planes of Closest Fit to Systems of Points in Space. Philos. Mag. Ser. 6 1901, 2, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Cheng, W.-C.; Chang, Y.-N.; Wang, W.-C. Structural Basis for Shikimate-Binding Specificity of Helicobacter pylori Shikimate Kinase. J. Bacteriol. 2005, 187, 8156–8163. [Google Scholar] [CrossRef] [Green Version]
- Ishida, T. Effects of Point Mutation on Enzymatic Activity: Correlation between Protein Electronic Structure and Motion in Chorismate Mutase Reaction. J. Am. Chem. Soc. 2010, 132, 7104–7118. [Google Scholar] [CrossRef]
- Dehury, B.; Raina, V.; Misra, N.; Suar, M. Effect of Mutation on Structure, Function and Dynamics of Receptor Binding Domain of Human SARS-CoV-2 with Host Cell Receptor ACE2: A Molecular Dynamics Simulations Study. J. Biomol. Struct. Dyn. 2021, 39, 7231–7245. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Abeysinghe, T.; Kohen, A. Linking Protein Motion to Enzyme Catalysis. Molecules 2015, 20, 1192–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, W.; Karabencheva-Christova, T.G.; Black, G.W.; Ainsley, J.; Dover, L.; Christov, C.Z. Conformational Dynamics, Ligand Binding and Effects of Mutations in NirE an S-Adenosyl-L-Methionine Dependent Methyltransferase. Sci. Rep. 2016, 6, 20107. [Google Scholar] [CrossRef] [Green Version]
- Watt, E.D.; Shimada, H.; Kovrigin, E.L.; Loria, J.P. The Mechanism of Rate-Limiting Motions in Enzyme Function. Proc. Natl. Acad. Sci. USA 2007, 104, 11981–11986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Wang, X.; Pang, L.; Zhang, J.Z.H.; Zhu, T. Effect of Mutations on Binding of Ligands to Guanine Riboswitch Probed by Free Energy Perturbation and Molecular Dynamics Simulations. Nucleic Acids Res. 2019, 47, 6618–6631. [Google Scholar] [CrossRef]
- Ojeda-May, P.; Mushtaq, A.U.; Rogne, P.; Verma, A.; Ovchinnikov, V.; Grundström, C.; Dulko-Smith, B.; Sauer, U.H.; Wolf-Watz, M.; Nam, K. Dynamic Connection between Enzymatic Catalysis and Collective Protein Motions. Biochemistry 2021, 60, 2246–2258. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ojeda-May, P. Exploring the Dynamics of Shikimate Kinase through Molecular Mechanics. Biophysica 2022, 2, 194-202. https://doi.org/10.3390/biophysica2030020
Ojeda-May P. Exploring the Dynamics of Shikimate Kinase through Molecular Mechanics. Biophysica. 2022; 2(3):194-202. https://doi.org/10.3390/biophysica2030020
Chicago/Turabian StyleOjeda-May, Pedro. 2022. "Exploring the Dynamics of Shikimate Kinase through Molecular Mechanics" Biophysica 2, no. 3: 194-202. https://doi.org/10.3390/biophysica2030020
APA StyleOjeda-May, P. (2022). Exploring the Dynamics of Shikimate Kinase through Molecular Mechanics. Biophysica, 2(3), 194-202. https://doi.org/10.3390/biophysica2030020