
The Amyloidogenic Peptide Amyloid Beta(16–22) Displays Facet Dependent Conformation on Metal Surfaces


Abstract
:1. Introduction
2. Materials and Methods
3. Results
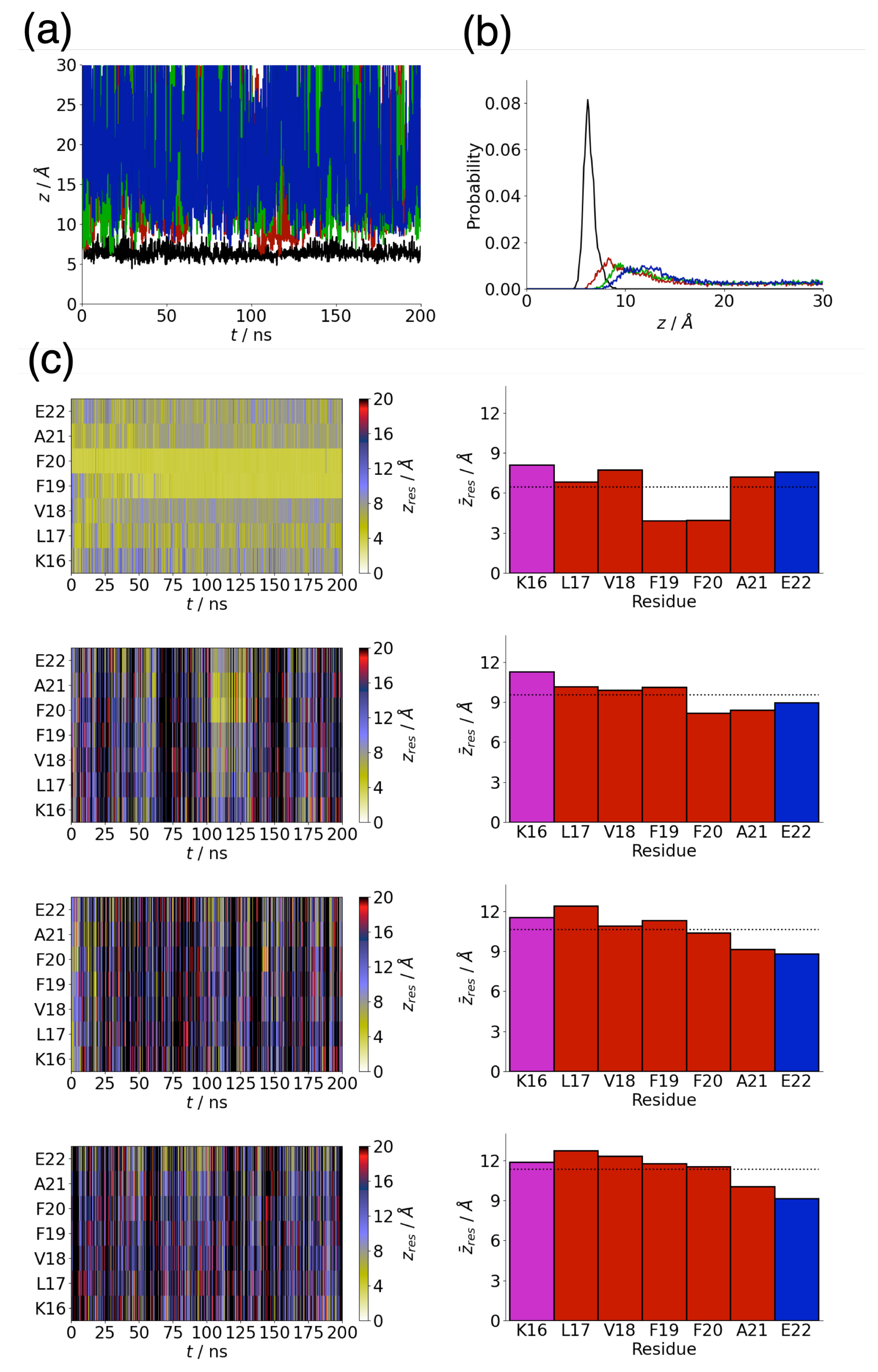
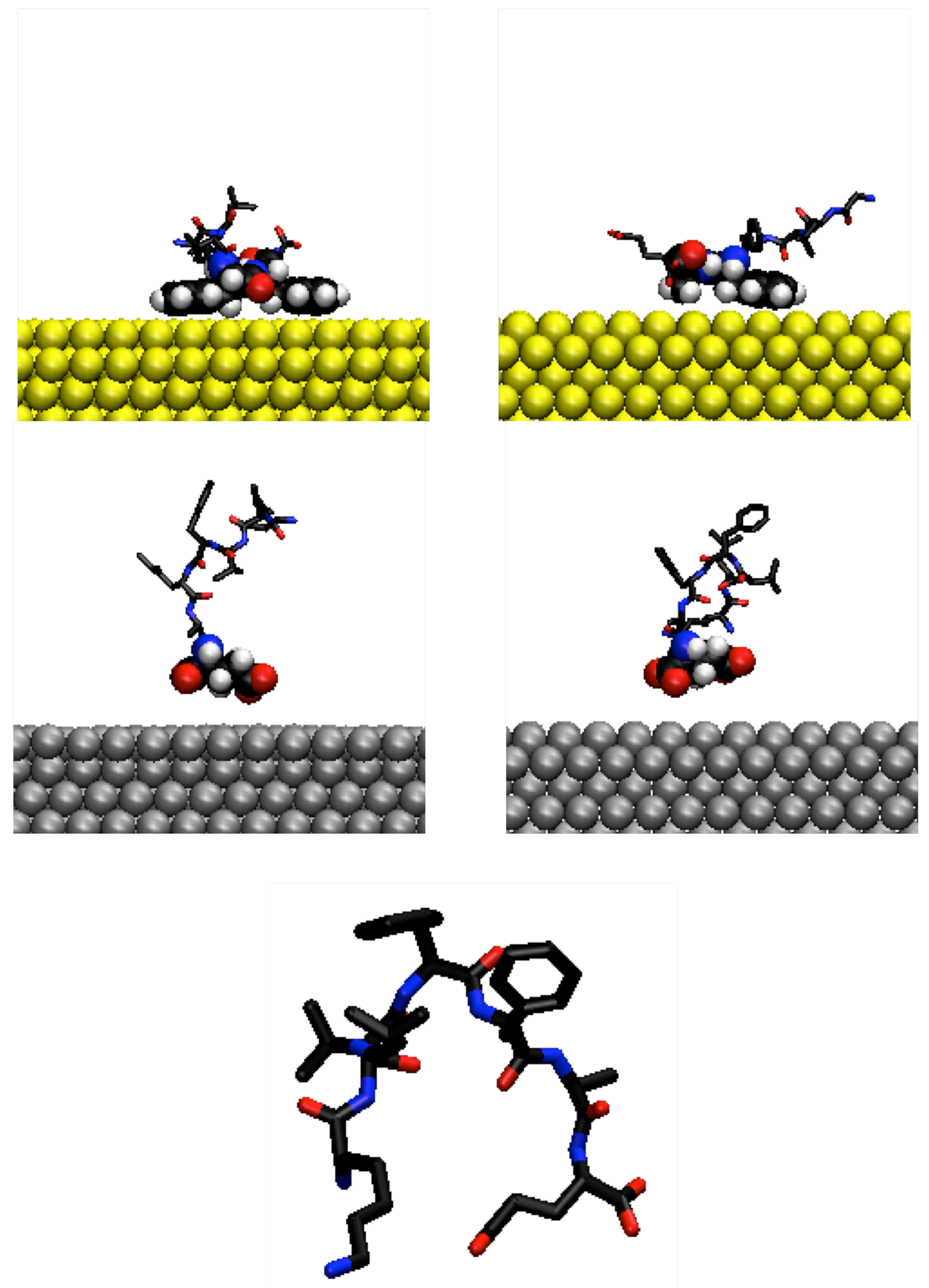
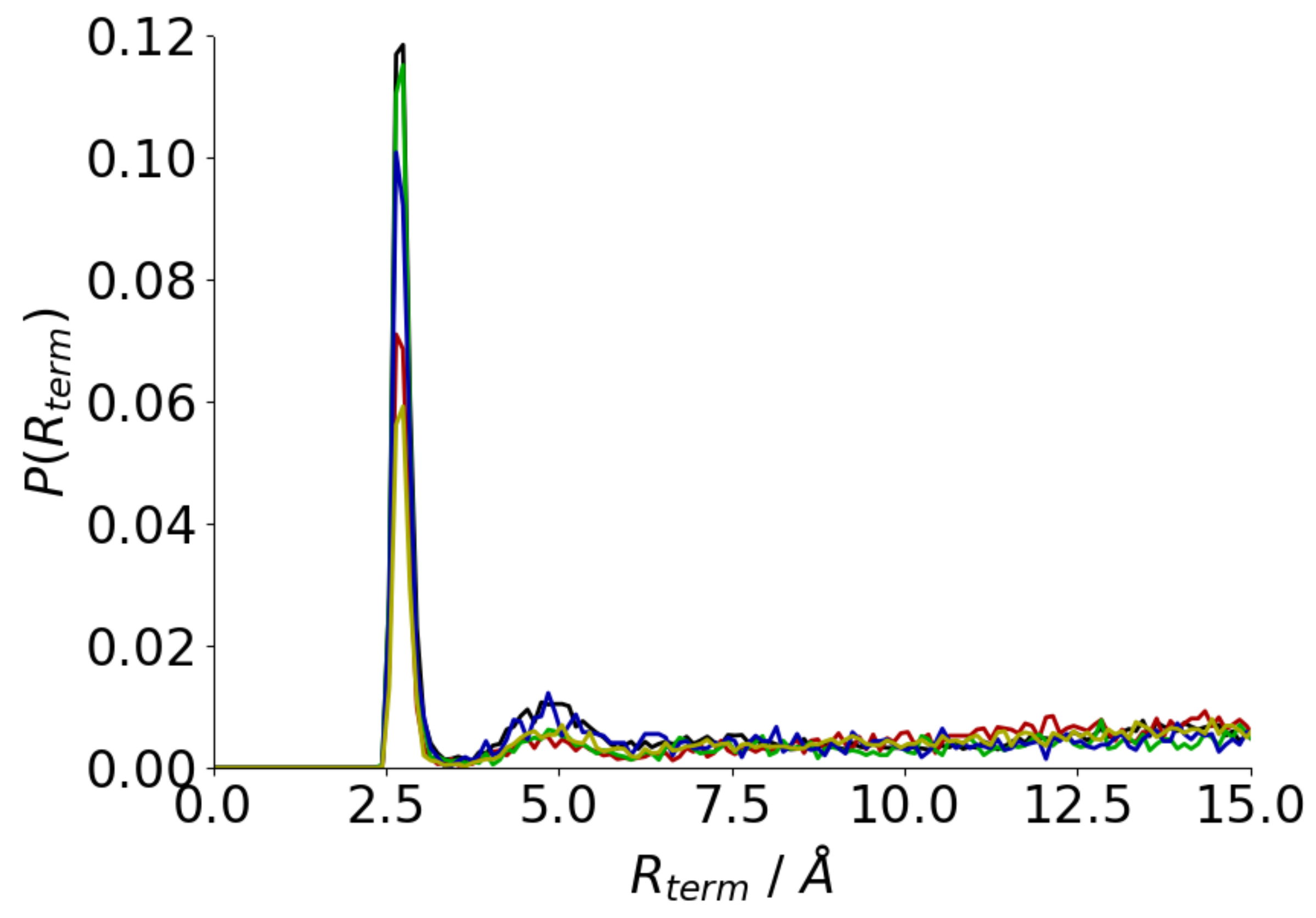
3.1. Adsorption Behaviour of Aβ(16–22) on Gold and Silver Surfaces
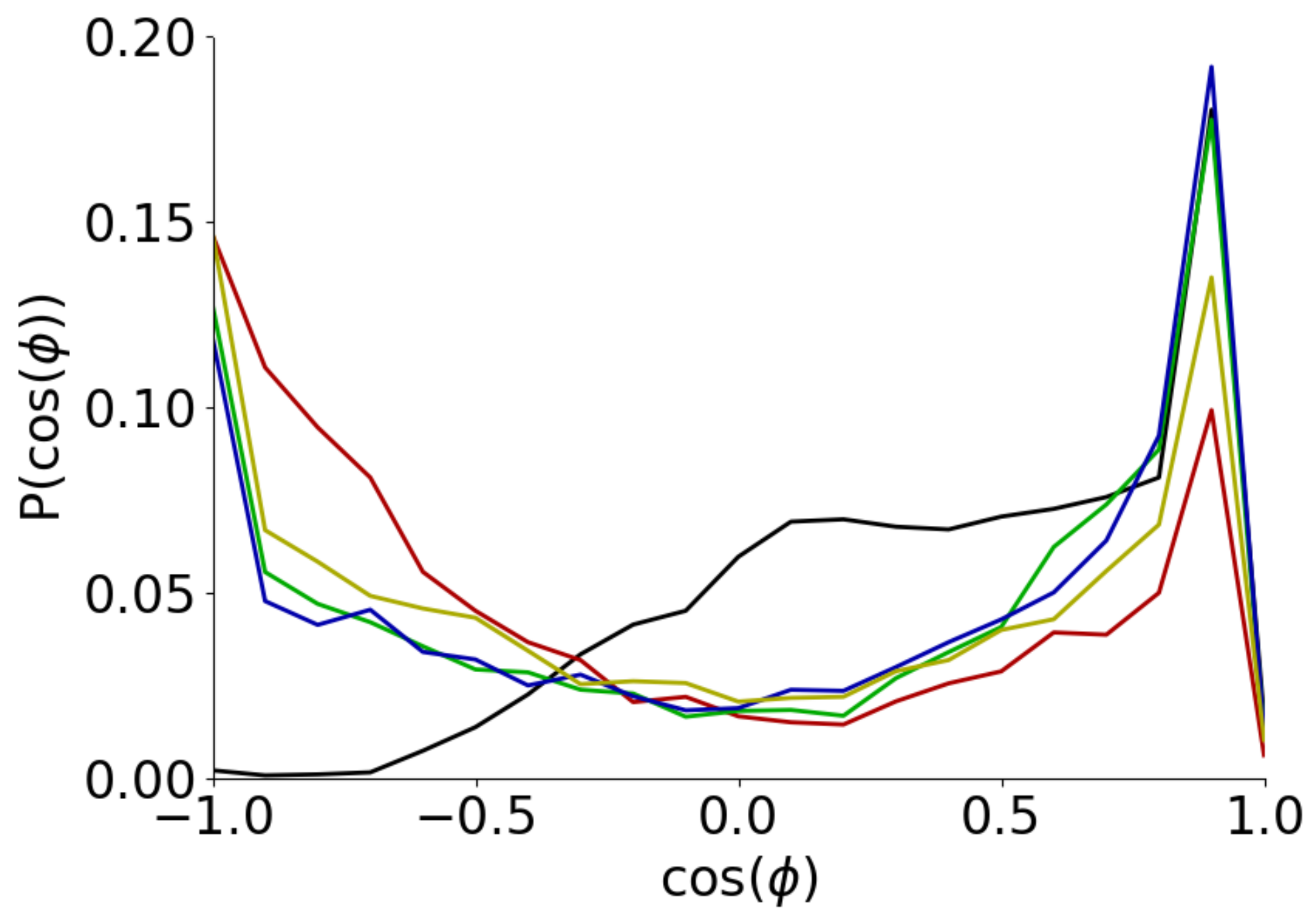
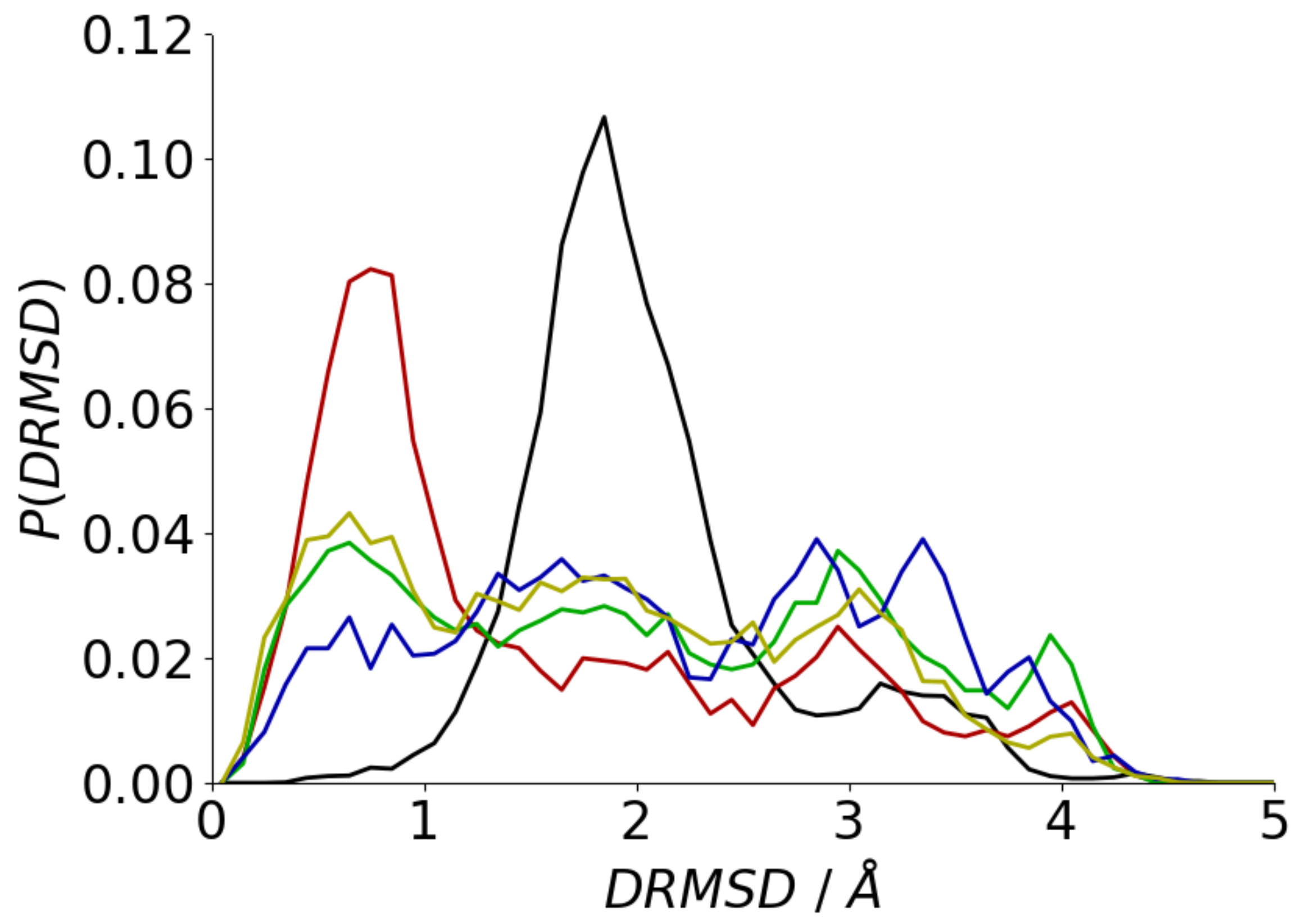
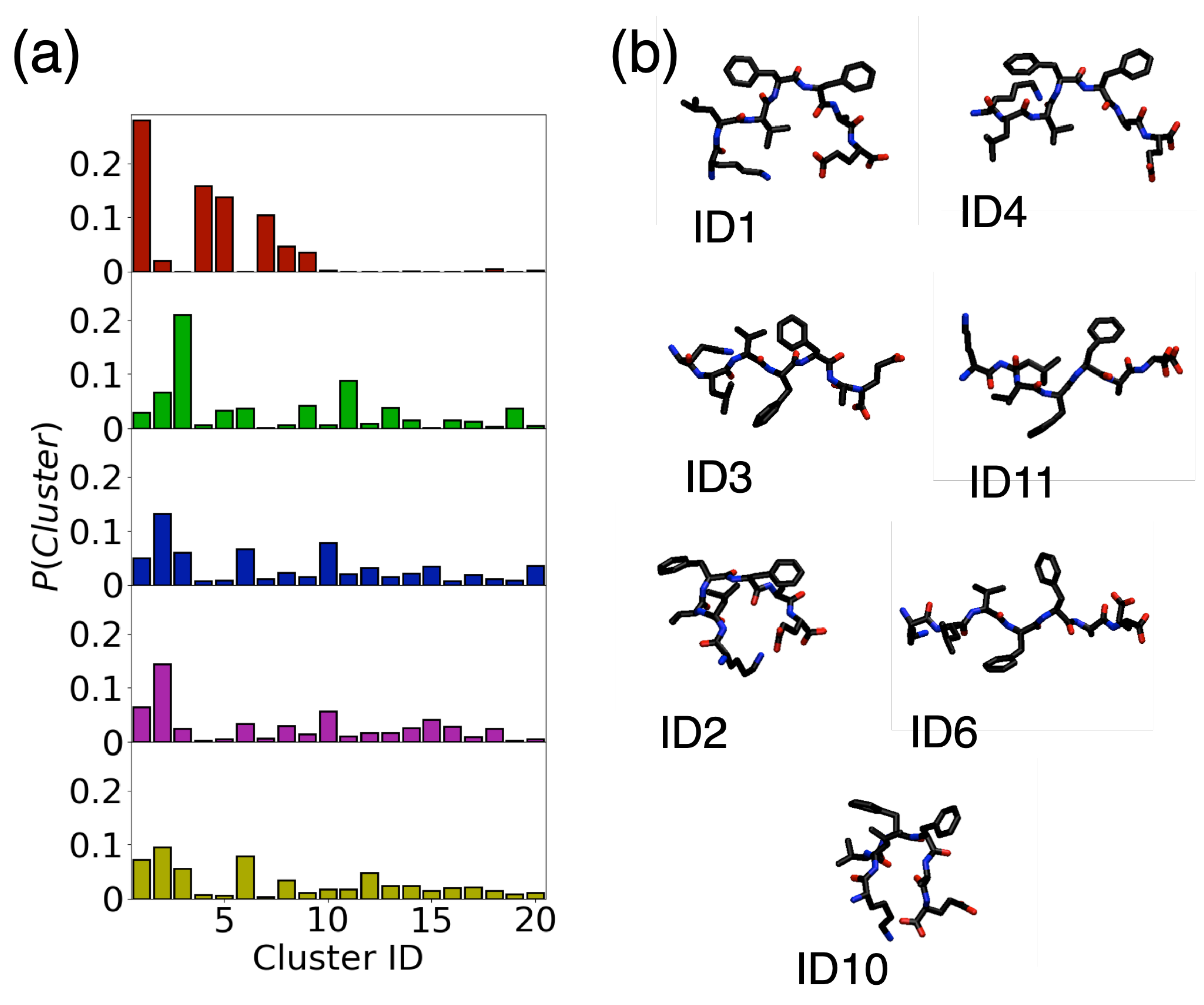
3.2. Conformation of Aβ(16–22) on Metal Surfaces
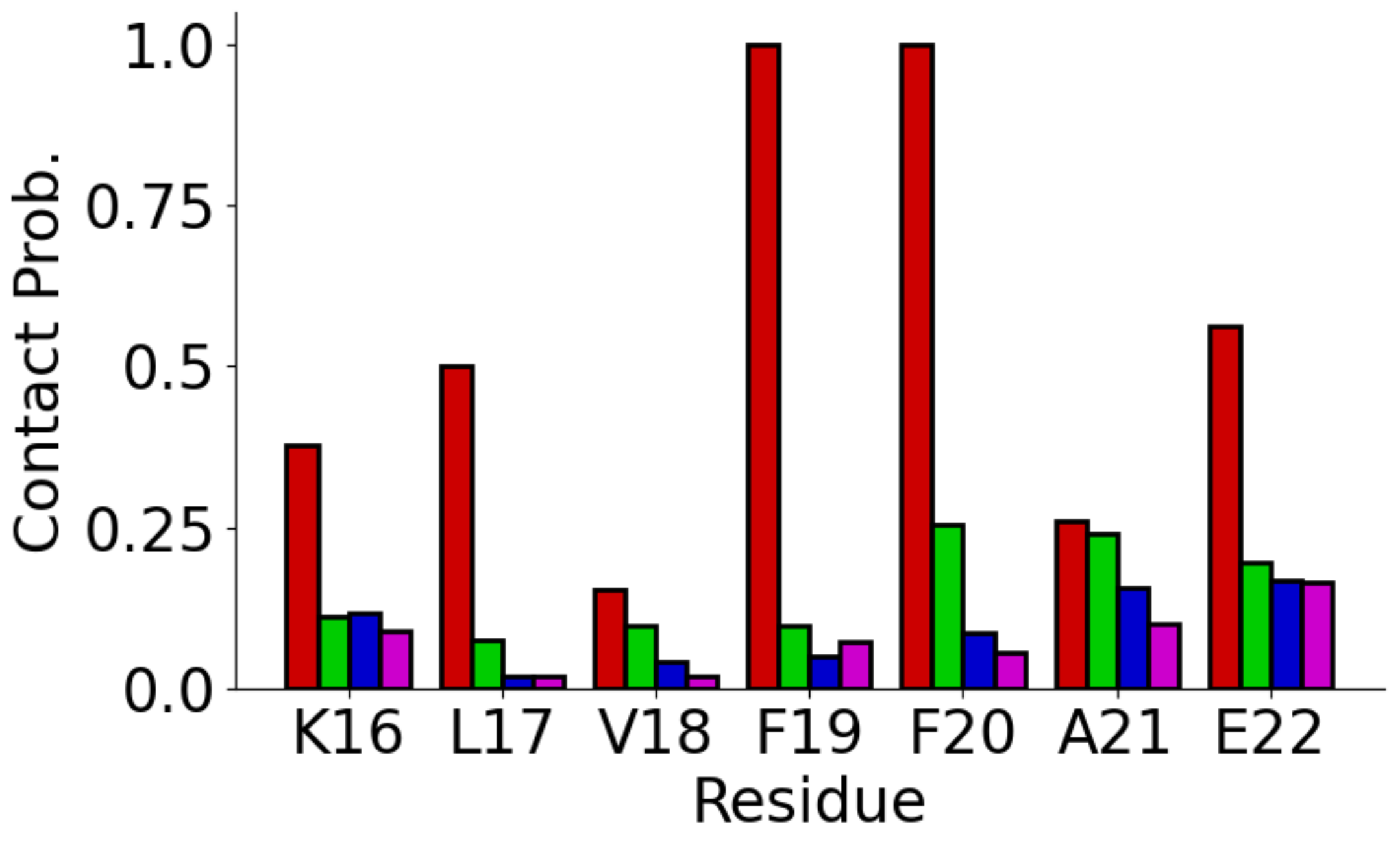
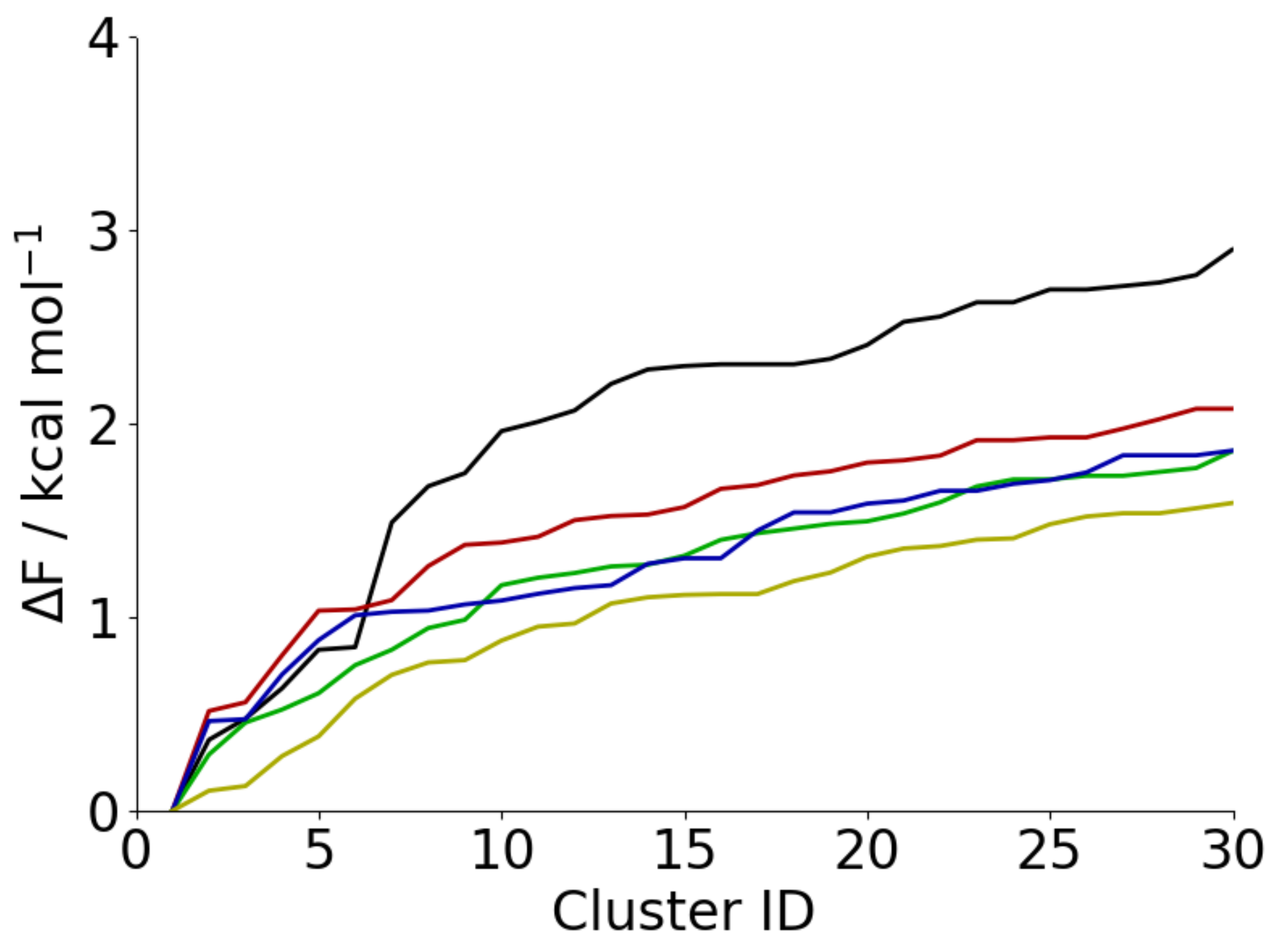
3.3. Adsorption onto Metal Surfaces Affects the Conformational Ensemble of Aβ(16–22)
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
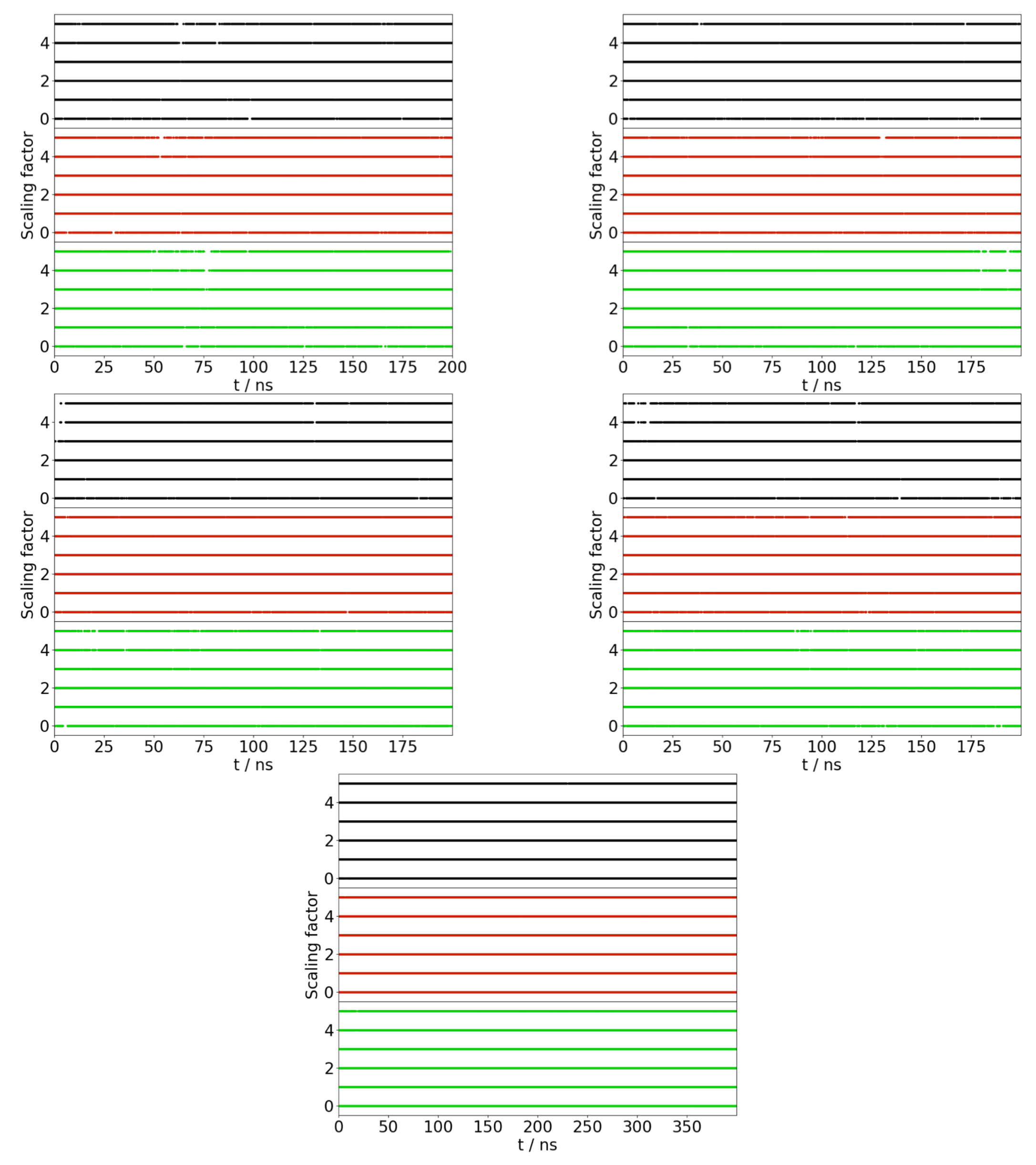
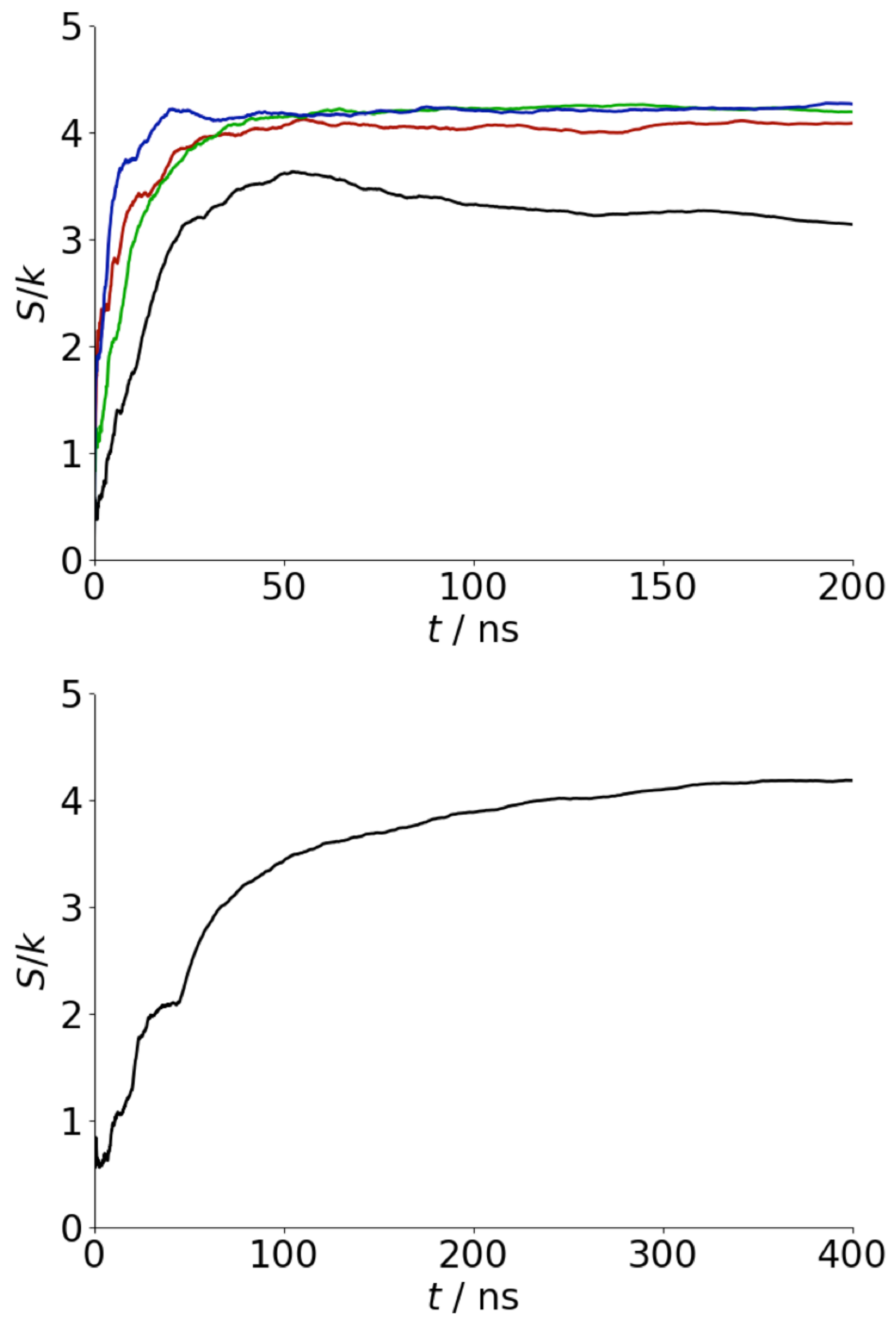
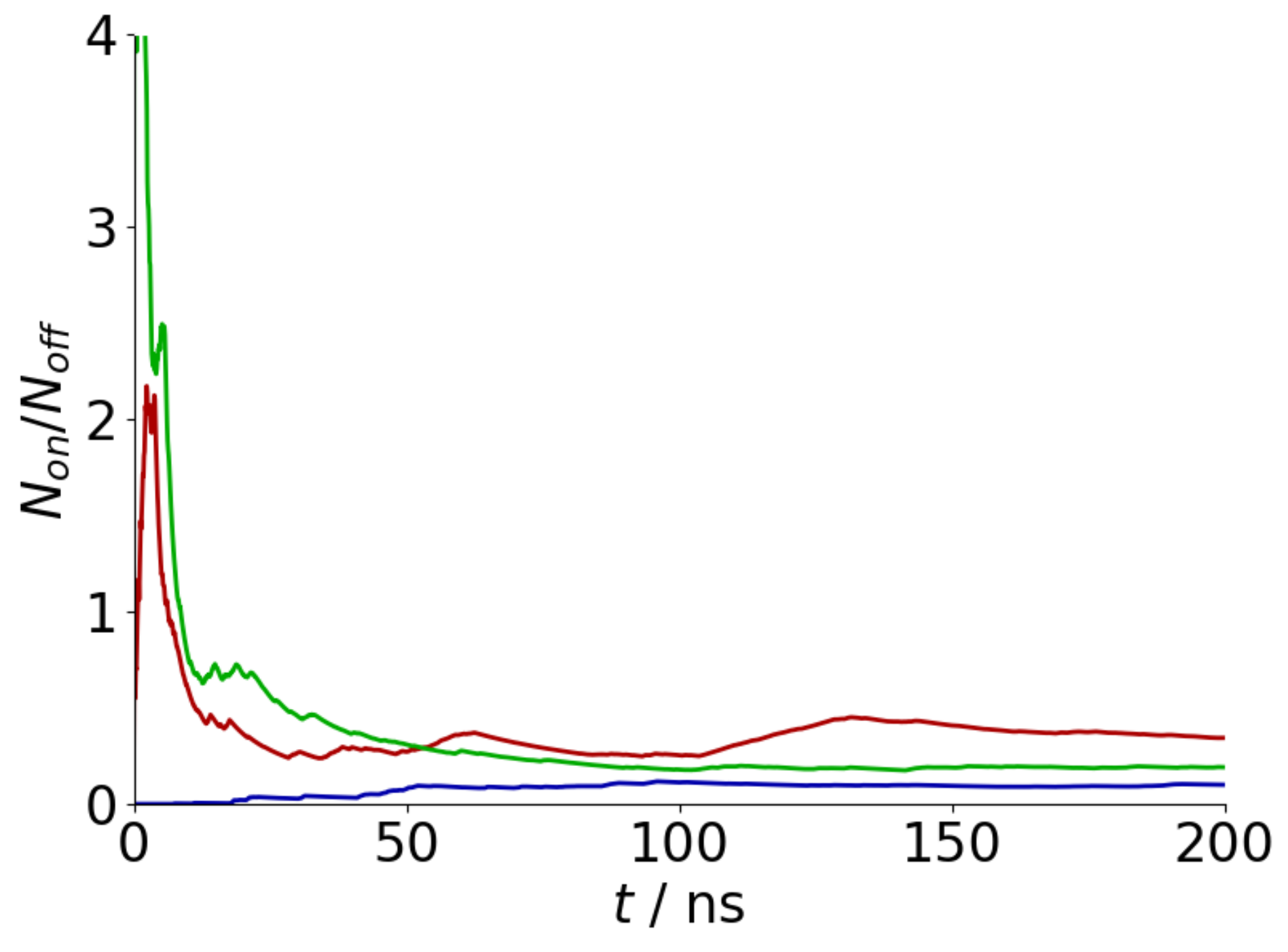
Appendix A. Convergence of REST Simulations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | 0 ↔ 1 | 1 ↔ 2 | 2 ↔ 3 | 3 ↔ 4 | 4 ↔ 5 |
|---|---|---|---|---|---|
| Au111 | 0.262 | 0.283 | 0.330 | 0.326 | 0.309 |
| Au100 | 0.298 | 0.309 | 0.331 | 0.361 | 0.355 |
| Ag111 | 0.308 | 0.307 | 0.328 | 0.356 | 0.357 |
| Ag100 | 0.284 | 0.297 | 0.295 | 0.324 | 0.339 |
| Solution | 0.312 | 0.300 | 0.317 | 0.363 | 0.359 |
References
- Iadanza, M.G.; Jackson, M.P.; Hewitt, E.W.; Ranson, N.A.; Radford, S.E. A new era for understanding amyloid structures and disease. Nat. Rev. Mol. Cell. Biol. 2018, 19, 755–773. [Google Scholar] [CrossRef] [PubMed]
- Ke, P.C.; Zhou, R.; Serpell, L.C.; Riek, R.; Knowles, T.P.J.; Lashuel, H.A.; Gazit, E.; Hamley, I.W.; Davis, T.P.; Fandrich, M.; et al. Half a century of amyloids: Past, present and future. Chem. Soc. Rev. 2020, 49, 5473–5509. [Google Scholar] [CrossRef] [PubMed]
- Maresova, P.; Mohelska, H.; Dolejs, J.; Kuca, K. Socio-economic Aspects of Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 12, 903–911. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Gurry, T.; Cheng, A.A.; Downey, J.; Deng, Z.; Stultz, C.M.; Lu, T.K. Strong underwater adhesives made by self-assembling multi-protein nanofibres. Nat. Nanotech. 2014, 9, 858–866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erskine, E.E.; MacPhee, C.E.; Stanley-Wall, N.R. Functional Amyloid and Other Protein Fibres in the Biofilm Matrix. J. Mol. Biol. 2018, 430, 3642–3656. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.J.; Mezzenga, R. Amyloid Fibrils as Building Blocks for Natural and Artificial Functional Materials. Adv. Mater. 2016, 28, 6546–6561. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Qin, R.; Liu, R.; Miao, S.; Yang, P. Functional amyloid materials at surfaces/interfaces. Biomater. Sci. 2018, 6, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Burke, K.A.; Yates, E.A.; Legleiter, J. Biophysical Insights into How Surfaces, Including Lipid Membranes, Modulate Protein Aggregation Related to Neurodegeneration. Front. Neurol. 2013, 4, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, A.; Grundmeier, G. Amyloid aggregation at solid-liquid interfaces: Perspectives of studies using model surfaces. Appl. Surf. Sci. 2020, 506, 144991. [Google Scholar] [CrossRef]
- Stefani, M. Generic Cell Dysfunction in Neurodegenerative Disorders: Role of Surfaces in Early Protein Misfolding, Aggregation, and Aggregate Cytotoxicity. Neuroscientist 2007, 13, 519–531. [Google Scholar] [CrossRef]
- Terzi, E.; Hozlemann, G.; Seelig, J. Interaction of Alzheimer beta-Amyloid Peptide(1-40) with Lipid Membranes. Biochemistry 1997, 36, 14845–14852. [Google Scholar] [CrossRef]
- Koppaka, V.; Axelsen, P.H. Accelerated Accumulation of Amyloid-beta Proteins on Oxidatively Damaged Lipid Membranes. Biochemistry 2000, 39, 10011–10016. [Google Scholar] [CrossRef]
- Bokvist, M.; Lindstrom, F.; Watts, A.; Grabner, G. Two Types of Alzheimer’s Amyloid (1-40) Peptide Membrane Interactions: Aggregation Preventing Transmembrane Anchoring Versus Accelerated Surface Fibril Formation. J. Mol. Biol. 2004, 335, 1039–1049. [Google Scholar] [CrossRef]
- Verdier, Y.; Zarandi, M.; Penke, B. Amyloid-beta peptide interactions with neuronal and glial cell plasma membrane: Binding sites and implications for Alzheimer’s disease. J. Pept. Sci. 2004, 10, 229–248. [Google Scholar] [CrossRef]
- Jayasinghe, S.A.; Langen, R. Lipid Membranes Modulate the Structure of Islet Amyloid Polypeptide. Biochemistry 2005, 44, 12113–12119. [Google Scholar] [CrossRef]
- Jean, L.; Lee, C.F.; Lee, C.; Shaw, M.; Vaux, D.J. Competing discrete interfacial effects are critical for amyloidogenesis. FASEB J. 2010, 24, 309–317. [Google Scholar] [CrossRef]
- Bolisetty, S.; Mezzenga, R. Amyloid-beta-carbon hybrid membranes for universal water purification. Nat. Nanotechnol. 2016, 11, 365–371. [Google Scholar] [CrossRef]
- Hauser, C.A.E.; Maurer-Stroh, S.; Martins, I.C. Amyloid-based nanosensors and nanodevices. Chem. Soc. Rev. 2014, 43, 5326–5345. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.; Bolisetty, S.; Cao, Y.; Handschin, S.; Adamcik, J.; Peng, Q.; Mezzenga, R. Selective and Efficient Removal of Fluoride from Water: In Situ Engineered Amyloid Fibril/ZrO2 Hybrid Membranes. Angew. Chem. Int. Ed. 2019, 58, 6012–6016. [Google Scholar] [CrossRef]
- Cedervall, T.; Lynch, I.; Lindman, S.; Berggard, T.; Thulin, E.; Nilsson, H.; Dawson, K.a.; Linse, S. Understanding the nanoparticle-protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc. Natl. Acad. Sci. USA 2007, 104, 2050–2055. [Google Scholar] [CrossRef] [Green Version]
- Cabaleiro-Lago, C.; Quinlan-Pluck, F.; Lynch, I.; Dawson, K.A.; Linse, S. Dual effect of amino modified polystyrene nanoparticles on amyloid-beta protein fibrillation. ACS Chem. Neurosci. 2010, 1, 279–287. [Google Scholar] [CrossRef]
- Gladytz, A.; Wagner, M.; Haupl, T.; Elsner, C.; Abel, B. Structure-Making Effects of Metal Nanoparticles in Amyloid Peptide Fibrillation. Part. Part. Syst. Charact. 2015, 32, 573–582. [Google Scholar] [CrossRef]
- Gladytz, A.; Abel, B.; Risselada, H.J. Gold-Induced Fibril Growth: The Mechanism of Surface-Facilitated Amyloid Aggregation. Angew. Chem. Int. Ed. 2016, 55, 11242–11246. [Google Scholar] [CrossRef]
- Gao, G.; Zhang, M.; Gong, D.; Chen, R.; Hu, X.; Sun, T. The size-effect of gold nanoparticles and nanoclusters in the inhibition of amyloid-beta fibrillation. Nanoscale 2017, 9, 4107–4113. [Google Scholar] [CrossRef]
- John, T.; Gladytz, A.; Kubeil, C.; Martin, L.L.; Risselada, H.J.; Abel, B. Impact of nanoparticles on amyloid peptide and protein aggregation: A review with a focus on gold nanoparticles. Nanoscale 2018, 10, 20894–20913. [Google Scholar] [CrossRef]
- Arvizo, R.R.; Bhattacharyya, S.; Kudgus, R.A.; Giri, K.; Bhattacharya, R.; Mukherjee, P. Intrinsic therapeutic applications of noble metal nanoparticles: Past, present and future. Chem. Soc. Rev. 2012, 41, 2943–2970. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Du, Z.; Ma, S.; Liu, Y.; Li, D.; Huang, H.; Jiang, S.; Cheng, S.; Wu, W.; Zhang, K.; et al. Effects of green-synthesized silver nanoparticles on lung cancer cells in vitro and grown as xenograft tumors in vivo. Int. J. Nanomed. 2016, 11, 1879–1887. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Liu, Y. Multifunctional platinum-based nanoparticles for biomedical applications. WIREs Nanomed. Nanobiotechnol. 2017, 9, e1410. [Google Scholar] [CrossRef]
- Pedone, D.; Moglianetti, M.; Luca, E.D.; Bardi, G.; Paolo-Pompa, P. Platinum nanoparticles in nanobiomedicine. Chem. Soc. Rev. 2017, 46, 4951–4975. [Google Scholar] [CrossRef]
- Puja, P.; Kumar, P. A perspective on biogenic synthesis of platinum nanoparticles and their biomedical applications. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2019, 211, 94–99. [Google Scholar] [CrossRef]
- Sonawane, S.K.; Ahmad, A.; Chinnathambi, S. Protein-Capped Metal Nanoparticles Inhibit Tau Aggregation in Alzheimer‚ Äôs Disease. ACS Omega 2019, 4, 12833–12840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppage, R.; Slocik, J.M.; Briggs, B.D.; Frenkel, A.I.; Heinz, H.; Naik, R.R.; Knecht, M.R. Crystallographic Recognition Controls Peptide Binding for Bio-Based Nanomaterials. J. Am. Chem. Soc. 2011, 133, 12346–12349. [Google Scholar] [CrossRef] [PubMed]
- Hughes, Z.E.; Kochandra, R.; Walsh, T.R. Facet-Specific Adsorption of Tripeptides at Aqueous Au Interfaces: Open Questions in Reconciling Experiment and Simulation. Langmuir 2017, 33, 3742–3754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, J.; Colombi Ciacchi, L. Specific material recognition by small peptides mediated by the interfacial solvent structure. J. Am. Chem. Soc. 2012, 134, 2407–2413. [Google Scholar] [CrossRef]
- Dong, L.; Luo, Q.; Cheng, K.; Shi, H.; Wang, Q.; Weng, W.; Han, W.Q. Facet-Specific Assembly of Proteins on SrTiO3 Polyhedral Nanocrystals. Sci. Rep. 2014, 4, 5084. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Huang, Y. Morphology-Controlled Synthesis of Platinum Nanocrystals with Specific Peptides. Adv. Mater. 2010, 22, 1921–1925. [Google Scholar] [CrossRef]
- Chiu, C.Y.; Li, Y.; Ruan, L.; Ye, X.; Murray, C.B.; Huang, Y. Platinum nanocrystals selectively shaped using facet-specific peptide sequences. Nat. Chem. 2011, 3, 393–399. [Google Scholar] [CrossRef]
- Wang, S.T.; Lin, Y.; Todorova, N.; Xu, Y.; Mazo, M.; Rana, S.; Leonardo, V.; Amdursky, N.; Spicer, C.D.; Alexander, B.D.; et al. Facet-Dependent Interactions of Islet Amyloid Polypeptide with Gold Nanoparticles: Implications for Fibril Formation and Peptide-Induced Lipid Membrane Disruption. Chem. Mater. 2017, 29, 1550–1560. [Google Scholar] [CrossRef]
- Shen, L.; Adachi, T.; Vanden Bout, D.; Zhu, X.Y. A Mobile Precursor Determines Amyloid-beta Peptide Fibril Formation at Interfaces. J. Am. Chem. Soc. 2012, 134, 14172–14178. [Google Scholar] [CrossRef]
- Keller, A.; Fritzsche, M.; Yu, Y.P.; Liu, Q.; Li, Y.M.; Dong, M.; Besenbacher, F. Influence of hydrophobicity on the surface-catalyzed assembly of the islet amyloid polypeptide. ACS Nano 2011, 5, 2770–2778. [Google Scholar] [CrossRef]
- Hajiraissi, R.; Giner, I.; Grundmeier, G.; Keller, A. Self-Assembly, Dynamics, and Polymorphism of hIAPP(20-29) Aggregates at Solid-Liquid Interfaces. Langmuir 2016, 33, 372–381. [Google Scholar] [CrossRef]
- Hajiraissi, R.; Hanke, M.; Yang, Y.; Duderija, B.; Gonzalez Orive, A.; Grundmeier, G.; Keller, A. Adsorption and Fibrillization of Islet Amyloid Polypeptide at Self-Assembled Monolayers Studied by QCM-D, AFM, and PM-IRRAS. Langmuir 2018, 34, 3517–3524. [Google Scholar] [CrossRef]
- Ando, T. High-speed AFM imaging. Curr. Opin. Struct. Biol. 2014, 28, 63–68. [Google Scholar] [CrossRef]
- Hosseinpour, S.; Roeters, S.J.; Bonn, M.; Peukert, W.; Woutersen, S.; Weidner, T. Structure and Dynamics of Interfacial Peptides and Proteins from Vibrational Sum-Frequency Generation Spectroscopy. Chem. Rev. 2020, 120, 3420–3465. [Google Scholar] [CrossRef]
- Ozboyaci, M.; Kokh, D.B.; Corni, S.; Wade, R.C. Modeling and simulation of protein-surface interactions: Achievements and challenges. Q. Rev. Biophys. 2016, 49, 1–45. [Google Scholar] [CrossRef] [Green Version]
- Walsh, T.R. Pathways to Structure-Property Relationships of Peptide-Materials Interfaces: Challenges in Predicting Molecular Structures. Acc. Chem. Res. 2017, 50, 1617–1624. [Google Scholar] [CrossRef] [Green Version]
- Bellucci, L.; Ardèvol, A.; Parrinello, M.; Lutz, H.; Lu, H.; Weidner, T.; Corni, S. The interaction with gold suppresses fiber-like conformations of the amyloid beta (16–22) peptide. Nanoscale 2016, 8, 8737–8748. [Google Scholar] [CrossRef] [Green Version]
- Bellucci, L.; Bussi, G.; Di Felice, R.; Corni, S. Fibrillation-prone conformations of the amyloid-beta-42 peptide at the gold/water interface. Nanoscale 2017, 9, 2279–2290. [Google Scholar] [CrossRef]
- Tavanti, F.; Pedone, A.; Menziani, M.C. Disclosing the Interaction of Gold Nanoparticles with Abeta(1-40) Monomers through Replica Exchange Molecular Dynamics Simulations. Int. J. Mol. Sci. 2020, 22, 26. [Google Scholar] [CrossRef]
- Cheung, D.L. Effect of surface chemistry on islet amyloid polypeptide conformation. Biointerphases 2020, 15, 051001. [Google Scholar] [CrossRef]
- Brancolini, G.; Corazza, A.; Vuano, M.; Fogolari, F.; Mimmi, M.C.; Bellotti, V.; Stoppini, M.; Corni, S.; Esposito, G. Probing the Influence of Citrate-Capped Gold Nanoparticles on an Amyloidogenic Protein. ACS Nano 2015, 9, 2600–2613. [Google Scholar] [CrossRef]
- Earl, D.J.; Deem, M.W. Parallel tempering: Theory, applications, and new perspectives. Phys. Chem. Chem. Phys. 2005, 7, 3910–3916. [Google Scholar] [CrossRef] [Green Version]
- Laio, A.; Gervasio, F.L. Metadynamics: A method to simulate rare events and reconstruct the free energy in biophysics, chemistry and material science. Rep. Prog. Phys. 2008, 71, 126601. [Google Scholar] [CrossRef]
- Heinz, H.; Jha, K.C.; Luettmer-Strathmann, J.; Farmer, B.L.; Naik, R.R. Polarization at metal‚ Äìbiomolecular interfaces in solution. J. R. Soc. Interface 2011, 8, 220–232. [Google Scholar] [CrossRef]
- Iori, F.; Corni, S. Including image charge effects in the molecular dynamics simulations of molecules on metal surfaces. J. Comput. Chem. 2008, 29, 1656–1666. [Google Scholar] [CrossRef]
- Corni, S. Reply to ‘Molecular mechanics models for the image charge’. J. Comput. Chem. 2017, 38, 2130–2133. [Google Scholar] [CrossRef] [Green Version]
- Palafox-Hernandez, J.P.; Tang, Z.; Hughes, Z.E.; Li, Y.; Swihart, M.T.; Prasad, P.N.; Walsh, T.R.; Knecht, M.R. Comparative Study of Materials-Binding Peptide Interactions with Gold and Silver Surfaces and Nanostructures: A Thermodynamic Basis for Biological Selectivity of Inorganic Materials. Chem. Mater. 2014, 26, 4960–4969. [Google Scholar] [CrossRef]
- Wright, L.B.; Palafox-Hernandez, J.P.; Rodger, P.M.; Corni, S.; Walsh, T.R. Facet selectivity in gold binding peptides: Exploiting interfacial water structure. Chem. Sci. 2015, 6, 5204–5214. [Google Scholar] [CrossRef] [Green Version]
- Balbach, J.J.; Ishii, Y.; Antzutkin, O.N.; Leapman, R.D.; Rizzo, N.W.; Dyda, F.; Reed, J.; Tycko, R. Amyloid Fibril Formation by Amyloid Beta-22, a Seven-Residue Fragment of the Alzheimer’s Beta-Amyloid Peptide, and Structural Characterization by Solid State NMR. Biochemistry 2000, 39, 13748–13759. [Google Scholar] [CrossRef]
- Perfilieva, O.A.; Pyshnyi, D.V.; Lomzov, A.A. Molecular Dynamics Simulation of Polarizable Gold Nanoparticles Interacting with Sodium Citrate. J. Chem. Theory Comput. 2019, 15, 1278–1292. [Google Scholar] [CrossRef]
- Wright, L.B.; Rodger, P.M.; Walsh, T.R.; Corni, S. First-principles-based force field for the interaction of proteins with Au(100)(5 × 1): An extension of GolP-CHARMM. J. Phys. Chem. C 2013, 117, 24292–24306. [Google Scholar] [CrossRef]
- Hughes, Z.E.; Wright, L.B.; Walsh, T.R. Biomolecular Adsorption at Aqueous Silver Interfaces: First-Principles Calculations, Polarizable Force-Field Simulations, and Comparisons with Gold. Langmuir 2013, 29, 13217–13229. [Google Scholar] [CrossRef] [PubMed]
- MacKerell, A.D.; Bashford, D.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; Joseph-McCarthy, D.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Bjelkmar, P.; Larsson, P.; Cuendet, M.A.; Hess, B.; Lindahl, E. Implementation of the {CHARMM} Force Field in {GROMACS}: Analysis of Protein Stability Effects from Correction Maps, Virtual Interaction Sites, and Water Models. J. Chem. Theory Comput. 2010, 6, 459–466. [Google Scholar] [CrossRef]
- Piana, S.; Lindorff-Larsen, K.; Shaw, D. How Robust Are Protein Folding Simulations with Respect to Force Field Parameterization? Biophys. J. 2011, 100, L47–L49. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Man, V.H.; He, X.; Derreumaux, P.; Ji, B.; Xie, X.Q.; Nguyen, P.H.; Wang, J. Effects of All-Atom Molecular Mechanics Force Fields on Amyloid Peptide Assembly: The Case of Abeta 16-22 Dimer. J. Chem. Theory Comput. 2019, 15, 1440–1452. [Google Scholar] [CrossRef]
- Wright, L.B.; Rodger, P.M.; Corni, S.; Walsh, T.R. GolP-CHARMM: First-principles based force fields for the interaction of proteins with Au(111) and Au(100). J. Chem. Theory Comput. 2013, 9, 1616–1630. [Google Scholar] [CrossRef]
- Liu, P.; Kim, B.; Friesner, R.A.; Berne, B.J. Replica exchange with solute tempering: A method for sampling biological systems in explicit water. Proc. Natl. Acad. Sci. USA 2005, 102, 13749–13754. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Friesner, R.A.; Berne, B.J. Replica exchange with solute scaling: A more efficient version of replica exchange with solute tempering (REST2). J. Phys. Chem. B 2011, 115, 9431–9438. [Google Scholar] [CrossRef] [Green Version]
- Sugita, Y.; Okamoto, Y. Replica exchange molecular dynamics method for protein folding simulation. Chem. Phys. Lett. 1999, 314, 141–151. [Google Scholar] [CrossRef]
- Samantray, S.; Cheung, D.L. Effect of the air-water interface on the conformation of amyloid beta. Biointerphases 2020, 15, 061011. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G. Hamiltonian replica exchange in GROMACS: A flexible implementation. Mol. Phys. 2013, 112, 379–384. [Google Scholar] [CrossRef] [Green Version]
- Daura, X.; Gademann, K.; Jaun, B.; Seebach, D.; Van Gunsteren, W.F.; Mark, A.E. Peptide Folding: When Simulation Meets Experiment. Angew. Chem. Int. Ed. 1999, 38, 236–240. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A Toolkit for the Analysis of MolecularDynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Dalgicdir, C.; Sensoy, O.; Peter, C.; Sayar, M. A transferable coarse-grained model for diphenylalanine: How to represent an environment driven conformational transition. J. Chem. Phys. 2013, 139, 234115. [Google Scholar] [CrossRef] [Green Version]
- Hajiraissi, R.; Hanke, M.; Gonzalez Orive, A.; Duderija, B.; Hofmann, U.; Zhang, Y.; Grundmeier, G.; Keller, A. Effect of Terminal Modifications on the Adsorption and Assembly of hIAPP(20–29). ACS Omega 2019, 4, 2649–2660. [Google Scholar] [CrossRef]
- Colletier, J.P.; Laganowsky, A.; Landau, M.; Zhao, M.; Soriaga, A.B.; Goldschmidt, L.; Flot, D.; Cascio, D.; Sawaya, M.R.; Eisenberg, D. Molecular basis for amyloid- polymorphism. Proc. Natl. Acad. Sci. USA 2011, 108, 16938–16943. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Palafox-Hernandez, J.P.; Law, W.C.; Hughes, Z.E.; Swihart, M.T.; Prasad, P.N.; Knecht, M.R.; Walsh, T.R. Biomolecular Recognition Principles for Bionanocombinatorics: An Integrated Approach To Elucidate Enthalpic and Entropic Factors. ACS Nano 2013, 7, 9632–9646. [Google Scholar] [CrossRef]
- Koshland, D.E. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Ma, B.; Tsai, C.J.; Sinha, N.; Nussinov, R. Folding and binding cascades: Dynamic landscapes and population shifts. Protein Sci. 2008, 9, 10–19. [Google Scholar] [CrossRef]
- Barnard, A.S.; Chen, Y. Kinetic modelling of the shape-dependent evolution of faceted gold nanoparticles. J. Mater. Chem. 2011, 21, 12239. [Google Scholar] [CrossRef]
| Surface | /kJ mol |
|---|---|
| Au111 | −188 ± 28 |
| Au100 | −57 ± 48 |
| Ag111 | −26 ± 26 |
| Ag100 | −16 ± 10 |
| /Å | Hydrogen Bonds | |
|---|---|---|
| Au111 | 13.74 ± 3.3 | K16-E22 (0.693) |
| Au100 | 15.4 ± 4.1 | K16-E22 (0.211) |
| Ag111 | 14.2 ± 4.4 | K16-E22 (0.231), L17-F20 (0.202) |
| Ag100 | 13.6 ± 4.2 | K16-E22 (0.296), L17-F20 (0.228), V18-F20 (0.178) |
| Solution | 15.2 ± 4.1 | K16-E22 (0.128), L17-F20 (0.109) |
| Au111 | 93 | 2.54 ± 0.02 |
| Au100 | 157 | 3.54 ± 0.15 |
| Ag111 | 156 | 3.72 ± 0.05 |
| Ag100 | 163 | 3.84 ± 0.08 |
| Solution | 260 | 4.18 ± 0.02 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Somers, K.P.; Cheung, D.L. The Amyloidogenic Peptide Amyloid Beta(16–22) Displays Facet Dependent Conformation on Metal Surfaces. Biophysica 2022, 2, 135-153. https://doi.org/10.3390/biophysica2020015
Somers KP, Cheung DL. The Amyloidogenic Peptide Amyloid Beta(16–22) Displays Facet Dependent Conformation on Metal Surfaces. Biophysica. 2022; 2(2):135-153. https://doi.org/10.3390/biophysica2020015
Chicago/Turabian StyleSomers, Kieran P., and David L. Cheung. 2022. "The Amyloidogenic Peptide Amyloid Beta(16–22) Displays Facet Dependent Conformation on Metal Surfaces" Biophysica 2, no. 2: 135-153. https://doi.org/10.3390/biophysica2020015
APA StyleSomers, K. P., & Cheung, D. L. (2022). The Amyloidogenic Peptide Amyloid Beta(16–22) Displays Facet Dependent Conformation on Metal Surfaces. Biophysica, 2(2), 135-153. https://doi.org/10.3390/biophysica2020015