
Amyloid-β Oligomers: Multiple Moving Targets


Abstract
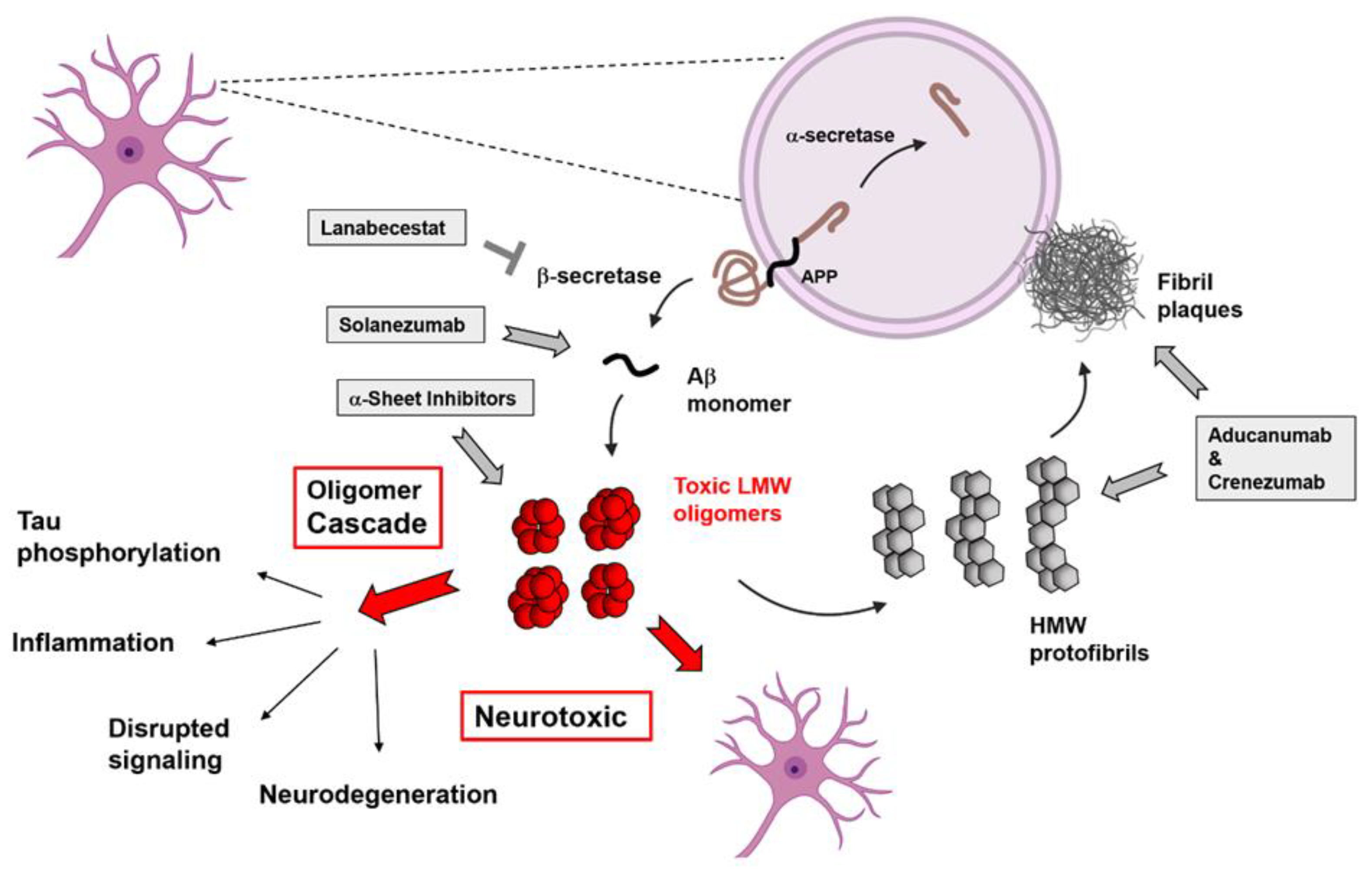
:1. Introduction
2. Aggregation and Characterization
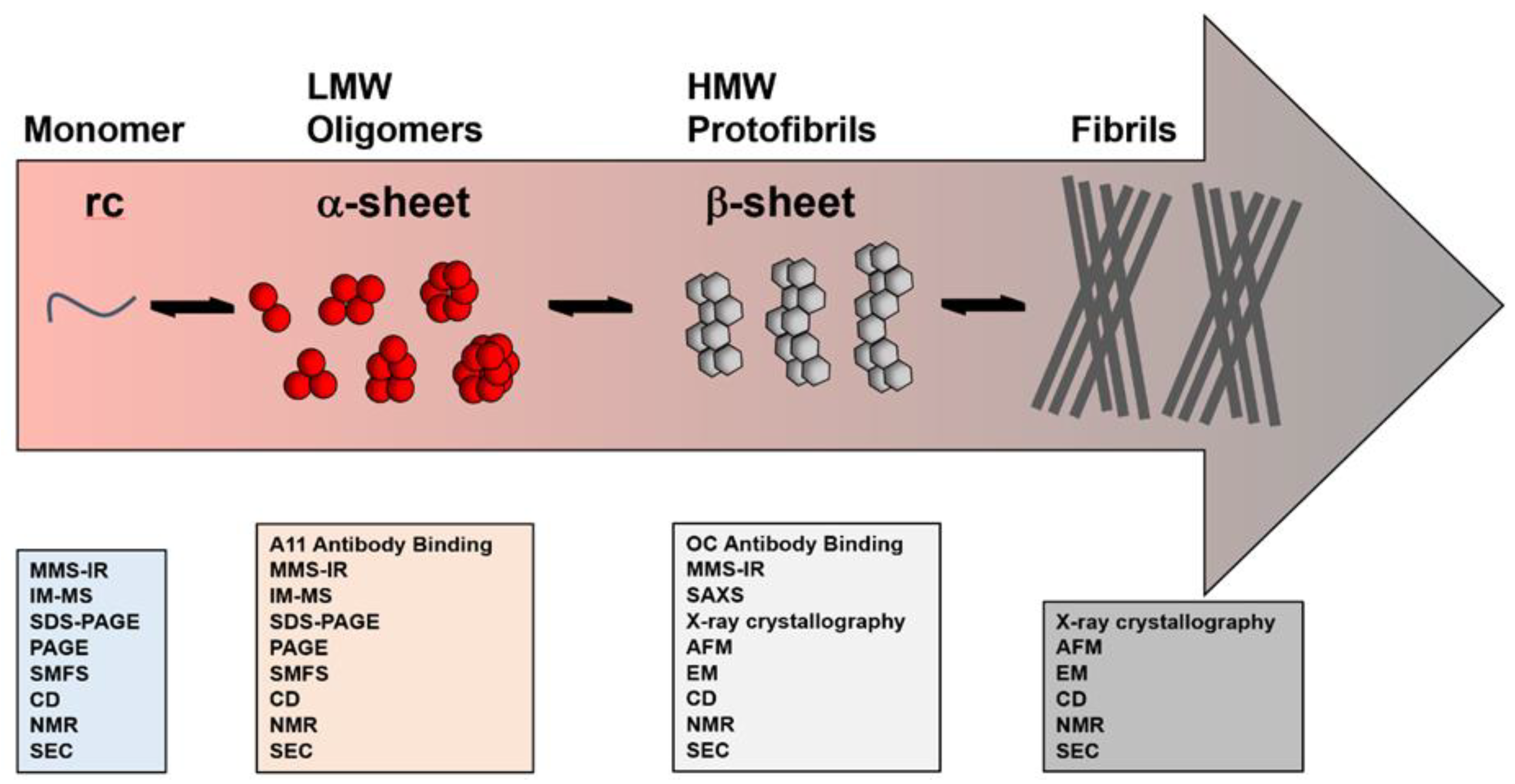
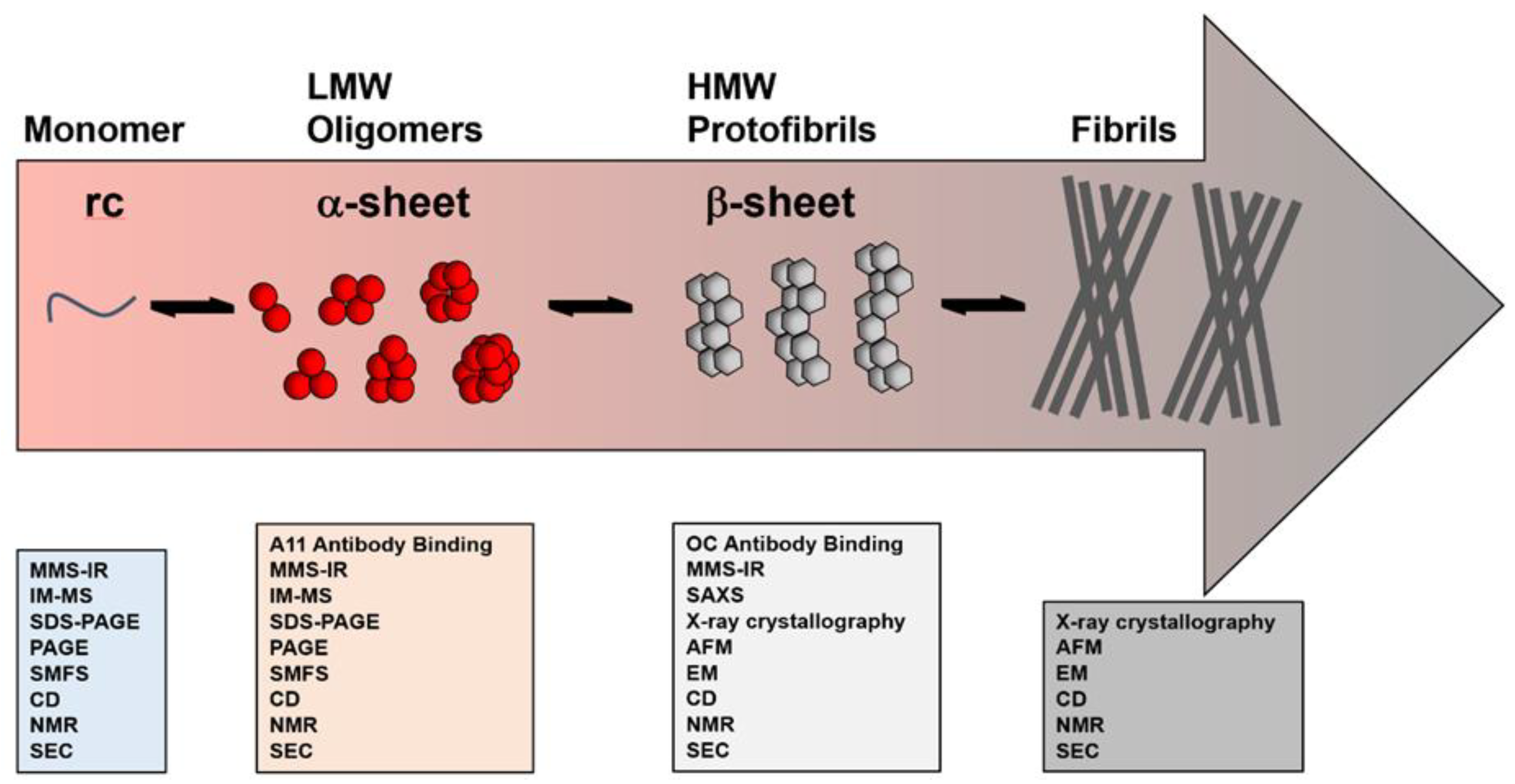
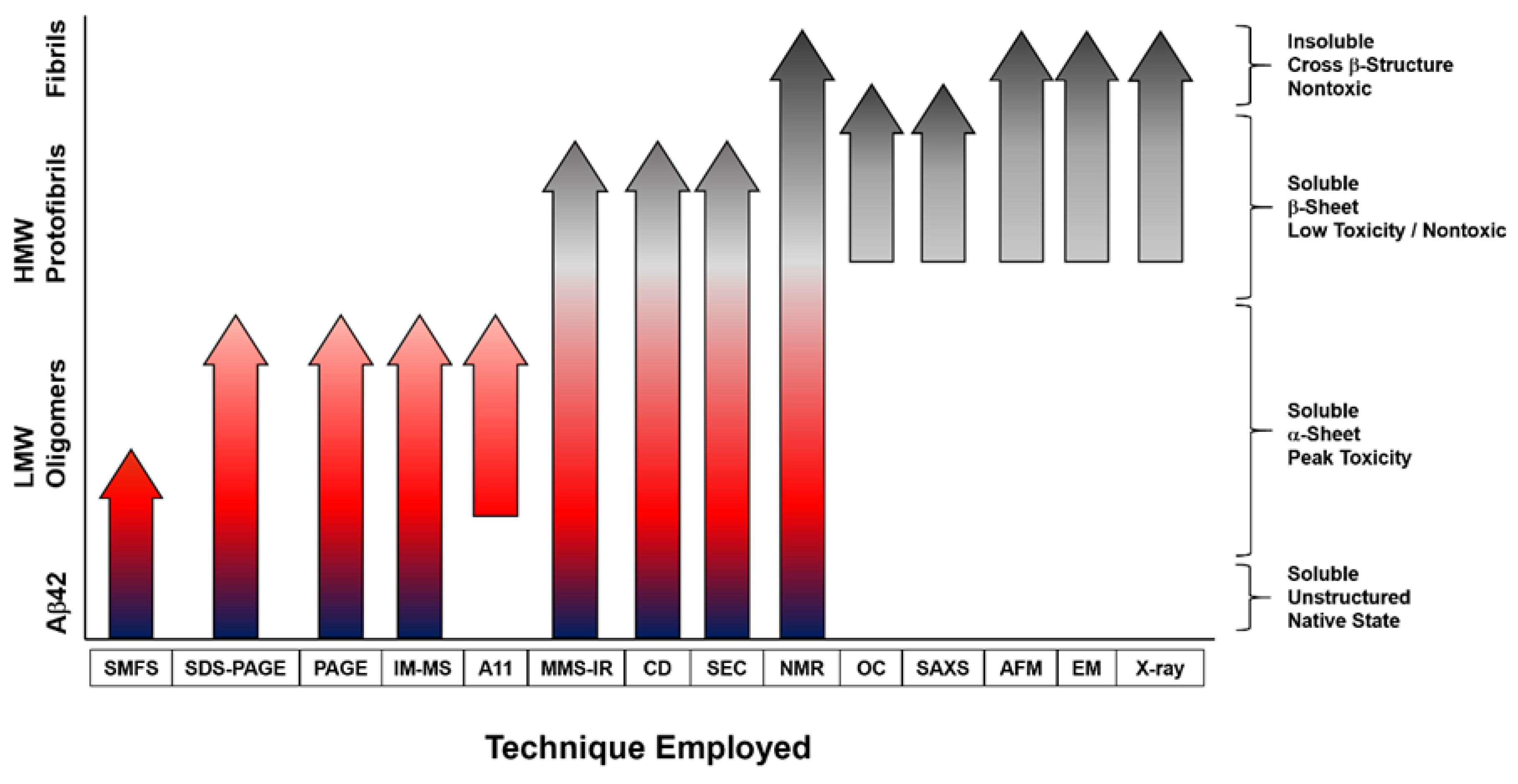
2.1. Characterization Based on Size
2.2. Conformational Insights Provide a Basis for Toxicity
2.3. Atomic Details of Toxic and Nontoxic Oligomer Conformations
2.4. Toward Characterization of Brain-Derived Oligomers
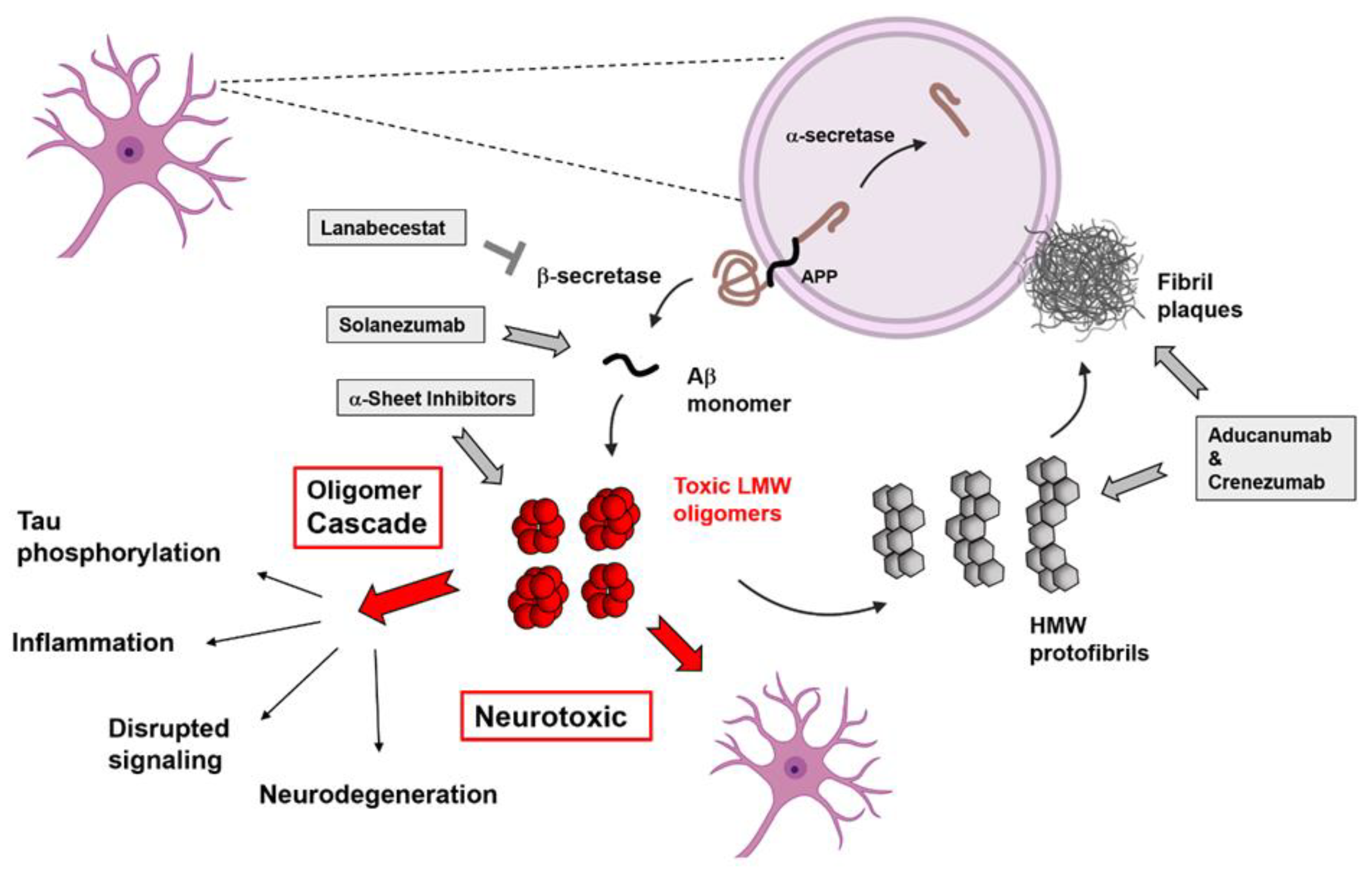
3. Mechanisms of Toxicity by Oligomers
3.1. Membrane Interactions Foster Aβ Toxicity
3.2. Intracellular Effects of Aβ Oligomers
4. Aβ-Based AD Treatments in Clinical Trials
5. Conclusions and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviation | Term |
| AD | Alzheimer’s Disease |
| Ab | Amyloid beta peptide |
| APP | Amyloid Precursor Protein |
| AFM | Atomic Force Microscopy |
| BACE Inhibitor | Beta-secretase inhibitor |
| CD | Circular Dichroism Spectroscopy |
| EM | Electron Microscopy |
| ER | Endoplasmic Reticulum |
| eNMDAR | Extrasynaptic N-methyl-D-aspartate Receptors |
| FTIR | Fourier Transform IR |
| Forster Resonance energy transfer | FRET |
| HMW | High Molecular Weight |
| IM-MS | Ion-Mobility Separation-Mass Spectrometry |
| LTP | Long Term Potentiation |
| LMW | Low Molecular Weight |
| MMS-IR | Microfluidic Modulation Spectroscopy |
| MCI | Mild Cognitive Impairment |
| MD | Molecular Dynamics |
| NMR | Nuclear Magentic Resonance Spectroscopy |
| NOEs | Nuclear Overhauser Effect Cross Peaks |
| PAGE | Polyacrylamide Gel Electrophoresis |
| PET | Positron Emission Tomography |
| SEC | Size Exclusion Chromatography |
| SAXS | Small-angle X-ray Scattering |
| SDS-PAGE | Sodium Dodecyl Sulfate Polycacrylamide Gel Electrophoresis |
References
- Alzheimer’s Association. Alzheimer’s Disease Facts and Figures. 2022. Available online: https://www.alz.org/alzheimers-dementia/facts-figures (accessed on 27 April 2022).
- Masters, C.L. Alzheimer’s disease. Nat. Rev. Dis. Primers 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, M.L.; Caraci, F.; De Bona, P.; Pappalardo, G.; Nicoletti, F.; Rizzarelli, E.; Copani, A. The monomer state of β-amyloid: Where the Alzheimer’s disease protein meets physiology. Rev. Neurosci. 2010, 21, 83–93. [Google Scholar] [CrossRef]
- Whitson, J.S.; Selkoe, D.J.; Cotman, C.W. Amyloid β Protein Enhances the Survival of Hippocampal Neurons in Vitro. Science 1989, 243, 1488–1490. [Google Scholar] [CrossRef]
- Morley, J.E.; Farr, S.A.; Banks, W.A.; Johnson, S.N.; Yamada, K.A.; Xu, L. A physiological role for amyloid-β protein: Enhancement of learning and memory. J. Alzheimer’s Dis. 2010, 19, 441–449. [Google Scholar] [CrossRef]
- Bishop, G.M.; Robinson, S.R. Physiological roles of amyloid-β and implications for its removal in Alzheimer’s disease. Drugs Aging 2004, 21, 621–630. [Google Scholar] [CrossRef]
- Hiltunen, M.; van Groen, T.; Jolkkonen, J. Functional roles of amyloid-beta protein precursor and amyloid-beta peptides: Evidence from experimental studies. J. Alz. Dis. 2009, 18, 401–412. [Google Scholar]
- Koudinov, A.R.; Berezov, T.T. Alzheimer’s amyloid-β (Aβ) is an essential synaptic protein, not neurotoxic junk. Acta Neurobiol. Exp. 2004, 64, 71–79. [Google Scholar]
- Jaunmuktane, Z.; Mead, S.; Ellis, M.; Wadsworth, J.D.; Nicoll, A.J.; Kenny, J.; Launchbury, F.; Linehan, J.; Richard-Loendt, A.; Walker, A.S.; et al. Evidence for human transmission of amyloid-pathology and cerebral amyloid angiopathy. Nature 2015, 525, 247–250. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar] [CrossRef] [PubMed]
- Mullan, M.; Crawford, F.; Axelman, K.; Houlden, H.; Lilius, L.; Winblad, B.; Lannfelt, L. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat. Genet. 1992, 1, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glenner, G.G.; Wong, C.W.; Quaranta, V.; Eanes, E.D. The amyloid deposits in Alzheimer’s disease: Their nature and pathogenesis. Appl. Pathol. 1984, 2, 357–369. [Google Scholar]
- McLean, C.A.; Cherny, R.A.; Fraser, F.W.; Fuller, S.J.; Smith, M.J.; Beyreuther, K.; Bush, A.I.; Masters, C.L. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1999, 46, 860–866. [Google Scholar] [CrossRef]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef]
- Lambert, M.P.; Viola, K.L.; Chromy, B.A.; Chang, L.; Morgan, T.E.; Yu, J.; Venton, D.L.; Krafft, G.A.; Finch, C.E.; Klein, W.L. Vaccination with soluble Abeta oligomers generates toxicity-neutralizing antibodies. J. Neurochem. 2001, 79, 595–605. [Google Scholar] [CrossRef]
- Wang, J.; Dickson, D.W.; Trojanowski, J.Q.; Lee, V.M. The levels of soluble versus insoluble brain Abeta distinguish Alzheimer’s disease from normal and pathologic aging. Exp. Neurol. 1999, 158, 328–337. [Google Scholar] [CrossRef]
- Yang, T.; Li, S.; Xu, H.; Walsh, D.M.; Selkoe, D.J. Large Soluble Oligomers of Amyloid β-Protein from Alzheimer Brain Are Far Less Neuroactive Than the Smaller Oligomers to Which They Dissociate. J. Neurosci. 2016, 37, 152–163. [Google Scholar] [CrossRef]
- Lambert, M.P.; Barlow, A.K.; Chromy, B.A.; Edwards, C.; Freed, R.; Liosatos, M.; Morgan, T.E.; Rozovsky, I.; Trommer, B.; Viola, K.L.; et al. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA 1998, 95, 6448–6453. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, M.; Davis, J.; Aucoin, D.; Sato, T.; Ahuja, S.; Aimoto, S.; Elliott, J.I.; Van Nostrand, W.E.; Smith, S.O. Structural conversion of neurotoxic amyloid-β(1-42) oligomers to fibrils. Nat. Struct. Mol. Biol. 2010, 17, 561–567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Sakono, M.; Zako, T. Amyloid oligomers: Formation and toxicity of Abeta oligomers. FEBS J. 2010, 277, 1348–1358. [Google Scholar] [CrossRef]
- Zahs, K.R.; Ashe, K.H. β-Amyloid oligomers in aging and Alzheimer’s disease. Front. Aging Neurosci. 2013, 5, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsia, A.Y.; Masliah, E.; McConlogue, L.; Yu, G.Q.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Malenka, R.C.; Nicoll, R.A.; Mucke, L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. USA 1999, 96, 3228–3233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomiyama, T.; Nagata, T.; Shimada, H.; Teraoka, R.; Fukushima, A.; Kanemitsu, H.; Takuma, H.; Kuwano, R.; Imagawa, M.; Ataka, S.; et al. A new amyloid beta variant favoring oligomerization in Alzheimer’s-type dementia. Ann. Neurol. 2008, 63, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Jack Jr, C.R.; Knopman, D.S.; Jagust, W.J.; Petersen, R.C.; Weiner, M.W.; Aisen, P.S.; Shaw, L.M.; Vemuri, P.; Wiste, H.J.; Weigand, S.D.; et al. Tracking pathophysiological processes in Alzheimer’s disease: An updated hypothetical model of dynamic biomarkers. Neurology 2013, 12, 207–216. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Selkoe, D.J. A mechanistic hypothesis for the impairment of synaptic plasticity by soluble Abeta oligomers from Alzheimer’s brain. J. Neurochem. 2020, 154, 583–597. [Google Scholar] [CrossRef]
- Ashe, K.H. The biogenesis and biology of amyloid β-oligomers in the brain. Alz. Dement. 2020, 16, 1561–1567. [Google Scholar] [CrossRef]
- Mucke, L.; Masliah, E.; Yu, G.Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of Abeta (1–42) in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef] [Green Version]
- Klein, W.L. Synaptotoxic amyloid-β oligomers: A molecular basis for the cause, diagnosis, and treatment of Alzheimer’s disease? J. Alz. Dis. 2013, 33, S49–S65. [Google Scholar] [CrossRef] [PubMed]
- Leuzy, A.; Heurling, K.; Ashton, N.J.; Scholl, M.; Zimmer, E.R. In vivo detection of Alzheimer’s disease. Yale J. Biol. Med. 2018, 91, 291–300. [Google Scholar] [PubMed]
- Counts, S.E.; Ikonomovic, M.D.; Mercado, N.; Vega, I.E.; Mufson, E.J. Biomarkers for the early detection and progression of Alzheimer’s Disease. Neurotherapeutics 2017, 14, 35–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, J.L.; Morris, J.C. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann. Neurol. 1999, 45, 358–368. [Google Scholar] [CrossRef]
- Herukka, S.K.; Simonsen, A.H.; Andreasen, N.; Baldeiras, I.; Bjerke, M.; Blennow, K.; Engelborghs, S.; Frisoni, G.B.; Gabryelewicz, T.; Galluzzi, S.; et al. Recommendations for cerebrospinal fluid Alzheimer’s disease biomarkers in the diagnostic evaluation of mild cognitive impairment. Alz. Dem. 2017, 13, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Diniz, B.S.; Pinto Junior, J.A.; Forlenza, O.V. Do CSF total tau, phosphorylated tau, and beta-amyloid 42 help to predict progression of mild cognitive impairment to Alzheimer’s disease? A systematic review and meta-analysis of the literature. World J. Biol. Psychiatry 2008, 9, 172–182. [Google Scholar] [CrossRef]
- Ferreira, L.; Santos-Galduroz, R.F.; Ferri, C.P.; Fernandes Galduroz, J.C. Rate of cognitive decline in relation to sex after 60 years-of-age: A systematic review. Geratr. Gerontol. Int. 2014, 14, 23–31. [Google Scholar] [CrossRef]
- Ferreira, D.; Rivero-Santana, A.; Perestelo-Pérez, L.; Westman, E.; Wahlund, L.O.; Sarría, A.; Serrano-Aguilar, P. Improving CSF biomarkers’ performance for predicting progression from mild cognitive impairment to Alzheimer’s disease by considering different confounding factors: A metaanalysis. Front. Aging Neurosci. 2014, 6, 287. [Google Scholar] [CrossRef] [Green Version]
- Van Rossum, I.A.; Vos, S.; Handels, R.; Visser, P.J. Biomarkers as predictors for conversion from mild cognitive impairment to Alzheimer-type dementia: Implications for trial design. J. Alz. Dis. 2010, 20, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, C.; Smailagic, N.; Noel-Storr, A.H.; Takwoingi, Y.; Flicker, L.; Mason, S.E.; McShane, R. Plasma and cerebrospinal fluid amyloid beta for the diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane Database Syst. Rev. 2014, 6, 8782. [Google Scholar] [CrossRef] [PubMed]
- Noel-Storr, A.H.; Flicker, L.; Ritchie, C.W.; Nguyen, G.H.; Gupta, T.; Wood, P.; Walton, J.; Desai, M.; Solomon, D.F.; Molena, E.; et al. Systematic review of the body of evidence for the use of biomarkers in the diagnosis of dementia. Alz. Dement. 2013, 9, e96–e105. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Su, B.; Zheng, H.; Kim, J.R. A peptide probe for detection of various beta-amyloid oligomers. Mol. Biosyst. 2012, 8, 2741–2752. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhong, Y.; Gui, J.; Wang, X.; Zhuang, X.; Weng, J. A hydrogel biosensor for high selective and sensitive detection of amyloid-beta oligomers. Int. J. Nanomed. 2018, 13, 843–856. [Google Scholar] [CrossRef] [Green Version]
- Laske, C.; Sohrabi, H.R.; Frost, S.M.; López-de-Ipiña, K.; Garrard, P.; Buscema, M.; Dauwels, J.; Soekadar, S.R.; Mueller, S.; Linnemann, C.; et al. Innovative diagnostic tools for early detection of Alzheimer’s disease. Alz. Dement. 2015, 11, 561–578. [Google Scholar] [CrossRef]
- More, S.S.; Beach, J.M.; McClelland, C.; Mokhtarzadeh, A.; Vince, R. In vivo assessment of retinal biomarkers by hyperspectral imaging: Early detection of Alzheimer’s disease. ACS Chem. Neurosci. 2019, 10, 4492–4501. [Google Scholar] [CrossRef]
- Forlenza, O.V.; Radanovic, M.; Talib, L.L.; Aprahamian, I.; Diniz, B.S.; Zetterberg, H.; Gattaz, W.F. Cerebrospinal fluid biomarkers in Alzheimer’s disease: Diagnostic accuracy and prediction of dementia. Alz. Dem. Diagn. Assess. Dis. Monit. 2015, 1, 455–463. [Google Scholar] [CrossRef]
- Nabers, A.; Hafermann, H.; Wiltfang, J.; Gerwert, K. Abeta and tau structure-based biomarkers for a blood- and CSF-based two-step recruitment strategy to identify patients with dementia due to Alzheimer’s disease. Alz. Dem. Diagn. Assess. Dis. Monit. 2019, 11, 257–263. [Google Scholar] [CrossRef]
- Palmqvist, S.; Janelidze, S.; Quiroz, Y.T.; Zetterberg, H.; Lopera, F.; Stomrud, E.; Su, Y.; Chen, Y.; Serrano, G.E.; Leuzy, A.; et al. Discriminative accuracy of plasma phospho-tau217 for Alzheimer disease vs other neurodegenerative disorders. J. Am. Med. Assoc. 2020, 324, 772–781. [Google Scholar] [CrossRef]
- Barthelemy, N.R.; Horie, K.; Sato, C.; Bateman, R.J. Blood plasma phosphortylated-tau isoforms track CNS change in Alzheimer’s disease. J. Exp. Med. 2020, 217, 861. [Google Scholar] [CrossRef]
- Toledo, J.B.; Xia, S.X.; Trojanowski, J.Q.; Shaw, L.M. Longitudinal change in CSF Tau and Abeta biomarkers for up to 48 months in ADNI. Acta Neuropathol. 2013, 126, 659–670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchhave, P.; Minthon, L.; Zetterberg, H.; Wallin, A.K.; Blennow, K.; Hansson, O. Cerebrospinal fluid levels of beta-amyloid 1-42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch. Gen. Psychiatry 2012, 69, 98–106. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Wu, D.; Lambert, M.P.; Fernandez, S.J.; Velasco, P.T.; Lacor, P.N.; Bigio, E.H.; Jerecic, J.; Acton, P.J.; Shughrue, P.J.; et al. Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by Abeta oligomers. Neurobio. Aging 2008, 29, 1334–1347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amar, F.; Sherman, M.A.; Rush, T.; Larson, M.; Boyle, G.; Chang, L.; Götz, J.; Buisson, A.; Lesné, S.E. Amyloid-beta oligomer Aβ*56 induces specific alterations of tau phosphorylation and neuronal signaling. Sci. Signal. 2017, 10, eaal2021. [Google Scholar] [CrossRef] [Green Version]
- Zempel, H.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Abeta oligomers cause localized Ca2+ elevation, missorting of endogenous tau into dendrites, tau phosphorylation, and destruction of microtubules and spines. J. Neurosci. 2010, 30, 11938–11950. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, E.H.; La Joie, R.; Wolf, A.; Strom, A.; Wang, P.; Iaccarino, L.; Bourakova, V.; Cobigo, Y.; Heuer, H.; Spina, S.; et al. Diagnostic value of plasma phosphorylated tau181 in Alzheimer’s disease and frontotemporal lobar degeneration. Nat. Med. 2020, 26, 387–397. [Google Scholar] [CrossRef]
- Janelidze, S.; Stomrud, E.; Smith, R.; Palmqvist, S.; Mattsson, N.; Airey, D.C.; Proctor, N.K.; Chai, X.; Shcherbinin, S.; Sims, J.R.; et al. Cerebrospinal fluid p-tau217 performs better than p-tau181 as a biomarker of Alzheimer’s disease. Nat. Commun. 2020, 11, 1683. [Google Scholar] [CrossRef] [Green Version]
- Musiek, E.S.; Holtzman, D.M. Three dimensions of the amyloid hypothesis: Time, space and ‘wingmen’. Nat. Neurosci. 2015, 18, 800–806. [Google Scholar] [CrossRef]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef]
- Miller, Y.; Ma, B.; Nussinov, R. Polymorphism in Alzheimer Abeta amyloid organization reflects conformation selection in a rugged energy landscape. Chem. Rev. 2010, 110, 4820–4838. [Google Scholar] [CrossRef]
- Ono, K.; Condron, M.M.; Teplow, D.B. Structure-neurotoxicity relationships of amyloid beta-protein oligomers. Proc. Natl. Acad. Sci. USA 2009, 106, 14745–14750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesne, S.; Koh, M.T.; Kotilinek, L.; Kayed, R.; Glabe, C.G.; Yang, A.; Gallagher, M.; Ashe, K.H. A specific amyloid-beta protein assembly in the brain impairs memory. Nature 2006, 440, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Townsend, M.; Shankar, G.M.; Mehta, T.; Walsh, D.M.; Selkoe, D.J. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: A potent role for trimers. J. Physiol. 2006, 572, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [Green Version]
- Quist, A.; Doudevski, I.; Lin, H.; Azimova, R.; Ng, D.; Frangione, B.; Kagan, B.; Ghiso, J.; & Lal, R. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA 2005, 102, 10427–10432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, D.M.; Klyubin, I.; Fadeeva, J.V.; Cullen, W.K.; Anwyl, R.; Wolfe, M.S.; Rowan, M.J.; Selkoe, D.J. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar] [CrossRef]
- Wang, H.W.; Pasternak, J.F.; Kuo, H.; Ristic, H.; Lambert, M.P.; Chromy, B.; Viola, K.L.; Klein, W.L.; Stine, W.B.; Krafft, G.A.; et al. Soluble oligomers of beta amyloid (1-42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002, 924, 133–140. [Google Scholar] [CrossRef]
- Ehrnhoefer, D.E.; Bieschke, J.; Boeddrich, A.; Herbst, M.; Masino, L.; Lurz, R.; Engemann, S.; Pastore, A.; Wanker, E.E. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat. Struct. Mol. Biol. 2008, 15, 558–566. [Google Scholar] [CrossRef]
- Ladiwala, R.A.; Dordick, J.S.; Tessier, P.M. Aromatic small molecules remodel toxic soluble oligomers of amyloid beta through three independent pathways. J. Biol. Chem. 2011, 286, 3209–3218. [Google Scholar] [CrossRef] [Green Version]
- Glabe, C.G. Structural classification of toxic amyloid oligomers. J. Biol. Chem. 2008, 283, 29639–29643. [Google Scholar] [CrossRef] [Green Version]
- Roychaudhuri, R.; Yang, M.; Hoshi, M.M.; Teplow, D.B. Amyloid beta-protein assembly and Alzheimer disease. J. Biol. Chem. 2009, 284, 4749–4753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pujol-Pina, R.; Vilaprinyó-Pascual, S.; Mazzucato, R.; Arcella, A.; Vilaseca, M.; Orozco, M.; Carulla, N. SDS-PAGE analysis of Aβ oligomers is disserving research into Alzheimer’s disease: Appealing for ESI-IM-MS. Sci. Rep. 2015, 5, 14809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinoshita, A.; Fukumoto, H.; Shah, T.; Whelan, C.M.; Irizarry, M.C.; Hyman, B.T. Demonstration by FRET of BACE interaction with the amyloid precursor protein at the cell surface and in early endosomes. J. Cell Sci. 2003, 116, 3339–3346. [Google Scholar] [CrossRef] [Green Version]
- De Strooper, B. Proteases and proteolysis in Alzheimer disease: A multifactorial view on the disease process. Physiol. Rev. 2010, 90, 465–494. [Google Scholar] [CrossRef]
- Kumar, S.; Rezaei-Ghaleh, N.; Terwel, D.; Thal, D.R.; Richard, M.; Hoch, M.; Mc Donald, J.M.; Wüllner, U.; Glebov, K.; Heneka, M.T.; et al. Extracellular phosphorylation of the amyloid beta-peptide promotes formation of toxic aggregates during the pathogenesis of Alzheimer’s disease. EMBO J. 2011, 30, 2255–2265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarrett, J.T.; Berger, E.P.; Lansbury, J.P.T. The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer’s disease. Biochemistry 1993, 32, 4693–4697. [Google Scholar] [CrossRef]
- Portelius, E.; Andreasson, U.; Ringman, J.M.; Buerger, K.; Daborg, J.; Buchhave, P.; Hansson, O.; Harmsen, A.; Gustavsson, M.K.; Hanse, E.; et al. Distinct cerebrospinal fluid amyloid beta peptide signatures in sporadic and PSEN1 A431E-associated familial Alzheimer’s disease. Mol. Neurodegener. 2010, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Culyba, E.K.; Powers, E.T.; Kelly, J.W. Amyloid-beta forms fibrils by nucleated conformational conversion of oligomers. Nat. Chem. Biol. 2011, 7, 602–609. [Google Scholar] [CrossRef]
- Bernstein, S.L.; Dupuis, N.F.; Lazo, N.D.; Wyttenbach, T.; Condron, M.M.; Bitan, G.; Teplow, D.B.; Shea, J.E.; Ruotolo, B.T.; Robinson, C.V.; et al. Amyloid-beta protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer’s disease. Nat. Chem. 2009, 1, 326–331. [Google Scholar] [CrossRef] [Green Version]
- Economou, N.J.; Giammona, M.J.; Do, T.D.; Zheng, X.; Teplow, D.B.; Buratto, S.K.; Bowers, M.T. Amyloid beta-protein assembly and Alzheimer’s disease: Dodecamers of Aβ42, but not of Aβ40, seed fibril formation. J. Am. Chem. Soc. 2016, 138, 1772–1775. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.I.; Linse, S.; Luheshi, L.M.; Hellstrand, E.; White, D.A.; Rajah, L.; Otzen, D.E.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P. Proliferation of amyloid-β42 aggregates occurs through a secondary nucleation mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 9758–9763. [Google Scholar] [CrossRef] [Green Version]
- Bitan, G.; Fradinger, E.A.; Spring, S.M.; Teplow, D.B. Neurotoxic protein oligomers—What you see is not always what you get. Amyloid 2005, 12, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Hepler, R.W.; Grimm, K.M.; Nahas, D.D.; Breese, R.; Dodson, E.C.; Acton, P.; Keller, P.M.; Yeager, M.; Wang, H.; Shughrue, P.; et al. Solution state characterization of amyloid beta-derived diffusible ligands. Biochemistry 2006, 45, 15157–15167. [Google Scholar] [CrossRef] [PubMed]
- Shea, D.; Hsu, C.C.; Bi, T.M.; Paranjapye, N.; Childers, M.C.; Cochran, J.; Tomberlin, C.P.; Wang, L.; Paris, D.; Zonderman, J.; et al. α-sheet secondary structure in amyloid-peptide drives aggregation and toxicity in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2019, 118, 8895–8900. [Google Scholar] [CrossRef] [Green Version]
- Shea, D.; Colasurdo, E.; Smith, A.; Paschall, C.; Shofer, J.B.; Jayadev, S.; Keene, C.D.; Galasko, D.; Ko, A.; Li, G.; Peskind, E.; Daggett, V. SOBA: Development and testing of a soluble oligomer binding assay for detection of amyloidogenic toxic oligomers. Proc. Natl. Acad. Sci. USA, 2022; submitted. [Google Scholar]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 5618, 486–489. [Google Scholar] [CrossRef] [Green Version]
- Glabe, C.G.; Kayed, R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology 2006, 66, 74–78. [Google Scholar] [CrossRef]
- Kayed, R.; Canto, I.; Breydo, L.; Rasool, S.; Lukacsovich, T.; Wu, J.; Albay III, R.; Pensalfini, A.; Yeung, S.; Head, E.; et al. Conformation dependent monoclonal antibodies distinguish different replicating strains or conformers of prefibrillar Aβ oligomers. Mol. Neurodeg. 2010, 5, 57. [Google Scholar] [CrossRef] [Green Version]
- Paravastu, A.K.; Leapman, R.D.; Yau, W.M.; Tycko, R. Molecular structural basis for polymorphism in Alzheimer’s beta-amyloid fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 18349–18354. [Google Scholar] [CrossRef] [Green Version]
- Klein, W.L.; Krafft, G.A.; Finch, C.E. Targeting small Aβ oligomers: The solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001, 24, 219–224. [Google Scholar] [CrossRef]
- Kuo, Y.M.; Emmerling, M.R.; Vigo-Pelfrey, C.; Kasunic, T.C.; Kirkpatrick, J.B.; Murdoch, G.H.; Ball, M.J.; Roher, A.E. Water-soluble Aβ (N-40, N-42) oligomers in normal and Alzheimer disease brains. J. Biol. Chem. 1996, 271, 4077–4081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, H.; Wong, P.T.; Lee, E.L.; Gafni, A.; Steel, D.G. Determination of the oligomer size of amyloidogenic protein beta-amyloid(1-40) by single-molecule spectroscopy. Biophys. J. 2009, 97, 912–921. [Google Scholar] [CrossRef] [Green Version]
- Ryan, T.M.; Kirby, N.; Mertens, H.D.T.; Roberts, B.; Barnham, K.J.; Cappai, R.; Pham, C.L.L.; Masters, C.L.; Curtain, C.C. Small angle x-ray scattering analysis of Cu2+-induced oligomers of the Alzheimer’s amyloid β peptide. Metallomics 2015, 7, 536–543. [Google Scholar] [CrossRef] [Green Version]
- Miller, Y.; Ma, B.; Tsai, C.J.; Nussinov, R. Hollow core of Alzheimer’s Aβ42 amyloid observed by cryoEM is relevant at physiological pH. Proc. Natl. Acad. Sci. USA 2010, 107, 14128–14133. [Google Scholar] [CrossRef] [Green Version]
- Hyung, S.J.; DeToma, A.S.; Brender, J.R.; Lee, S.; Vivekanandan, S.; Kochi, A.; Choi, J.S.; Ramamoorthy, A.; Ruotolo, B.T.; Lim, M.H. Insights into antiamyloidogenic properties of the green tea extract (−)-epigallocatechin-3-gallate toward metal-associated amyloid-β species. Proc. Natl. Acad. Sci. USA 2013, 110, 3743–3748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derrick, J.S.; Kerr, R.A.; Nam, Y.; Oh, S.B.; Lee, H.J.; Earnest, K.G.; Suh, N.; Peck, K.L.; Ozbil, M.; Korshavn, K.J.; et al. A redox-active, compact molecule for cross-linking amyloidogenic peptides into nontoxic, off-pathway aggregates: In vitro and in vivo efficacy and molecular mechanisms. Am. Chem. Soc. 2015, 137, 14785–14797. [Google Scholar] [CrossRef] [Green Version]
- Armen, R.; Alonso, D.; Daggett, V. Anatomy of an amyloidgenic intermediate: Conversion of β-sheet to α-pleated sheet structure in transthyretin at acidic pH. Structure 2004, 12, 1847–1863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armen, R.; DeMarco, M.; Alonso, D.; Daggett, V. Pauling and Corey’s α-pleated sheet structure may define the prefibrillar amyloidogenic intermediate in amyloid disease. Proc. Natl. Acad. Sci. USA 2004, 101, 11622–11627. [Google Scholar] [CrossRef] [Green Version]
- Daggett, V. α-sheet: The toxic conformer in amyloid diseases? Acc. Chem. Res. 2006, 39, 594–602. [Google Scholar] [CrossRef]
- Hopping, G.; Kellock, J.; Barnwal, R.P.; Law, P.; Bryers, J.; Varani, G.; Caughey, B.; Daggett, V. Designed α-sheet peptides inhibit amyloid formation by targeting toxic oligomers. eLife 2014, 3, e01681. [Google Scholar] [CrossRef]
- Kellock, J.; Hopping, G.; Caughey, B.; Daggett, V. Peptides composed of alternating L- and D-amino acids inhibit amyloidogenesis in three distinct amyloid systems independent of sequence. J. Mol. Biol. 2016, 428, 2317–2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maris, N.; Shea, D.; Bleem, A.; Bryers, J.D.; Daggett, V. Chemical and physical variability in structural isomers of an L/D α-sheet peptide designed to inhibit amyloidogenesis. Biochemistry 2018, 57, 507–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleem, A.; Francisco, R.; Bryers, J.D.; Daggett, V. Designed alpha-sheet peptides suppress amyloid formation in Staphylococcus aureus biofilms. Nat. Biofilms Microbiomes 2017, 3, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bi, T.M.; Daggett, V. The role of alpha-sheet in amyloid oligomer aggregation and toxicity. Yale J. Biol. Med. 2018, 91, 247–255. [Google Scholar]
- Daggett, V.; Fersht, A. The present view of the mechanism of protein folding. Nat. Rev. Mol. Cell Biol. 2003, 4, 497–502. [Google Scholar] [CrossRef]
- Daggett, V.; Fersht, A.R. Is there a unifying mechanism for protein folding? Trends Biochem. Sci. 2003, 28, 18–25. [Google Scholar] [CrossRef]
- Sciacca, M.F.M.; Kotler, S.A.; Brender, J.R.; Chen, J.; Lee, D.K.; Ramamoorthy, A. Two-step mechanism of membrane disruption by Aβ through membrane fragmentation and pore formation. Biophys. J. 2012, 103, 702–710. [Google Scholar] [CrossRef] [Green Version]
- Chimon, S.; Shaibat, M.A.; Jones, C.R.; Calero, D.C.; Aizezi, B.; Ishii, Y. Evidence of fibril-like β-sheet structures in a neurotoxic amyloid intermediate of Alzheimer’s β-amyloid. Nat. Struct. Mol. Biol. 2007, 14, 1157–1164. [Google Scholar] [CrossRef]
- Pham, J.D.; Chim, N.; Goulding, C.W.; Nowick, J.S. Structures of oligomers of a peptide from β-amyloid. J. Am. Chem. Soc. 2013, 135, 12460–12467. [Google Scholar] [CrossRef] [Green Version]
- Petkova, A.T.; Yau, W.M.; Tycko, R. Experimental constraints on quaternary structure Alzheimer’s beta-amyloid fibrils. Biochem. 2006, 45, 498–512. [Google Scholar] [CrossRef] [Green Version]
- McDonald, J.M.; Savva, G.M.; Brayne, C.; Welzel, A.T.; Forster, G.; Shankar, G.M.; Selkoe, D.J.; Ince, P.G.; Walsh, D.M. The presence of sodium dodecyl sulphate-stable Abeta dimers is strongly associated with Alzheimer-type dementia. Brain 2010, 133, 1328–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Nuallain, B.; Freir, D.B.; Nicoll, A.J.; Risse, E.; Ferguson, N.; Herron, C.E.; Collinge, J.; Walsh, D.M. Amyloid beta-protein dimers rapidly form stable synaptotoxic protofibrils. J. Neurosci. 2010, 30, 14411–14419. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, A.; Matsumura, S.; Dezawa, M.; Tada, M.; Yanazawa, M.; Ito, A.; Akioka, M.; Kikuchi, S.; Sato, M.; Ideno, S.; et al. Isolation and characterization of patient-derived, toxic, high mass amyloid beta-protein (Abeta) assembly from Alzheimer disease brains. J. Biol. Chem. 2009, 284, 32895–32905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, S.; Shinoda, K.; Yamada, M.; Yokojima, S.; Inoue, M.; Ohnishi, T.; Shimada, T.; Kikuchi, K.; Masui, D.; Hashimoto, S.; et al. Two distinct amyloid β-protein (Aβ) assembly pathways leading to oligomers and fibrils identified by combined fluorescence correlation spectroscopy, morphology and toxicity analyses. J. Biol. Chem. 2011, 286, 11555–11562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatanpaa, K.; Isaacs, K.R.; Shirao, T.; Brady, D.R.; Rapoport, S.I. Loss of proteins regulating synaptic plasticity in normal aging of the human brain and in Alzheimer disease. J. Neuropath. Exp. Neur. 1999, 58, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Kawarabayashi, T.; Shoji, M.; Younkin, L.H.; Wen-Lang, L.; Dickson, D.W.; Murakami, T.; Matsubara, E.; Abe, K.; Ashe, K.H.; Younkin, S.G. Dimeric amyloid β protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 2004, 24, 3801–3809. [Google Scholar] [CrossRef]
- Hong, S.; Ostaszewski, B.L.; Yang, T.; O’Malley, T.T.; Jin, M.; Yanagisawa, K.; Li, S.; Bartels, T.; Selkoe, D.J. Soluble Aβ oligomers are rapidly sequestered from brain ISF in vivo and bind GM1 ganglioside on cellular membranes. Neuron 2014, 82, 308–319. [Google Scholar] [CrossRef] [Green Version]
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 2005, 280, 17294–17300. [Google Scholar] [CrossRef] [Green Version]
- Cerf, E.; Sarroukh, R.; Tamamizu-Kato, S.; Breydo, L.; Derclaye, S.; Dufrene, Y.F.; Narayanaswami, V.; Goormaghtigh, E.; Ruysschaert, J.M.; Raussens, V. Antiparallel beta-sheet: A signature structure of the oligomeric amyloid beta-peptide. Biochem. J. 2009, 421, 415–423. [Google Scholar] [CrossRef]
- Fonte, V.; Dostal, V.; Roberts, C.M.; Gonzales, P.; Lacor, P.; Magrane, J.; Dingwell, N.; Fan, E.Y.; Silverman, M.A.; Stein, G.H.; et al. A glycine zipper motif mediates the formation of toxic β-amyloid oligomers in vitro and in vivo. Mol. Neurodeg. 2011, 6, 61. [Google Scholar] [CrossRef] [Green Version]
- Hung, L.W.; Ciccotosto, G.D.; Giannakis, E.; Tew, D.J.; Perez, K.; Masters, C.L.; Cappai, R.; Wade, J.D.; Barnham, K.J. Amyloid-β peptide (Aβ) neurotoxicity is modulated by the rate of peptide aggregation: Aβ dimers and trimers correlate with neurotoxicity. J. Neuro. 2008, 28, 11950–11958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, D.A.; Cabral, J.M.; Pfuetzner, R.A.; Kuo, A.; Gulbis, J.M.; Cohen, S.L.; Chait, B.T.; MacKinnon, R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science 1998, 280, 69–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talantova, M.; Sanz-Blasco, S.; Zhang, X.; Xia, P.; Akhtar, M.W.; Okamoto, S.; Dziewczapolski, G.; Nakamura, T.; Cao, G.; Pratt, A.E.; et al. Aβ induces astrocytic glutamate release, extrasynaptic NMDA receptor activation, and synaptic loss. Proc. Natl. Acad. Sci. USA 2013, 110, 2518–2527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Um, J.W.; Nygaard, H.B.; Heiss, J.K.; Kostylev, M.A.; Stagi, M.; Vortmeyer, A.; Wisniewski, T.; Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, A.; McLaurin, J. Mechanisms of amyloid-beta peptide uptake by neurons: The role of lipid rafts and lipid raft-associated proteins. Int. J. Alz. Dis. 2010, 2011, 548380. [Google Scholar] [CrossRef] [Green Version]
- Umeda, T.; Tomiyama, T.; Sakama, N.; Tanaka, S.; Lambert, M.P.; Klein, W.L.; Mori, H. Intraneuronal amyloid oligomers cause cell death via endoplasmic reticulum stress, endosomal/lysosomal leakage, and mitochondrial dysfunction in vivo. Neurosci. Res. 2011, 89, 1031–1042. [Google Scholar] [CrossRef]
- Resende, R.; Ferreiro, E.; Pereira, C.; de Oliveira, C.R. Neurotoxic effect of oligomeric and fibrillar species of amyloid-beta peptide 1–42: Involvement of endoplasmic reticulum calcium release in oligomer-induced cell death. Neuroscience 2008, 155, 725–737. [Google Scholar] [CrossRef]
- Cho, D.H.; Nakamura, T.; Fang, J.; Cieplak, P.; Godzik, A.; Gu, Z.; Lipton, S.A. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science 2009, 324, 102–105. [Google Scholar] [CrossRef] [Green Version]
- Wogulis, M.; Wright, S.; Cunningham, D.; Chilcote, T.; Powell, K.; Rydel, R.E. Nucleation-dependent polymerization is an essential component of amyloid-mediated neuronal cell death. J. Neurosci. 2005, 25, 1071–1080. [Google Scholar] [CrossRef] [Green Version]
- Sciacca, M.F.; Lolicato, F.; Tempra, C.; Scollo, F.; Sahoo, B.R.; Watson, M.D.; Garcia-Vinuales, S.; Milardi, D.; Raudino, A.; Lee, J.C.; et al. Lipid-chaperone hypothesis: A common molecular mechanism of membrane disruption by intrinsically disordered proteins. ACS Chem. Neurosci. 2020, 11, 4336–4350. [Google Scholar] [CrossRef]
- Cummings, J.; Lee, G.; Mortsdorf, T.; Ritter, A.; Zhong, K. Alzheimer’s disease drug development pipeline: 2019. Alz. Dem. Trans. Res. Clin. Int. 2019, 5, 272–293. [Google Scholar] [CrossRef] [PubMed]
- Sevigny, J.; Chiao, P.; Bussiere, T.; Weinreb, P.H.; Williams, L.; Maier, M.; Dunstan, R.; Salloway, S.; Chen, T.; Ling, Y.; et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature 2016, 537, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Haeberlein, S.H.; O’Gorman, J.; Chiao, P.; Bussiere, T.; von Rosenstiel, P.; Tian, Y.; Zhu, Y.; von Hehn, C.; Gheuens, S.; Skordos, L.; et al. Clinical development of Aducanumab, an anti-Aβ human monoclonal antibody being investigated for the treatment of early Alzheimer’s disease. J. Prev. Alz. Dis. 2017, 4, 255–263. [Google Scholar]
- Imbimbo, B.P.; Ottonello, S.; Frisardi, V.; Solfrizzi, V.; Greco, A.; Seripa, D.; Pilotto, A.; Panza, F. Solanezumab for the treatment of mild-to-moderate Alzheimer’s disease. Expert Rev. Clin. Immunol. 2012, 8, 135–149. [Google Scholar] [CrossRef]
- Samadi, H.; Sultzer, D. Solanezumab for Alzheimer’s disease. Drug Eval. 2011, 11, 787–798. [Google Scholar] [CrossRef]
- Meilandt, W.; Maloney, J.; Imperio, J.; Bainbridge, T.; Reichelt, M.; Mandikian, D.; Lu, Y.; Ernst, J.; Fuji, R.; Atwal, J. Characterization of the selective in vivo and in vitro binding properties of Crenezumab: Insights into Crenezumab’s unique mechanism of action. Neurology 2018, 90, 174. [Google Scholar]
- Cummings, J.L.; Cohen, S.; van Dyck, C.H.; Brody, M.; Curtis, C.; Cho, W.; Ward, M.; Friesenhahn, M.; Rabe, C.; Brunstein, F.; et al. A phase 2 randomized trial of crenezumab in mild to moderate Alzheimer disease. Neurology. 2018, 90, 1889–1897. [Google Scholar] [CrossRef] [Green Version]
- Adolfsson, O.; Pihlgren, M.; Toni, N.; Varisco, Y.; Buccarello, A.L.; Antoniello, K.; Lohmann, S.; Piorkowska, K.; Gafner, V.; Atwal, J.K.; et al. An effector-reduced anti-amyloid (Aβ) antibody with unique Aβ binding properties promotes neuroprotection and glial engulfment of Aβ. J. Neuro. 2012, 32, 9677–9689. [Google Scholar] [CrossRef]
- Sakamoto, K.; Matsuki, S.; Matsuguma, K.; Yoshihara, T.; Uchida, N.; Azuma, F.; Russell, M.; Hughes, G.; Haeberlein, S.B.; Alexander, R.C.; et al. BACE1 inhibitor Lanabecestat (AZD3293) in a phase 1 study of healthy Japanese subjects: Pharmacokinetics and effects on plasma and cerebrospinal fluid Aβ peptides. J. Clin. Pharmacol. 2017, 57, 1460–1471. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Lesne, S.E.; Sherman, M.A.; Grant, M.; Kuskowski, M.; Schneider, J.A.; Bennett, D.A.; Ashe, K.H. Brain amyloid-β oligomers in ageing and Alzheimer’s disease. Brain 2013, 136, 1383–1398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zott, B.; Simon, M.M.; Hong, W.; Unger, F.; Chen-Engerer, H.J.; Frosch, M.P.; Sakmann, B.; Walsh, D.M.; Konnerth, A. A vicious cycle of β amyloid-dependent neuronal hyperactivation. Science 2019, 365, 559–565. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Species | Size & Characterizations | Isolation Technique (Source, Ie In Vitro) |
|---|---|---|
| SDS-stable dimers and synthetic dimers [65] | Dimers, 8–12 kDa, 3–4 nm height, no detected secondary structure | SDS-PAGE (brain derived or synthetic in vitro Aβ40Ser26Cys mutant) |
| SDS-stable dimers/trimers [67] | Dimer/trimer, 6–12 kDa, Aβ40/Aβ42 with Arg5 N-term truncated species | SDS-PAGE (transfected CHO cell culture medium) |
| Trimers from mutant human APPV717F [64] | Trimers, 12 kDa, unstructured | SEC and PAGE (7PA2 cells) |
| Tetramers (Aβ42 and Aβ40) [80] | Tetramer, 18 kDa, ring-shaped (Aβ40) or bent (Aβ42), non-aggregation prone (Aβ40) or aggregation prone (Aβ42) | IM-MS (synthetic in vitro) |
| Pentamer [22] | Pentamer, compact pentagonal shape with C-termini buried | ssNMR (synthetic in vitro kinetically trapped) |
| Hexamer/dodecamer [85,86] | Hexamer-dodecamer, 27–56 kDa, α-sheet secondary structure, A11 positive | SEC (synthetic in vitro, human CSF) |
| Aβ*56 [63] | Dodecamer, ~56 kDa, globular, A11 positive | SEC and PAGE (Human brain isolates) |
| Aβo [87,88,89] | 15–20 mer, spherical vesicles 2–5 nm diameter, A11 positive | SEC (synthetic in vitro) |
| ADDLs [21] | Trimer-24 mer, 17 kDa tetramer major, globular, 2–5 nm height, A11-positive | Nondenaturing electrophoresis (synthetic in vitro) |
| ASPD (amylospheroids) [90] | 32–150 mers, spheroids, 10–15 nm diameter assemblies, A11 negative | SDS-PAGE (brain derived and synthetic in vitro) |
| Aβ42 Ellipsoids [66] | High molecular weight, ellipsoidal and annular | SAXS (Cu(II)-guided Aβ42 oligomerization in vitro) |
| Aβ40 Protofibrils [61] | High molecular weight, protofibrillar | SAXS (Cu(II)-guided Aβ40 oligomerization in vitro) |
| HMW soluble oligomers [66] | Large, circular, 8–12 nm, form membrane-permeable pore at lipid bilayers | AFM (synthetic in vitro) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shea, D.; Daggett, V. Amyloid-β Oligomers: Multiple Moving Targets. Biophysica 2022, 2, 91-110. https://doi.org/10.3390/biophysica2020010
Shea D, Daggett V. Amyloid-β Oligomers: Multiple Moving Targets. Biophysica. 2022; 2(2):91-110. https://doi.org/10.3390/biophysica2020010
Chicago/Turabian StyleShea, Dylan, and Valerie Daggett. 2022. "Amyloid-β Oligomers: Multiple Moving Targets" Biophysica 2, no. 2: 91-110. https://doi.org/10.3390/biophysica2020010
APA StyleShea, D., & Daggett, V. (2022). Amyloid-β Oligomers: Multiple Moving Targets. Biophysica, 2(2), 91-110. https://doi.org/10.3390/biophysica2020010