
The Dynamics of Subunit Rotation in a Eukaryotic Ribosome

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Multi-Basin Structure-Based Model
2.2. Simulation Details
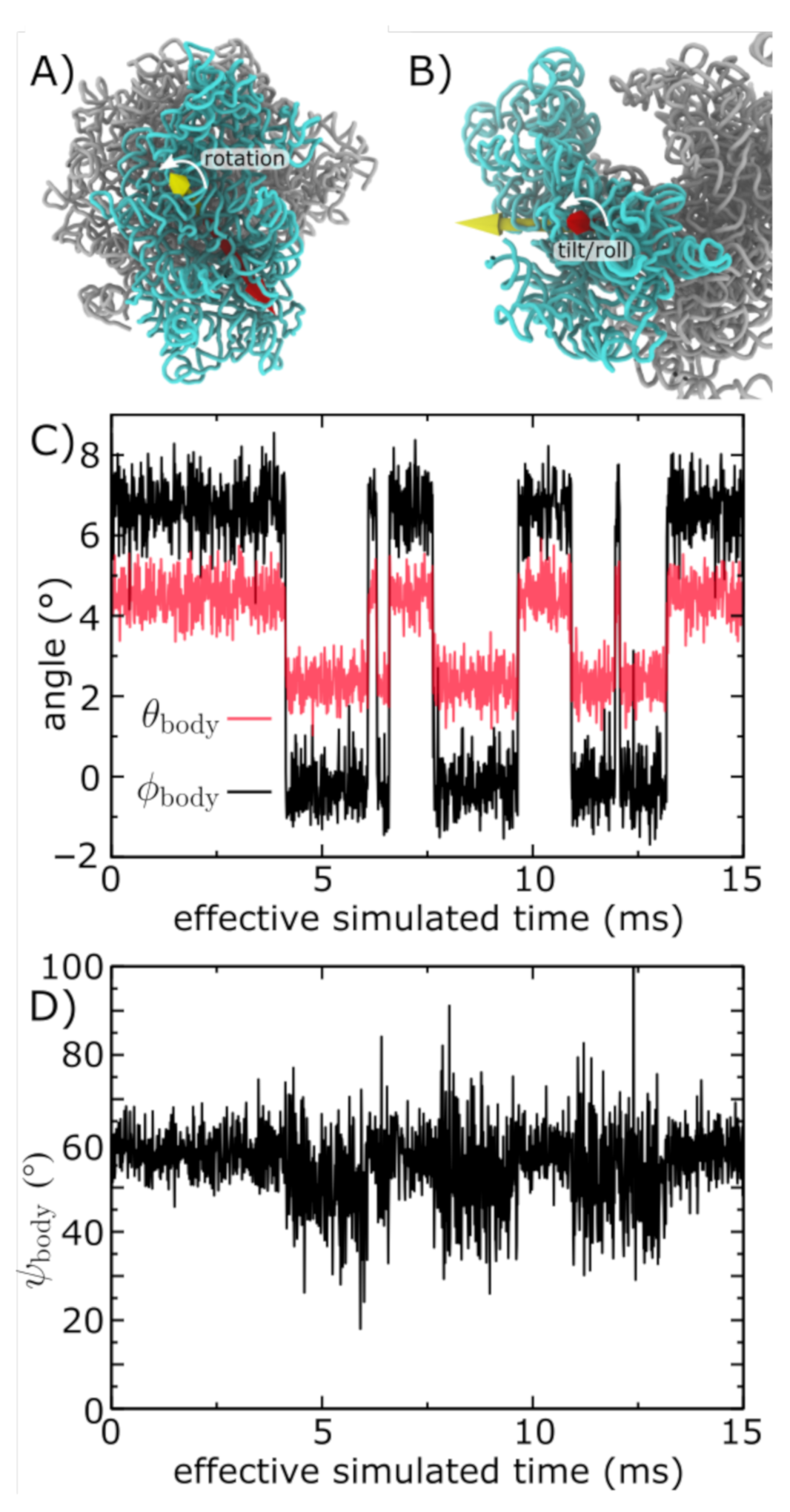
2.3. SSU Rotation Measures
3. Results
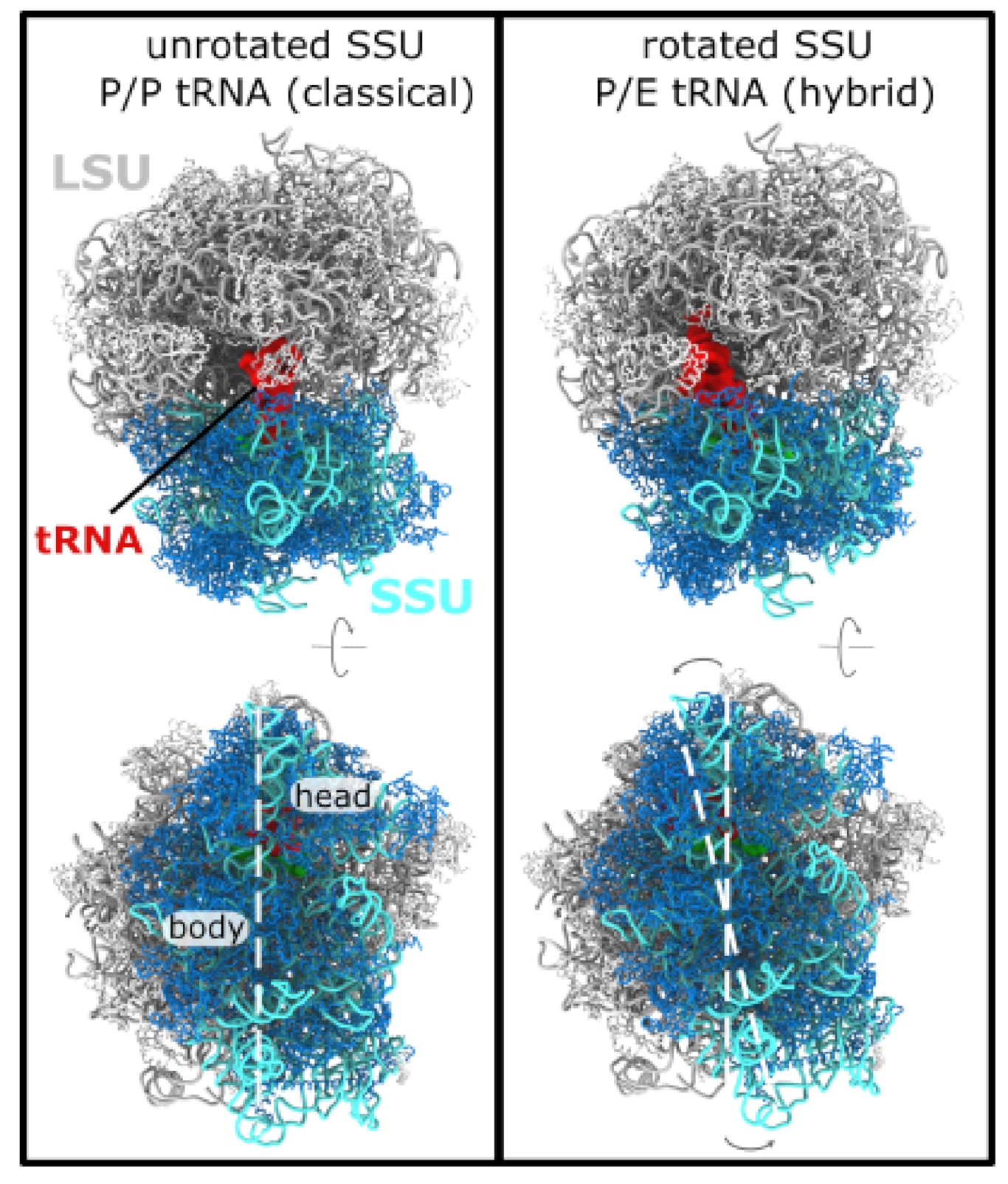
3.1. Simulating Spontaneous Subunit Rotation Events in a Complete Ribosome
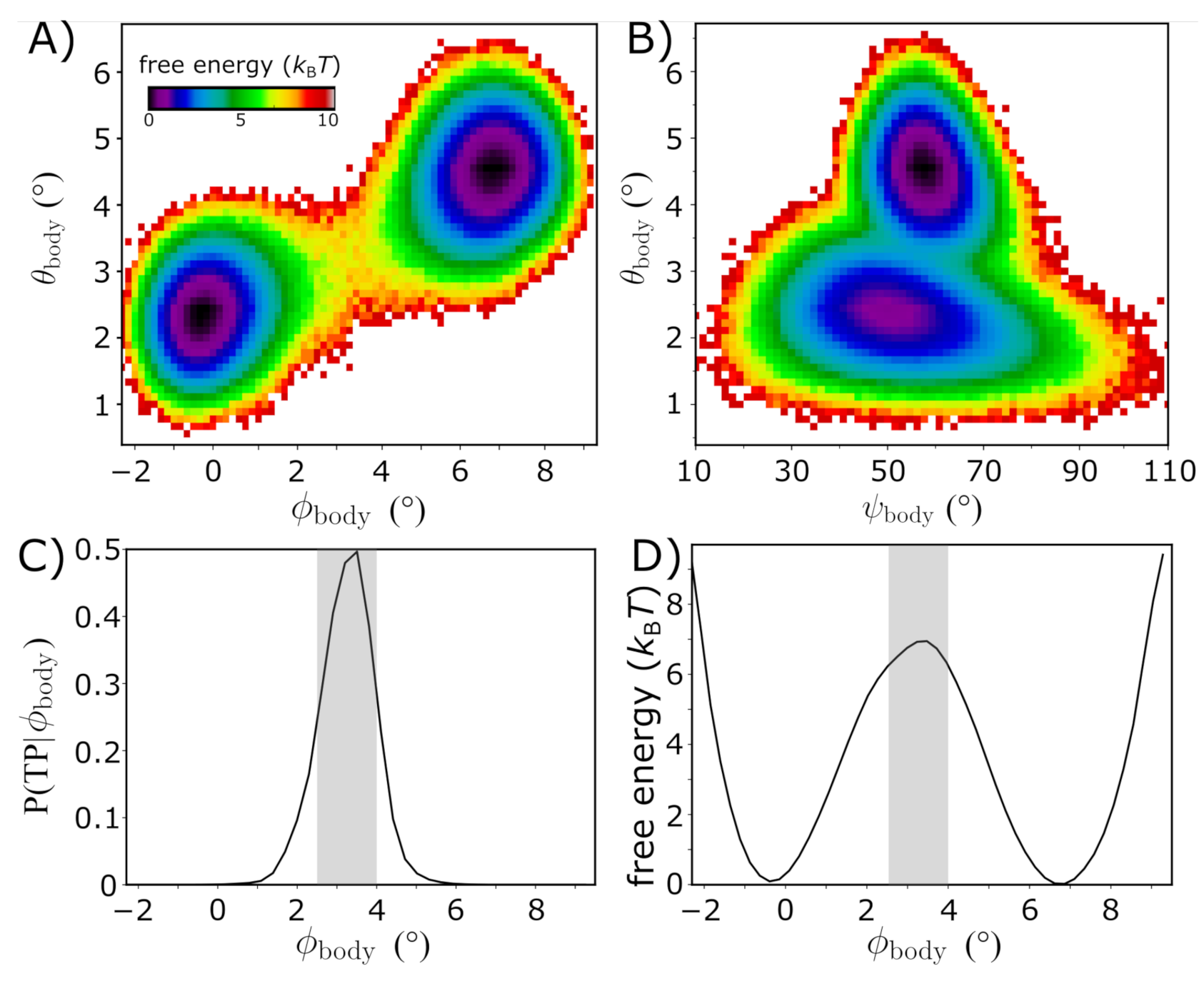
3.2. Quantifying the Energy Landscape of Rotation
3.3. Simulations Implicate Millisecond-Scale Dynamics of Rotation
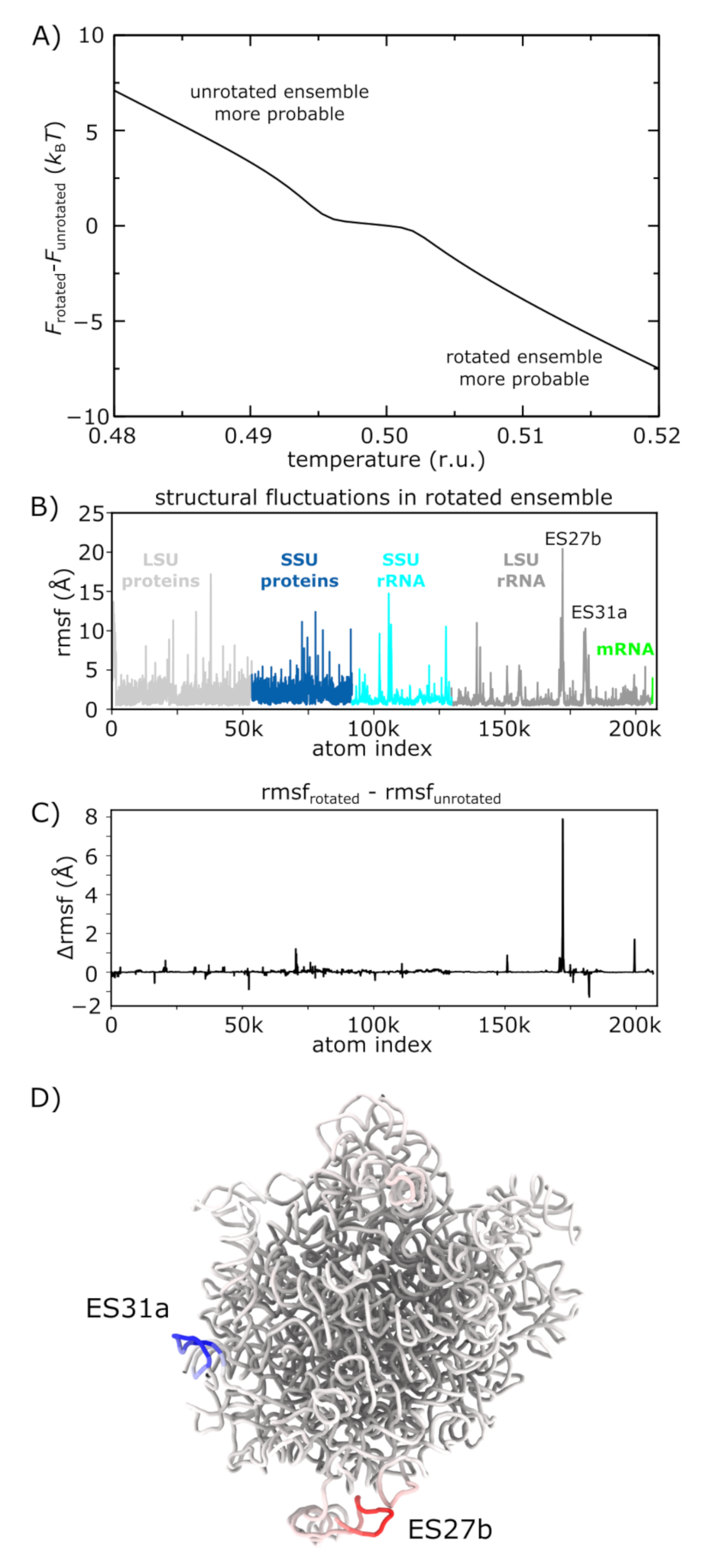
3.4. Asynchronous Dynamics of Subunit Bridge Interactions during Rotation
3.5. Molecular Flexibility Leads to Temperature-Induced Population Shift
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| rmsf | root mean-squared fluctuations |
| LSU | large subunit |
| SSU | small subunit |
| mfpt | mean first-passage time |
References
- Korostelev, A.; Noller, H.F. The ribosome in focus: New structures bring new insights. Trends Biochem. Sci. 2007, 32, 434–441. [Google Scholar] [CrossRef] [PubMed]
- Frank, J.; Gonzalez, R.L., Jr. Structure and Dynamics of a Processive Brownian Motor: The Translating Ribosome. Annu. Rev. Biochem. 2010, 79, 381–412. [Google Scholar] [CrossRef] [PubMed]
- Schmeing, T.M.; Ramakrishnan, V. What recent ribosome structures have revealed about the mechanism of translation. Nature 2009, 461, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Rodnina, M.V.; Wintermeyer, W. The ribosome as a molecular machine: The mechanism of tRNA-mRNA movement in translocation. Biochem. Soc. Trans. 2011, 39, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Valle, M.; Zavialov, A.; Sengupta, J.; Rawat, U.; Ehrenberg, M.; Frank, J. Locking and unlocking of ribosomal motions. Cell 2003, 114, 123–134. [Google Scholar] [CrossRef]
- Dunkle, J.; Wang, L.; Feldman, M.B.; Pulk, A.; Chen, V.B.; Kapral, G.J.; Noeske, J.; Richardson, J.S.; Blanchard, S.C.; Cate, J.H.D. Structures of the bacterial ribosome in classical and hybrid states of tRNA binding. Science 2011, 332, 981–984. [Google Scholar] [CrossRef] [PubMed]
- Ratje, A.; Loerke, J.; Mikolajka, A.; Brünner, M.; Hildebrand, P.W.; Starosta, A.; Dönhöfer, A.; Connell, S.; Fucini, P.; Mielke, T.; et al. Head swivel on the ribosome facilitates translocation by means of intra-subunit tRNA hybrid sites. Nature 2010, 468, 713–716. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.; Donohue, J.P.; Noller, H.F. Molecular mechanics of 30S subunit head rotation. Proc. Natl. Acad. Sci. USA 2014, 111, 13325–13330. [Google Scholar] [CrossRef] [PubMed]
- Ramrath, D.J.F.; Yamamoto, H.; Rother, K.; Wittek, D.; Pech, M.; Mielke, T.; Loerke, J.; Scheerer, P.; Ivanov, P.; Teraoka, Y.; et al. The complex of tmRNA–SmpB and EF-G on translocating ribosomes. Nature 2013, 485, 526–529. [Google Scholar] [CrossRef] [PubMed]
- Horan, L.H.; Noller, H.F. Intersubunit movement is required for ribosomal translocation. Proc. Natl. Acad. Sci. USA 2007, 104, 4881–4885. [Google Scholar] [CrossRef]
- Ermolenko, D.N.; Majumdar, Z.K.; Hickerson, R.P.; Spiegel, P.C.; Clegg, R.M.; Noller, H.F. Observation of intersubunit movement of the ribosome in solution using FRET. J. Mol. Biol. 2007, 370, 530–540. [Google Scholar] [CrossRef]
- Cornish, P.V.; Ermolenko, D.N.; Noller, H.F.; Ha, T. Spontaneous intersubunit rotation in single ribosomes. Mol. Cell 2008, 30, 578–588. [Google Scholar] [CrossRef]
- Cornish, P.V.; Ermolenko, D.N.; Staple, D.W.; Hoang, L.; Hickerson, R.P.; Noller, H.F.; Ha, T. Following movement of the L1 stalk between three functional states in single ribosomes. Proc. Natl. Acad. Sci. USA 2009, 106, 2571–2576. [Google Scholar] [CrossRef]
- Fei, J.; Bronson, J.E.; Hofman, J.M.; Srinivas, R.L.; Wiggins, C.H.; Gonzalez, R.L. Allosteric collaboration between elongation factor G and the ribosomal L1 stalk directs tRNA movements during translation. Proc. Natl. Acad. Sci. USA 2009, 106, 15702–15707. [Google Scholar] [CrossRef]
- Marshall, R.; Dorywalska, M.; Puglisi, J. Irreversible chemical steps control intersubunit dynamics during translation. Proc. Nat. Acad. Sci. USA 2008, 105, 15364–15369. [Google Scholar] [CrossRef]
- Ferguson, A.; Wang, L.; Altman, R.B.; Terry, D.S.; Juette, M.F.; Burnett, B.J.; Alejo, J.L.; Dass, R.A.; Parks, M.M.; Vincent, C.T.; et al. Functional Dynamics within the Human Ribosome Regulate the Rate of Active Protein Synthesis. Mol. Cell 2015, 60, 475–486. [Google Scholar] [CrossRef]
- Flis, J.; Holm, M.; Rundlet, E.J.; Loerke, J.; Hilal, T.; Dabrowski, M.; Bürger, J.; Mielke, T.; Blanchard, S.C.; Spahn, C.M.T.; et al. tRNA Translocation by the Eukaryotic 80S Ribosome and the Impact of GTP Hydrolysis. Cell Rep. 2018, 25, 2676–2688.e7. [Google Scholar] [CrossRef]
- Svidritskiy, E.; Brilot, A.F.; Koh, C.S.; Grigorieff, N.; Korostelev, A.A. Structures of yeast 80S ribosome-tRNA complexes in the rotated and nonrotated conformations. Structure 2014, 22, 1210–1218. [Google Scholar] [CrossRef]
- Budkevich, T.V.; Giesebrecht, J.; Behrmann, E.; Loerke, J.; Ramrath, D.J.; Mielke, T.; Ismer, J.; Hildebrand, P.W.; Tung, C.S.; Nierhaus, K.H.; et al. Regulation of the mammalian elongation cycle by subunit rolling: A eukaryotic-specific ribosome rearrangement. Cell 2014, 158, 121–131. [Google Scholar] [CrossRef]
- Itoh, Y.; Naschberger, A.; Mortezaei, N.; Herrmann, J.M.; Amunts, A. Analysis of translating mitoribosome reveals functional characteristics of translation in mitochondria of fungi. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Tama, F.; Valle, M.; Frank, J.; Brooks, C.L. Dynamic reorganization of the functionally active ribosome explored by normal mode analysis and cryo-electron microscopy. Proc. Natl. Acad. Sci. USA 2003, 100, 9319–9323. [Google Scholar] [CrossRef]
- Wang, Y.; Rader, A.J.; Bahar, I.; Jernigan, R.L. Global ribosome motions revealed with elastic network model. J. Struct. Biol. 2004, 147, 302–314. [Google Scholar] [CrossRef]
- Whitford, P.C.; Blanchard, S.C.; Cate, J.H.D.; Sanbonmatsu, K.Y. Connecting the Kinetics and Energy Landscape of tRNA Translocation on the Ribosome. PLoS Comput. Biol. 2013, 9, e1003003. [Google Scholar] [CrossRef]
- Bock, L.V.; Blau, C.; Schröder, G.F.; Davydov, I.I.; Fischer, N.; Stark, H.; Rodnina, M.V.; Vaiana, A.C.; Grubmüller, H. Energy barriers and driving forces in tRNA translocation through the ribosome. Nat. Struct. Mol. Biol. 2013, 20, 1390–1396. [Google Scholar] [CrossRef] [PubMed]
- Bock, L.V.; Blau, C.; Vaiana, A.C.; Grubmüller, H. Dynamic contact network between ribosomal subunits enables rapid large-scale rotation during spontaneous translocation. Nucleic Acid Res. 2015, 43, 6747–6760. [Google Scholar] [CrossRef]
- Levi, M.; Whitford, P.C. Dissecting the energetics of subunit rotation in the ribosome. J. Phys. Chem. B 2019, 123, 2812–2923. [Google Scholar] [CrossRef] [PubMed]
- Whitford, P.C.; Noel, J.K.; Gosavi, S.; Schug, A.; Sanbonmatsu, K.Y.; Onuchic, J.N. An all-atom structure-based potential for proteins: Bridging minimal models with all-atom empirical forcefields. Prot. Struct. Func. Bioinfo. 2009, 75, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Noel, J.K.; Levi, M.; Raghunathan, M.; Lammert, H.; Hayes, R.L.; Onuchic, J.N.; Whitford, P.C. SMOG 2: A Versatile Software Package for Generating Structure-Based Models. PLoS Comput. Biol. 2016, 12, e1004794. [Google Scholar] [CrossRef]
- Whitford, P.C.; Jiang, W.; Serwer, P. Simulations of Phage T7 Capsid Expansion Reveal the Role of Molecular Sterics on Dynamics. Viruses 2020, 12, 1273. [Google Scholar] [CrossRef]
- Duan, Y.; Wu, C.; Chowdhury, S.; Lee, M.C.; Xiong, G.; Zhang, W.; Yang, R.; Cieplak, P.; Luo, R.; Lee, T.; et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comp. Chem. 2003, 24, 1999–2012. [Google Scholar] [CrossRef]
- Noel, J.K.; Whitford, P.C.; Onuchic, J.N. The shadow map: A general contact definition for capturing the dynamics of biomolecular folding and function. J. Phys. Chem. B 2012, 116, 8692–8702. [Google Scholar] [CrossRef]
- Nguyen, K.; Whitford, P.C. Steric interactions lead to collective tilting motion in the ribosome during mRNA-tRNA translocation. Nat. Commun. 2016, 7, 10586. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; van der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Mod. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Yang, H.; Bandarkar, P.; Horne, R.; Leite, V.B.; Chahine, J.; Whitford, P.C. Diffusion of tRNA inside the ribosome is position-dependent. J. Chem. Phys. 2019, 151. [Google Scholar] [CrossRef]
- Hoffer, E.D.; Hong, S.; Sunita, S.; Maehigashi, T.; Gonzalez, R.L.J.; Whitford, P.C.; Dunham, C.M. Structural insights into mRNA reading frame regulation by tRNA modification and slippery codon–anticodon pairing. eLife 2020, 9, e51898. [Google Scholar] [CrossRef]
- Russell, R.B.; Barton, G.J. Multiple protein sequence alignment from tertiary structure comparison. Proteins 1992, 14, 309–323. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Okazaki, K.; Koga, N.; Takada, S.; Onuchic, J.N.; Wolynes, P.G. Multiple-basin energy landscapes for large-amplitude conformational motions of proteins: Structure-based molecular dynamics simulations. Proc. Nat. Acad. Sci. USA 2006, 103, 11844–11849. [Google Scholar] [CrossRef]
- Hyeon, C.; Thirumalai, D. Capturing the essence of folding and functions of biomolecules using coarse-grained models. Nat. Commun. 2011, 2, 487. [Google Scholar] [CrossRef]
- Pierro, M.D.; Potoyan, D.A.; Wolynes, P.G.; Onuchic, J.N. Anomalous diffusion, spatial coherence, and viscoelasticity from the energy landscape of human chromosomes. Proc. Natl. Acad. Sci. USA 2018, 115, 7753–7758. [Google Scholar] [CrossRef]
- Chan, H.S.; Zhang, Z.; Wallin, S.; Liu, Z. Cooperativity, local-nonlocal coupling, and nonnative interactions: Principles of protein folding from coarse-grained models. Annu. Rev. Phys. Chem. 2011, 62. [Google Scholar] [CrossRef]
- Noel, J.K.; Whitford, P.C. How EF-Tu can contribute to efficient proofreading of aa-tRNA by the ribosome. Nat. Commun. 2016, 7, 13314. [Google Scholar] [CrossRef]
- Nguyen, K.; Yang, H.; Whitford, P.C. How the Ribosomal A-Site Finger Can Lead to tRNA Species-Dependent Dynamics. J. Phys. Chem. B 2017, 121, 2767–2775. [Google Scholar] [CrossRef]
- Levi, M.; Walak, K.; Wang, A.; Mohanty, U.; Whitford, P.C. A steric gate controls P/E hybrid-state formation of tRNA on the ribosome. Nat. Commun. 2020, 11, 5706. [Google Scholar] [CrossRef]
- Levi, M.; Noel, J.K.; Whitford, P.C. Studying ribosome dynamics with simplified models. Methods 2019, 162-163, 128–140. [Google Scholar] [CrossRef]
- Best, R.B.; Hummer, G. Reaction coordinates and rates from transition paths. Proc. Natl. Acad. Sci. USA 2005, 102, 6732–6737. [Google Scholar] [CrossRef]
- Das, P.; Moll, M.; Stamati, H.; Kavraki, L.; Clementi, C. Low-dimensional, free-energy landscapes of protein-folding reactions by nonlinear dimensionality reduction. Proc. Natl. Acad. Sci. USA 2006, 103, 9885–9890. [Google Scholar] [CrossRef]
- Best, R.; Paci, E.; Hummer, G.; Dudko, O. Pulling direction as a reaction coordinate for the mechanical unfolding of single molecules. J. Phys. Chem. B 2008, 112, 5968–5976. [Google Scholar] [CrossRef]
- Krivov, S.V. On reaction coordinate optimality. J. Chem. Theory Comput. 2013, 9, 135–146. [Google Scholar] [CrossRef]
- Makarov, D.E. Barrier Crossing Dynamics from Single-Molecule Measurements. J. Phys. Chem. B 2021, 125, 2467–2476. [Google Scholar] [CrossRef] [PubMed]
- Hummer, G. Position-dependent diffusion coefficients and free energies from Bayesian analysis of equilibrium and replica molecular dynamics simulations. New J. Phys. 2005, 7, 34. [Google Scholar] [CrossRef]
- Lacroute, F. RNA and protein elongation rates in Saccharomyces cerevisiae. MGG Mol. Gen. Genet. 1973, 125, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Noel, J.K.; Chahine, J.; Leite, V.B.; Whitford, P.C. Capturing transition paths and transition states for conformational rearrangements in the ribosome. Biophys. J. 2014, 107, 2881–2890. [Google Scholar] [CrossRef]
- Atilgan, A.R.; Durell, S.R.; Jernigan, R.L.; Demirel, M.C.; Keskin, O.; Bahar, I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 2001, 80, 505–515. [Google Scholar] [CrossRef]
- Miyashita, O.; Onuchic, J.; Wolynes, P. Nonlinear elasticity, proteinquakes, and the energy landscapes of functional transitions in proteins. Proc. Natl. Acad. Sci. USA 2003, 100, 12570–12575. [Google Scholar] [CrossRef]
- Shan, Y.; Arkhipov, A.; Kim, E.T.; Pan, A.C.; Shawa, D.E. Transitions to catalytically inactive conformations in EGFR kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 7270–7275. [Google Scholar] [CrossRef]
- Jackson, J.; Nguyen, K.; Whitford, P.C. Exploring the balance between folding and functional dynamics in proteins and RNA. Int. J. Mol. Sci. 2015, 16, 6868–6889. [Google Scholar] [CrossRef]
- Müller, C.W.; Schlauderer, G.J.; Reinstein, J.; Schulz, G.E. Adenylate kinase motions during catalysis: An energetic counterweight balancing substrate binding. Structure 1996, 4, 147–156. [Google Scholar] [CrossRef]
- Munro, J.B.; Altman, R.B.; O’Connor, N.; Blanchard, S.C. Identification of two distinct hybrid state intermediates on the ribosome. Mol. Cell 2007, 25, 505–517. [Google Scholar] [CrossRef]
- Chen, J.; Petrov, A.; Tsai, A.; O’Leary, S.E.; Puglisi, J.D. Coordinated conformational and compositional dynamics drive ribosome translocation. Nat. Struct. Mol. Biol. 2013, 20, 718–727. [Google Scholar] [CrossRef]
- Wasserman, M.R.; Alejo, J.L.; Altman, R.B.; Blanchard, S.C. Multiperspective smFRET reveals rate-determining late intermediates of ribosomal translocation. Nat. Struct. Mol. Biol. 2016, 23, 333–341. [Google Scholar] [CrossRef]
- Levi, M.; Nguyen, K.; Dukaye, L.; Whitford, P.C. Quantifying the Relationship between Single-Molecule Probes and Subunit Rotation in the Ribosome. Biophys. J. 2017, 113, 2777–2786. [Google Scholar] [CrossRef]
- Whitford, P.C.; Ahmed, A.; Yu, Y.; Hennelly, S.P.; Tama, F.; Spahn, C.M.T.; Onuchic, J.N.; Sanbonmatsu, K.Y. Excited states of ribosome translocation revealed through integrative molecular modeling. Proc. Natl. Acad. Sci. USA 2011, 108, 18943–18948. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Freitas, F.C.; Fuchs, G.; de Oliveira, R.J.; Whitford, P.C. The Dynamics of Subunit Rotation in a Eukaryotic Ribosome. Biophysica 2021, 1, 204-221. https://doi.org/10.3390/biophysica1020016
Freitas FC, Fuchs G, de Oliveira RJ, Whitford PC. The Dynamics of Subunit Rotation in a Eukaryotic Ribosome. Biophysica. 2021; 1(2):204-221. https://doi.org/10.3390/biophysica1020016
Chicago/Turabian StyleFreitas, Frederico Campos, Gabriele Fuchs, Ronaldo Junio de Oliveira, and Paul Charles Whitford. 2021. "The Dynamics of Subunit Rotation in a Eukaryotic Ribosome" Biophysica 1, no. 2: 204-221. https://doi.org/10.3390/biophysica1020016
APA StyleFreitas, F. C., Fuchs, G., de Oliveira, R. J., & Whitford, P. C. (2021). The Dynamics of Subunit Rotation in a Eukaryotic Ribosome. Biophysica, 1(2), 204-221. https://doi.org/10.3390/biophysica1020016