
Probing Conformational Dynamics by Protein Contact Networks: Comparison with NMR Relaxation Studies and Molecular Dynamics Simulations



Abstract
1. Introduction
2. Materials and Methods
2.1. The Collection of PDB Structures
2.2. Construction and Centrality Measurements of Protein Structure Networks
2.3. NMR Spin Relaxation Measurements and MDs
3. Results
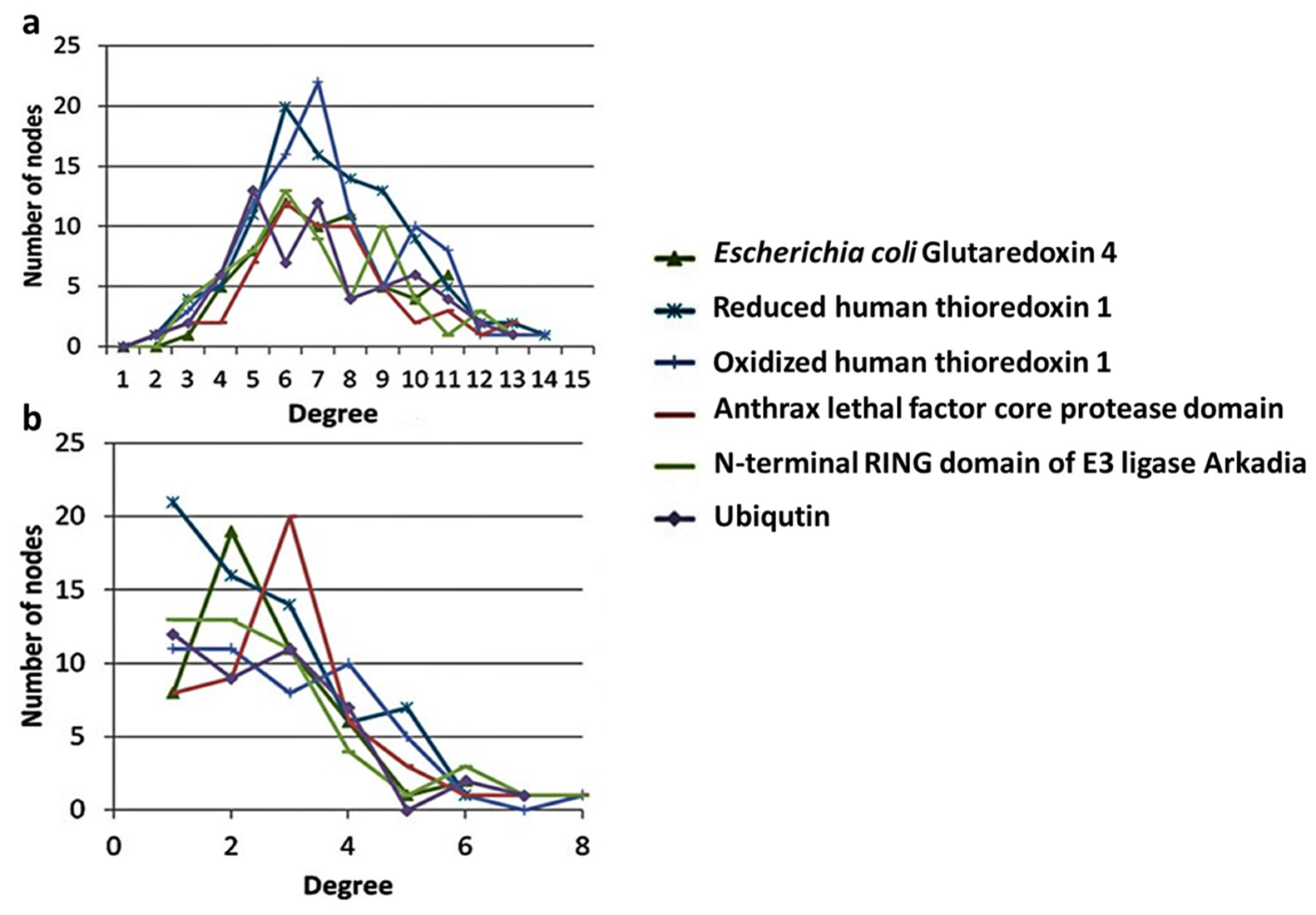
3.1. PCNs Followed a Non-Scale Free Connectivity Distribution
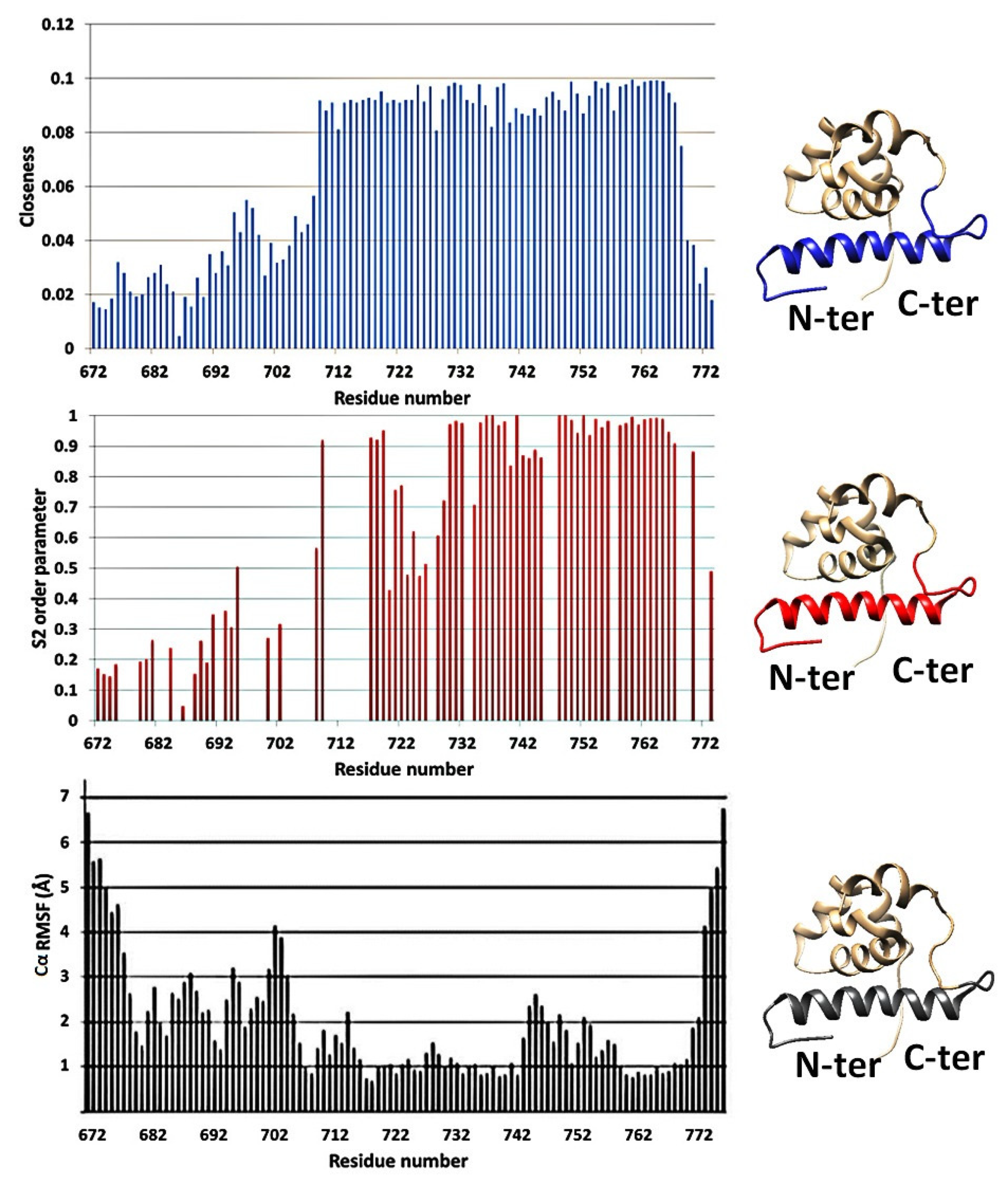
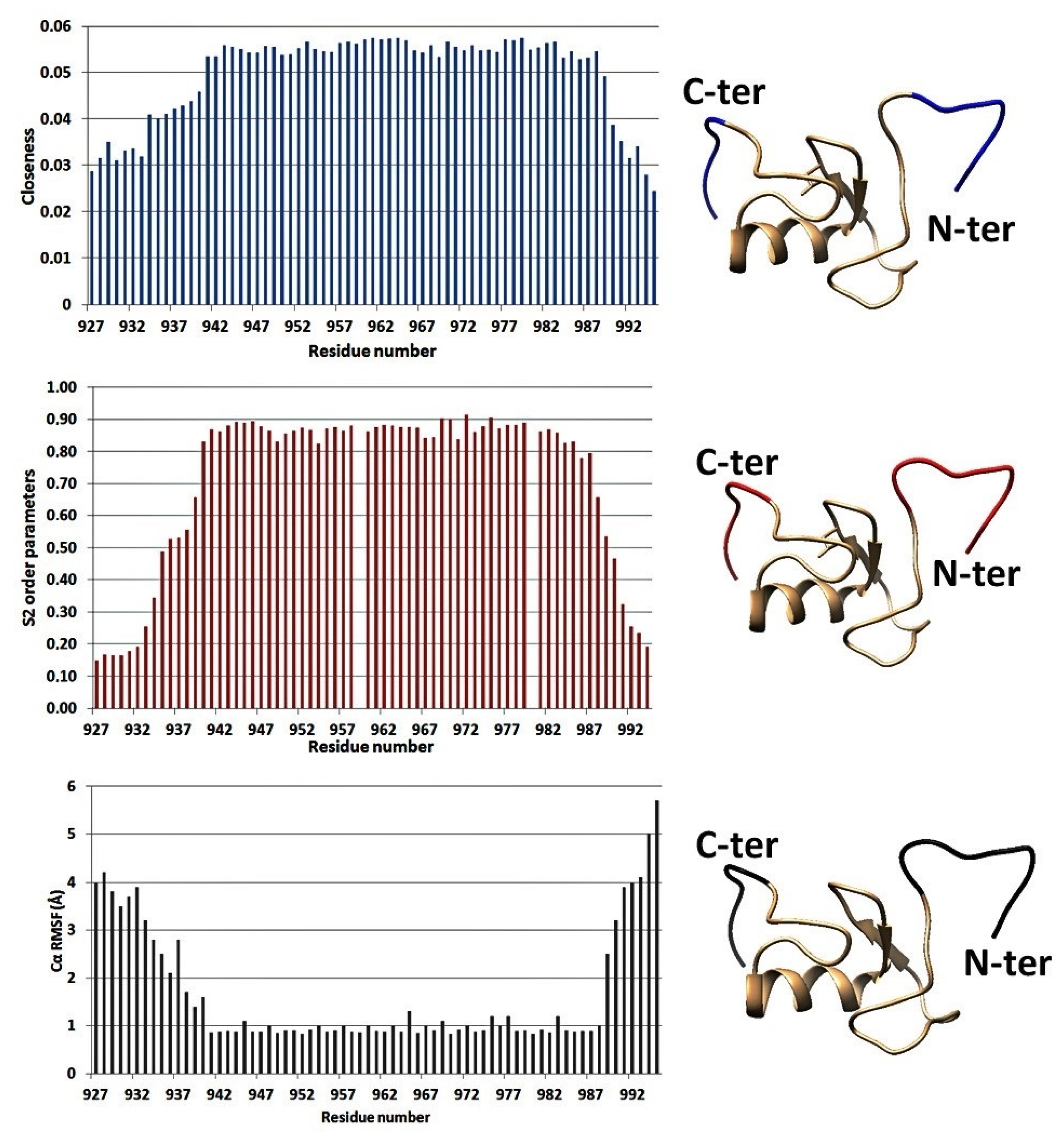
3.2. Protein Plasticity Correlated Inversely to Closeness Centrality
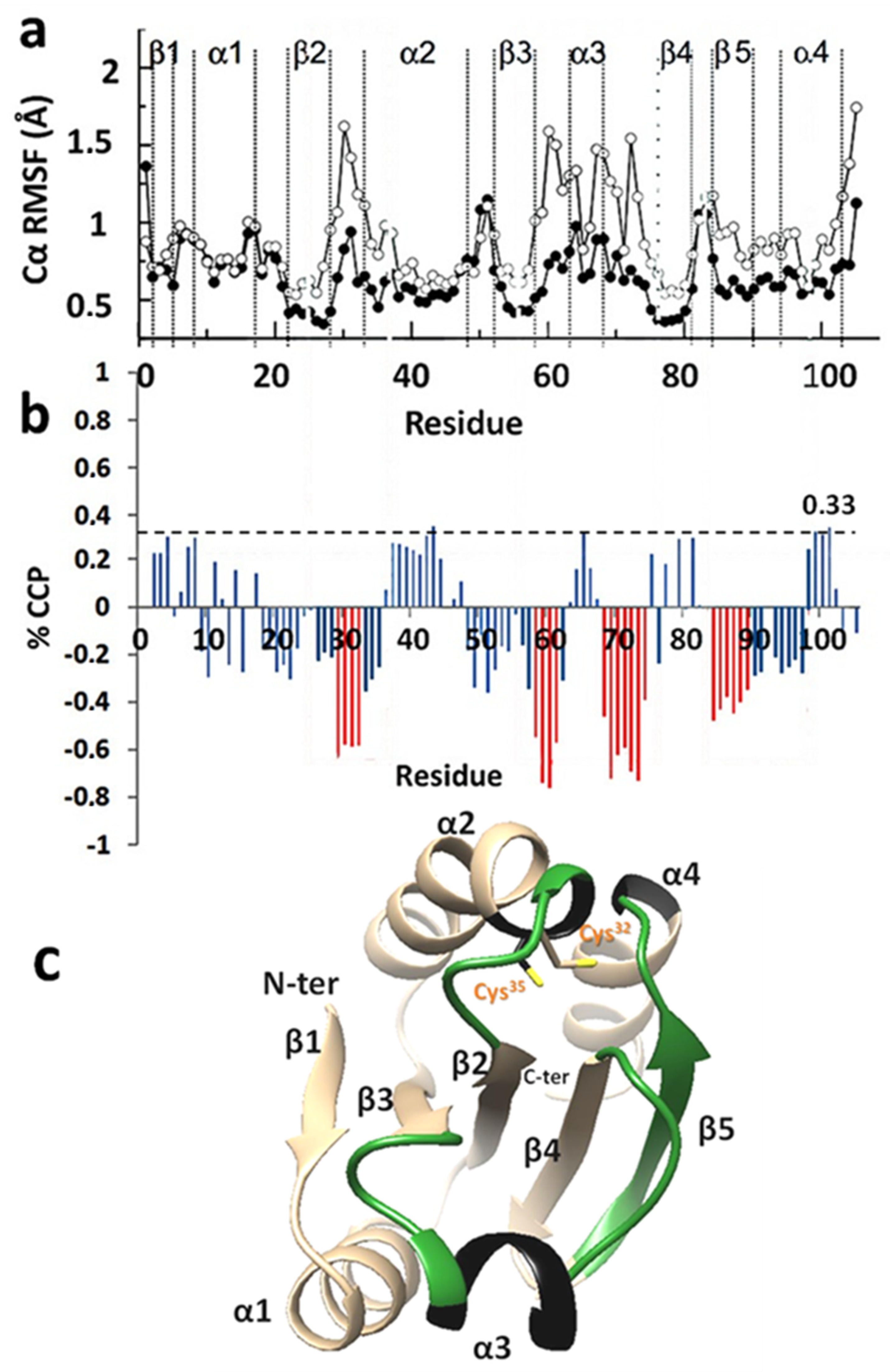
3.3. Percentage CCP Changes May Indicate Alterations in Plasticity upon Formation of Disulfides within A Protein Sequence
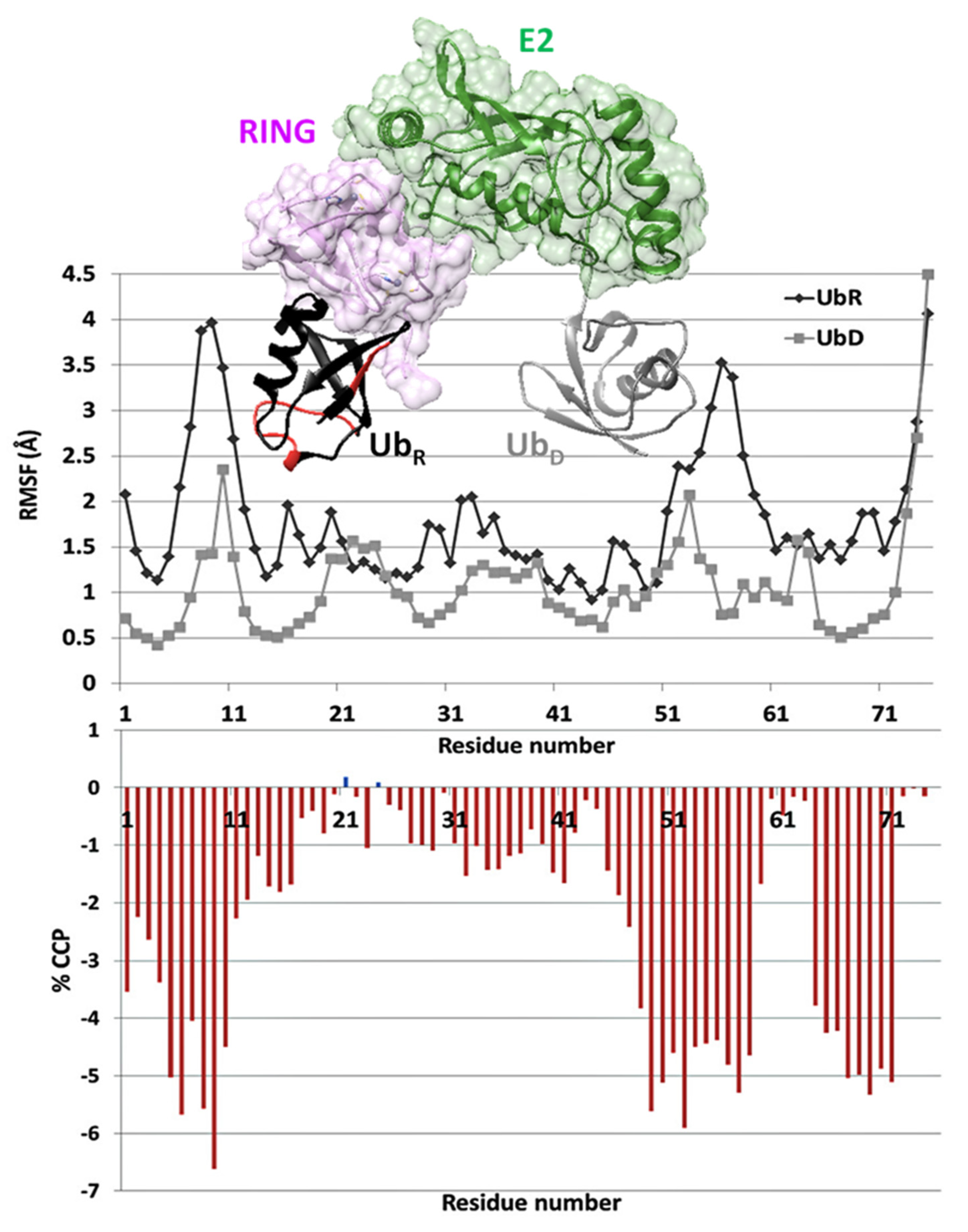
3.4. CCP Changes May Indicate Alterations in Plasticity upon Complexation
4. Discussion
General Outcomes from the Measurements of Centrality Parameters
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Case, D.A. Molecular Dynamics and NMR Spin Relaxation in Proteins. Acc. Chem. Res. 2002, 35, 325–331. [Google Scholar] [CrossRef]
- Gu, Y.; Li, D.-W.; Brüschweiler, R. NMR Order Parameter Determination from Long Molecular Dynamics Trajectories for Objective Comparison with Experiment. J. Chem. Theory Comput. 2014, 10, 2599–2607. [Google Scholar] [CrossRef]
- Kay, L.E. Solution NMR spectroscopy of supra-molecular systems, why bother? A methyl-TROSY view. J. Magn. Reson. 2011, 210, 159–170. [Google Scholar] [CrossRef]
- Van den Bedem, H.; Bhabha, G.; Yang, K.; Wright, P.E.; Fraser, J.S. Automated identification of functional dynamic contact networks from X-ray crystallography. Nat. Methods 2013, 10, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Hensen, U.; Meyer, T.; Haas, J.; Rex, R.; Vriend, G.; Grubmuller, H. Exploring protein dynamics space: The dynasome as the missing link between protein structure and function. PLoS ONE 2012, 7, e33931. [Google Scholar] [CrossRef]
- Sutto, L.; Marsili, S.; Valencia, A.; Gervasio, F.L. From residue coevolution to protein conformational ensembles and functional dynamics. Proc. Natl. Acad. Sci. USA 2015, 112, 13567–13572. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarty, B.; Parekh, N. NAPS: Network Analysis of Protein Structures. Nucleic Acids Res. 2016, 44, W375–W382. [Google Scholar] [CrossRef] [PubMed]
- Grewal, R.K.; Roy, S. Modeling proteins as residue interaction networks. Protein Pept. Lett. 2015, 22, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Zhou, J.; Sun, M.; Chen, J.; Hu, G.; Shen, B. The construction of an amino acid network for understanding protein structure and function. Amino Acids 2014, 46, 1419–1439. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, L.; De Ruvo, M.; Paci, P.; Santoni, D.; Giuliani, A. Protein contact networks: An emerging paradigm in chemistry. Chem. Rev. 2013, 113, 1598–1613. [Google Scholar] [CrossRef] [PubMed]
- Greene, L.H. Protein structure networks. Brief. Funct. Genom. 2012, 11, 469–478. [Google Scholar] [CrossRef]
- Taylor, N.R. Small world network strategies for studying protein structures and binding. Comput. Struct. Biotechnol. J. 2013, 5, e201302006. [Google Scholar] [CrossRef]
- Taketomi, H.; Ueda, Y.; Go, N. Studies on protein folding, unfolding and fluctuations by computer simulation. I. The effect of specific amino acid sequence represented by specific inter-unit interactions. Int. J. Pept. Protein Res. 1975, 7, 445–459. [Google Scholar] [CrossRef]
- Sobolev, V.; Sorokine, A.; Prilusky, J.; Abola, E.; Edelman, M. Automated analysis of interatomic contacts in proteins. Bioinformatics 1999, 15, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Atilgan, A.R.; Durell, S.R.; Jernigan, R.L.; Demirel, M.C.; Keskin, O.; Bahar, I. Anisotropy of Fluctuation Dynamics of Proteins with an Elastic Network Model. Biophys. J. 2001, 80, 505–515. [Google Scholar] [CrossRef]
- Togashi, Y.; Flechsig, H. Coarse-Grained Protein Dynamics Studies Using Elastic Network Models. Int. J. Mol. Sci. 2018, 19, 3899. [Google Scholar] [CrossRef]
- Patra, S.M.; Vishveshwara, S. Backbone cluster identification in proteins by a graph theoretical method. Biophys. Chem. 2000, 84, 13–25. [Google Scholar] [CrossRef]
- Paola, L.D.; Paci, P.; Santoni, D.; Ruvo, M.D.; Giuliani, A. Proteins as sponges: A statistical journey along protein structure organization principles. J. Chem. Inf. Modeling 2012, 52, 474–482. [Google Scholar] [CrossRef]
- Atilgan, A.R.; Akan, P.; Baysal, C. Small-world communication of residues and significance for protein dynamics. Biophys. J. 2004, 86 Pt 1, 85–91. [Google Scholar] [CrossRef]
- Bode, C.; Kovacs, I.A.; Szalay, M.S.; Palotai, R.; Korcsmaros, T.; Csermely, P. Network analysis of protein dynamics. FEBS Lett. 2007, 581, 2776–2782. [Google Scholar] [CrossRef]
- Atilgan, A.R.; Turgut, D.; Atilgan, C. Screened nonbonded interactions in native proteins manipulate optimal paths for robust residue communication. Biophys. J. 2007, 92, 3052–3062. [Google Scholar] [CrossRef]
- Amitai, G.; Shemesh, A.; Sitbon, E.; Shklar, M.; Netanely, D.; Venger, I.; Pietrokovski, S. Network analysis of protein structures identifies functional residues. J. Mol. Biol. 2004, 344, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Del Sol, A.; Fujihashi, H.; O’Meara, P. Topology of small-world networks of protein-protein complex structures. Bioinformatics 2005, 21, 1311–1315. [Google Scholar] [CrossRef]
- Cusack, M.P.; Thibert, B.; Bredesen, D.E.; Del Rio, G. Efficient identification of critical residues based only on protein structure by network analysis. PLoS ONE 2007, 2, e421. [Google Scholar] [CrossRef]
- Freeman, L.C. Centrality in social networks conceptual clarification. Soc. Netw. 1978, 1, 215–239. [Google Scholar] [CrossRef]
- Bonacich, P. Factoring and weighting approaches to status scores and clique identification. J. Math. Sociol. 1972, 2, 113–120. [Google Scholar] [CrossRef]
- Chasapis, C.T. Building Bridges between Structural and Network-Based Systems Biology. Mol. Biotechnol. 2019, 61, 221–229. [Google Scholar] [CrossRef]
- Vendruscolo, M.; Dokholyan, N.V.; Paci, E.; Karplus, M. Small-world view of the amino acids that play a key role in protein folding. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2002, 65 Pt 1, 061910. [Google Scholar] [CrossRef] [PubMed]
- Thibert, B.; Bredesen, D.E.; del Rio, G. Improved prediction of critical residues for protein function based on network and phylogenetic analyses. BMC Bioinform. 2005, 6, 213. [Google Scholar] [CrossRef]
- Chasapis, C.T. Hierarchical core decomposition of RING structure as a method to capture novel functional residues within RING-type E3 ligases: A structural systems biology approach. Comput. Biol. Med. 2018, 100, 86–91. [Google Scholar] [CrossRef]
- Li, H.; Chang, Y.Y.; Lee, J.Y.; Bahar, I.; Yang, L.W. DynOmics: Dynamics of structural proteome and beyond. Nucleic Acids Res. 2017, 45, W374–W380. [Google Scholar] [CrossRef]
- Di Paola, L.; Giuliani, A. Protein contact network topology: A natural language for allostery. Curr. Opin. Struct. Biol. 2015, 31, 43–48. [Google Scholar] [CrossRef]
- Brysbaert, G.; Mauri, T.; De Ruyck, J.; Lensink, M.F. Identification of key residues in proteins through centrality analysis and flexibility prediction with RINspector. Curr. Protoc. Bioinform. 2019, 65, e66. [Google Scholar] [CrossRef]
- Di Paola, L.; Mei, G.; Di Venere, A.; Giuliani, A. Disclosing Allostery through Protein Contact Networks. In Allostery; Humana: New York, NY, USA, 2021; Volume 2253, pp. 7–20. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Dalkas, G.A.; Chasapis, C.T.; Gkazonis, P.V.; Bentrop, D.; Spyroulias, G.A. Conformational dynamics of the anthrax lethal factor catalytic center. Biochemistry 2010, 49, 10767–10769. [Google Scholar] [CrossRef] [PubMed]
- Han, S. Molecular dynamics simulations of thioredoxin with S-glutathiolated cysteine-73. Biochem. Biophys. Res. Commun. 2007, 362, 532–537. [Google Scholar] [CrossRef]
- Mintis, D.G.; Chasapi, A.; Poulas, K.; Lagoumintzis, G.; Chasapis, C.T. Assessing the Direct Binding of Ark-Like E3 RING Ligases to Ubiquitin and Its Implication on Their Protein Interaction Network. Molecules 2020, 25, 4787. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Chasapis, C.T. Interactions between metal binding viral proteins and human targets as revealed by network-based bioinformatics. J. Inorg. Biochem. 2018, 186, 157–161. [Google Scholar] [CrossRef]
- Chasapis, C.T.; Konstantinoudis, G. Protein isoelectric point distribution in the interactomes across the domains of life. Biophys. Chem. 2020, 256, 106269. [Google Scholar] [CrossRef]
- Peana, M.; Chasapis, C.T.; Simula, G.; Medici, S.; Zoroddu, M.A. A Model for Manganese interaction with Deinococcus radiodurans proteome network involved in ROS response and defense. J. Trace Elem. Med. Biol. 2018, 50, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Chasapis, C.T. Shared gene-network signatures between the human heavy metal proteome and neurological disorders and cancer types. Metallomics 2018, 10, 1678–1686. [Google Scholar] [CrossRef]
- Barabási, A.-L.; Albert, R. Emergence of Scaling in Random Networks. Science 1999, 286, 509–512. [Google Scholar] [CrossRef] [PubMed]
- Amaral, L.A.; Scala, A.; Barthelemy, M.; Stanley, H.E. Classes of small-world networks. Proc. Natl. Acad. Sci. USA 2000, 97, 11149–11152. [Google Scholar] [CrossRef]
- Gkazonis, P.V.; Dalkas, G.A.; Chasapis, C.T.; Vlamis-Gardikas, A.; Bentrop, D.; Spyroulias, G.A. Purification and biophysical characterization of the core protease domain of anthrax lethal factor. Biochem. Biophys. Res. Commun. 2010, 396, 643–647. [Google Scholar] [CrossRef] [PubMed]
- Vourtsis, D.J.; Chasapis, C.T.; Pairas, G.; Bentrop, D.; Spyroulias, G.A. NMR conformational properties of an Anthrax Lethal Factor domain studied by multiple amino acid-selective labeling. Biochem. Biophys. Res. Commun. 2014, 450, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Chasapis, C.T.; Kandias, N.G.; Episkopou, V.; Bentrop, D.; Spyroulias, G.A. NMR-based insights into the conformational and interaction properties of Arkadia RING-H2 E3 Ub ligase. Proteins Struct. Funct. Bioinform. 2012, 80, 1484–1489. [Google Scholar] [CrossRef]
- Kandias, N.G.; Chasapis, C.T.; Bentrop, D.; Episkopou, V.; Spyroulias, G.A. High yield expression and NMR characterization of Arkadia E3 ubiquitin ligase RING-H2 finger domain. Biochem. Biophys. Res. Commun. 2009, 378, 498–502. [Google Scholar] [CrossRef]
- Birkou, M.; Chasapis, C.T.; Marousis, K.D.; Loutsidou, A.K.; Bentrop, D.; Lelli, M.; Herrmann, T.; Carthy, J.M.; Episkopou, V.; Spyroulias, G.A. A Residue Specific Insight into the Arkadia E3 Ubiquitin Ligase Activity and Conformational Plasticity. J. Mol. Biol. 2017, 429, 2373–2386. [Google Scholar] [CrossRef]
- Chasapis, C.T.; Loutsidou, A.K.; Orkoula, M.G.; Spyroulias, G.A. Zinc Binding Properties of Engineered RING Finger Domain of Arkadia E3 Ubiquitin Ligase. Bioinorg. Chem. Appl. 2010, 2010, 323152. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.D.; Mace, P.D.; Day, C.L. Secondary ubiquitin-RING docking enhances Arkadia and Ark2C E3 ligase activity. Nat. Struct. Mol. Biol. 2015, 23, 45–52. [Google Scholar] [CrossRef]
- Demchenko, A.P. Fluorescence and Dynamics in Proteins. In Topics in Fluorescence Spectroscopy; Springer: Boston, MA, USA, 2002; Volume 3, pp. 65–111. [Google Scholar]
- Van den Bedem, H.; Fraser, J.S. Integrative, dynamic structural biology at atomic resolution—It’s about time. Nat. Methods 2015, 12, 307–318. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| A/A | Protein Name | PDB ID |
|---|---|---|
| 1 | Escherichia coli glutaredoxin 4 | 1YKA |
| 2 | Reduced human thioredoxin 1 | 1ERT |
| 3 | Oxidized human thioredoxin 1 | 1ERU |
| 4 | Anthrax lethal factor core protease domain | 2L0R |
| 5 | N-terminal RING domain of E3 ligase Arkadia | 2KIZ |
| 6 | Ubiqutin | 1D3Z |
| 7 | UbR and UbD of the complex UbR-RING-E2-UbD | 5D0K |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chasapis, C.T.; Vlamis-Gardikas, A. Probing Conformational Dynamics by Protein Contact Networks: Comparison with NMR Relaxation Studies and Molecular Dynamics Simulations. Biophysica 2021, 1, 157-167. https://doi.org/10.3390/biophysica1020012
Chasapis CT, Vlamis-Gardikas A. Probing Conformational Dynamics by Protein Contact Networks: Comparison with NMR Relaxation Studies and Molecular Dynamics Simulations. Biophysica. 2021; 1(2):157-167. https://doi.org/10.3390/biophysica1020012
Chicago/Turabian StyleChasapis, Christos T., and Alexios Vlamis-Gardikas. 2021. "Probing Conformational Dynamics by Protein Contact Networks: Comparison with NMR Relaxation Studies and Molecular Dynamics Simulations" Biophysica 1, no. 2: 157-167. https://doi.org/10.3390/biophysica1020012
APA StyleChasapis, C. T., & Vlamis-Gardikas, A. (2021). Probing Conformational Dynamics by Protein Contact Networks: Comparison with NMR Relaxation Studies and Molecular Dynamics Simulations. Biophysica, 1(2), 157-167. https://doi.org/10.3390/biophysica1020012