



Concurrent AtC Multiscale Modeling of Material Coupled Thermo-Mechanical Behaviors: A Review



Abstract
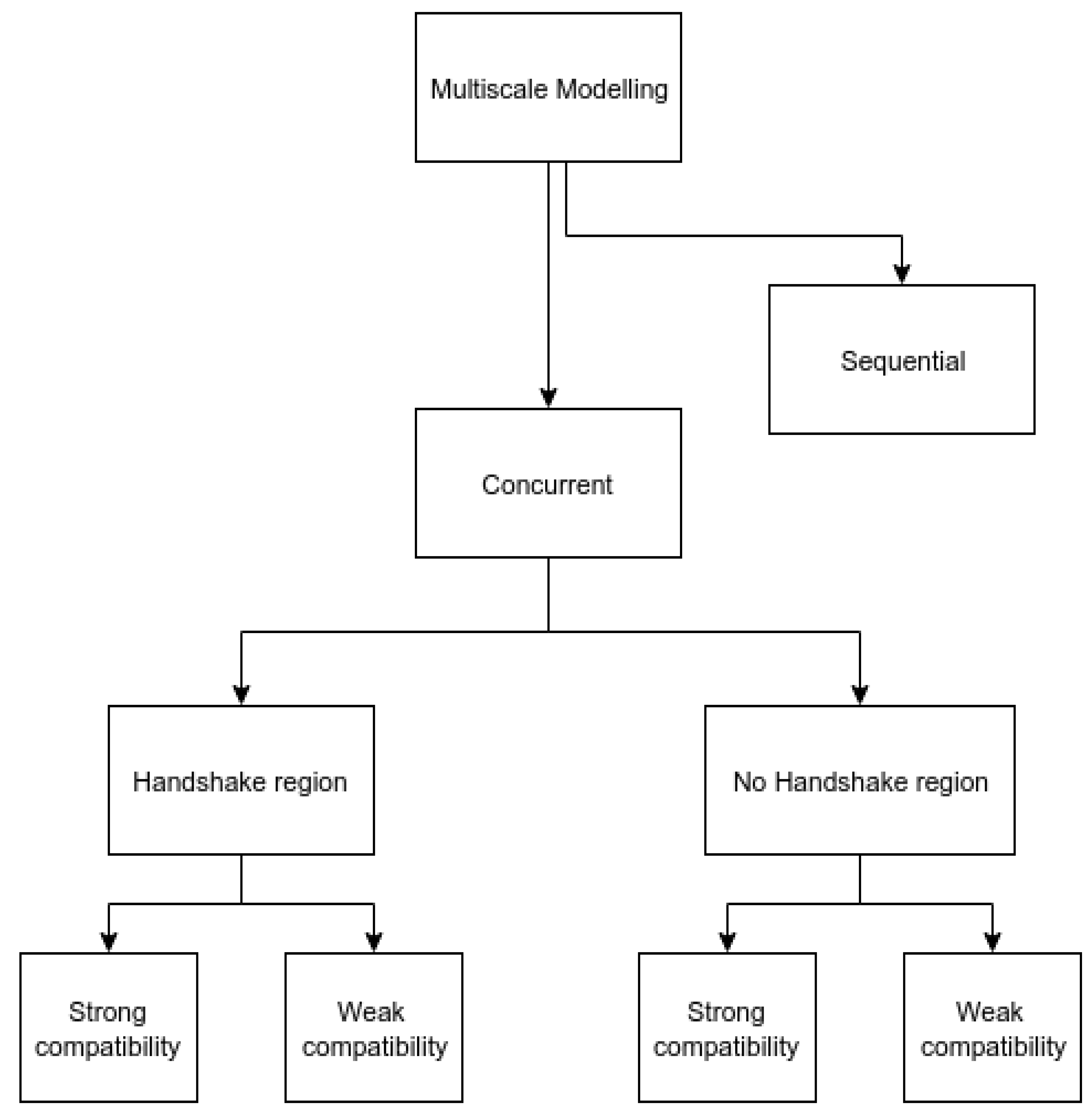
1. Introduction
2. Coarse-Graining Methods
2.1. Virial Theorem (VT)
2.2. Irving and Kirkwood Method (IK)
2.3. Hardy Method (HM)
2.4. Atomic Kinetic Temperature
2.5. Hardy Temperature (HT)
2.6. Equivalent Continuum (EC)
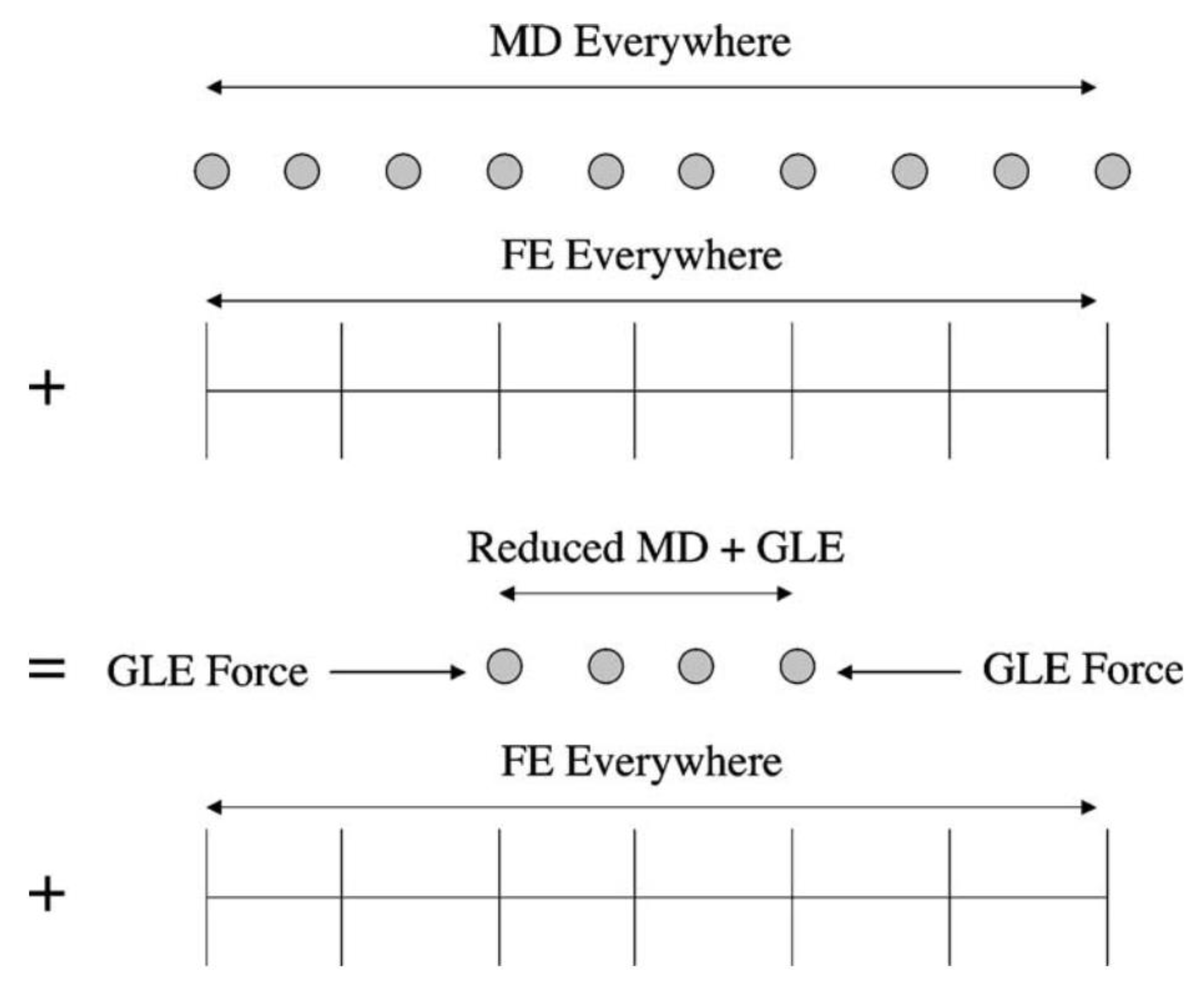
3. Downscaling Methods
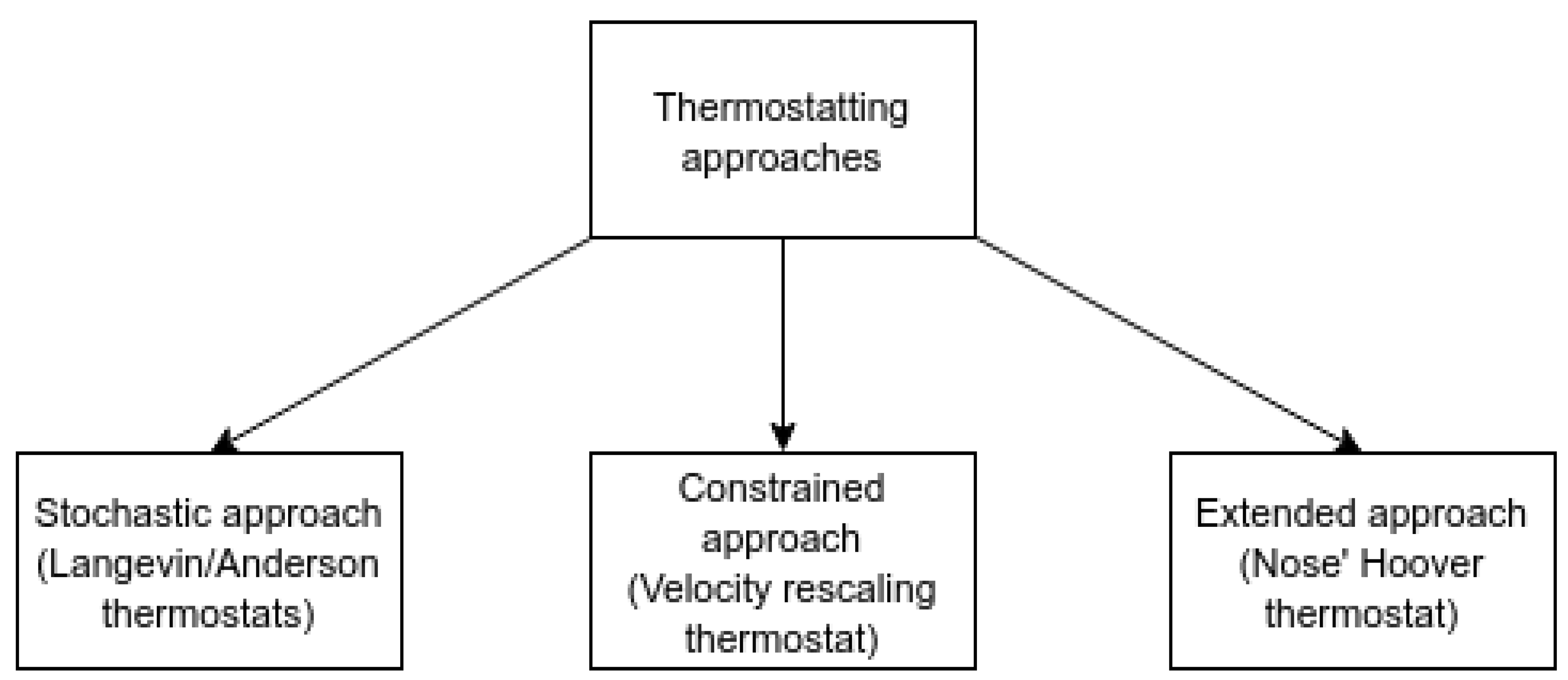
Thermostats
4. Mechanical Coupling Methods
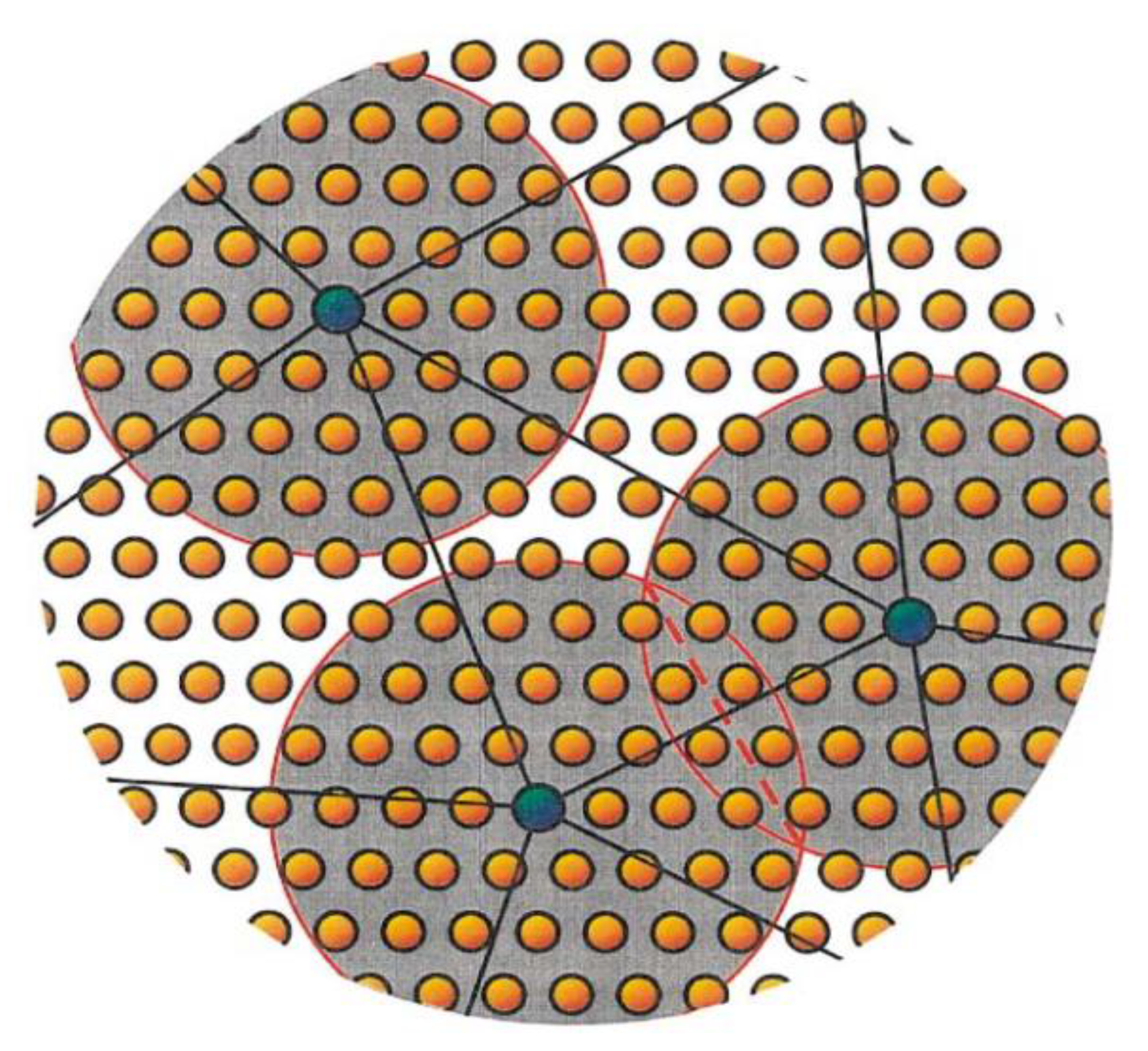
4.1. Quasicontinuum Method (QC)
4.2. Bridging Scale Decomposition Method (BSD)
4.3. Virtual Internal Bond (VIB)
5. Thermal Coupling Methods
5.1. BSD Uni-Directional Temperature Coupling (BUTC)
5.2. BSD Bi-Directional Temperature Coupling (BBTC)
5.3. BSD Spatially-Varying Thermostats (BST)
6. Thermomechanical Coupling Methods
6.1. Thermo-Mechanical Equivalent Continuum Theory (TMEC)
6.2. Filter and Restitution Method (FR)
6.3. Atom-Continuum Coupled Model (ACC)
6.4. Wavelet-Based Denoising (WBD)
6.5. Finite Temperatures Using Spatial Filters (SF)
6.6. Extended Irving-Kirkwood Statistical Mechanical (EIK)
7. Conclusive Remarks
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A. Tables

{kind=link}
{kind=link}
{kind=link}
{kind=link}
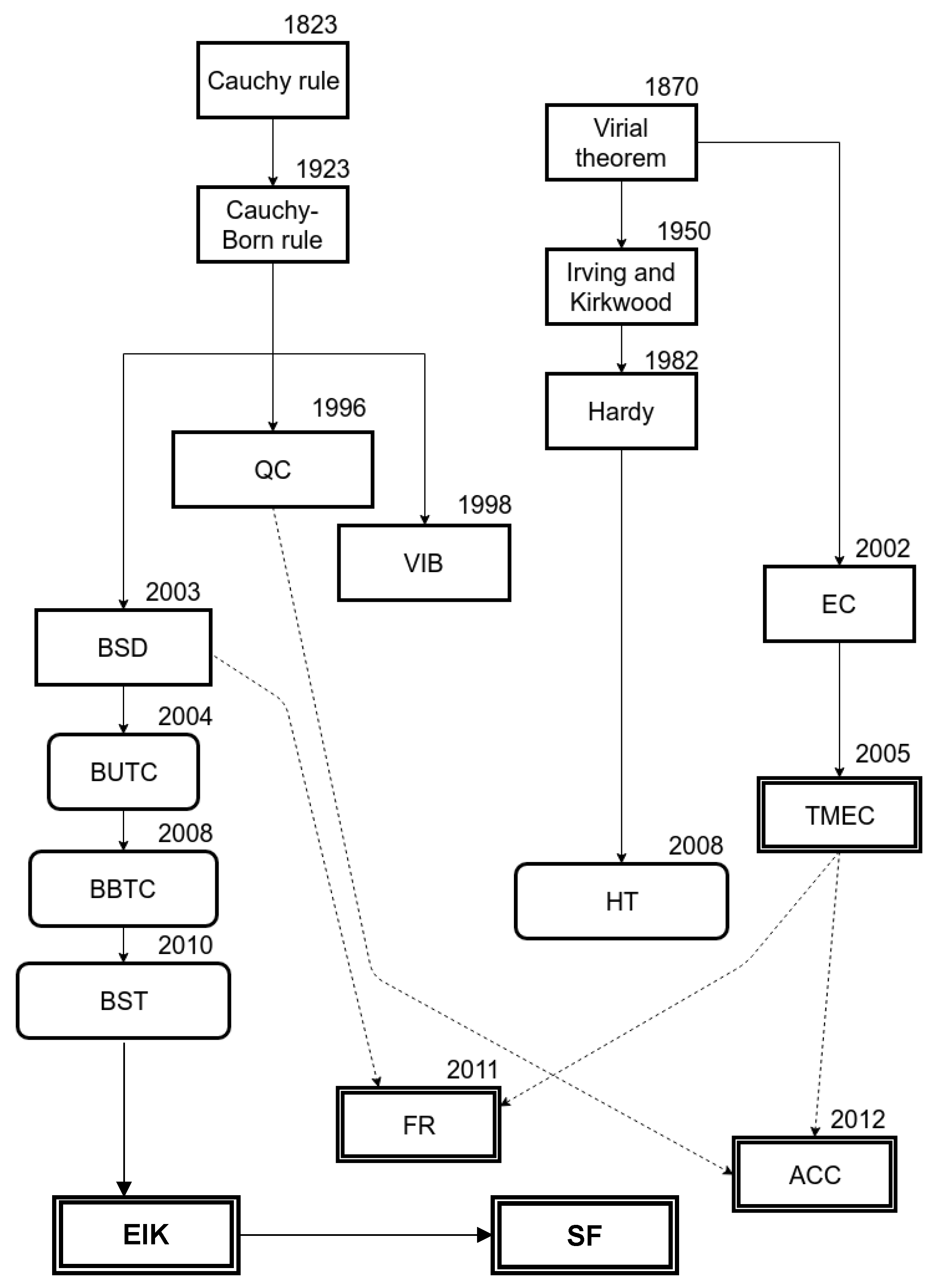{kind=link}
| Method | Features |
|---|---|
| VT [26] | Introduced the ability to map discrete atomic quantities to a continuum representation of stress as a point-wise function using summation over sub ensembles. |
| CB | Introduced a method to map uniform deformation at macroscale to atomic lattice deformation. |
| IK [31] | Summation over sub ensembles in VT is replaced by density of atomic functions in conjunction with a Dirac delta function as the localization function. |
| EC [41] | A purely mechanical theory which defines work-conjugate stress and deformation fields from non-local MD system. |
| HM [35] | Introduced localization functions with finite range (compact support) instead of using just the Dirac delta function as in IK. |
| QC [18] | Introduced the idea of deriving constitutive parameter for FE nodes (rep atoms) from a small set of atoms (crystallite) based on CB. |
| BSD [17] | Introduced the idea of decomposing total displacement can be MD displacement) into coarse and fine scale components which can and cannot be projected to FE shape functions respectively. |
| VIB [60] | Introduced the idea of filling continuum with virtual material points which are not atoms but obey a pseudo potential energy function based on experimental data. |
| HT [30] | Uses the relative atomic velocity derived in Hardy method to define a weighted average temperature. |
| BUTC [61] | Uses the projection matrix derived in BSD to project atomic temperature to FE shape functions, i.e., up-scaling only. |
| BBTC [62] | Extended BUTC by implemented down-scaling and introduced row-sum lumping as an approximation for the projection method used in BUTC and BSD. |
| BST [2] | Introduced the ability to prescribe spatially varying initial conditions, Neumann and Dirichlet boundary conditions to the MD-FE coupled material model using Gaussian isokinetic thermostats. |
| TMEC [64] | Introduced the idea of decomposing atomic motion into high frequency thermal motion and low frequency structural deformation motion using Fourier analysis. |
| FS [57] | Demonstrated 1D examples for thermo-mechanical coupling model derived from BSD and TMEC methods. |
| ACC [67] | Demonstrated 1D examples for thermo-mechanical coupling model derived from QC and TMEC methods but uses an averaging scheme to approximate the Fourier analysis method used in TMEC. |
| SF [69] | Demonstrated 2D example of a high-velocity impact test using selective spatial filters for damping the wavelength modes for coupling atomistic and continuum models at finite temperatures. |
| EIK [70] | Demonstrated 2D examples of fluxes that can be then obtained to quantify the flow of conserved quantities across the lattice cells as well as those that flow back and forth within the cells. |
References
- Yip, S.; Short, M.P. Multiscale materials modelling at the mesoscale. Nat. Mater. 2013, 12, 774–777. [Google Scholar] [CrossRef] [PubMed]
- Templeton, J.A.; Jones, R.E.; Wagner, G.J. Application of a field-based method to spatially varying thermal transport problems in molecular dynamics. Model. Simul. Mater. Sci. Eng. 2010, 18, 085007. [Google Scholar] [CrossRef]
- Trelewicz, J.R.; Schuh, C.A. The Hall-Petch breakdown in nanocrystalline metals: A crossover to glass-like deformation. Acta Mater. 2007, 55, 5948–5958. [Google Scholar] [CrossRef]
- Miller, R.E.; Rodney, D. On the nonlocal nature of dislocation nucleation during nanoindentation. J. Mech. Phys. Solids 2008, 56, 1203–1223. [Google Scholar] [CrossRef]
- Qian, D.; Wagner, G.J.; Liu, W.K. A multiscale projection method for the analysis of carbon nanotubes. Comput. Methods Appl. Mech. Eng. 2004, 193, 1603–1632. [Google Scholar] [CrossRef]
- Picinbono, G.; Lombardo, J.C.; Delingette, H.; Ayache, N. Improving realism of a surgery simulator: Linear anisotropic elasticity, complex interactions and force extrapolation. J. Vis. Comput. Animat. 2002, 13, 147–167. [Google Scholar] [CrossRef]
- Yu, W.; Blanchard, J.P. An elastic-plastic indentation model and its solutions. J. Mater. Res. 1996, 11, 2358–2367. [Google Scholar] [CrossRef]
- Tang, K.C.; Faulkner, A.; Schwarzer, N.; Arnell, R.D.; Richter, F. Comparison between an elastic-perfectly plastic finite element model and a purely elastic analytical model for a spherical indenter on a layered substrate. Thin Solid Film. 1997, 300, 177–188. [Google Scholar] [CrossRef]
- Diard, O.; Leclercq, S.; Rousselier, G.; Cailletaud, G. Evaluation of finite element based analysis of 3D multicrystalline aggregates plasticity Application to crystal plasticity model identification and the study of stress and strain fields near grain boundaries. Int. J. Plast. 2005, 21, 691–722. [Google Scholar] [CrossRef]
- van der Giessen, E.; Needleman, A. Discrete Dislocation Plasticity: {A} Simple Planar Model. Model. Simul. Mater. Sci. Eng. 1995, 3, 689–735. [Google Scholar] [CrossRef]
- Griggs, J.; Gamboa, E.; Lavigne, O. A review of modelling high pH stress corrosion cracking of high pressure gas pipelines. Mater. Corros. 2015, 61, 251–263. [Google Scholar] [CrossRef]
- Broughton, J.; Abraham, F.; Bernstein, N.; Kaxiras, E. Concurrent coupling of length scales: Methodology and application. Phys. Rev. B 1999, 60, 2391–2403. [Google Scholar] [CrossRef]
- Lu, G.; Tadmor, E.B.; Kaxiras, E. From electrons to finite elements: A concurrent multiscale approach for metals. Phys. Rev. B Condens. Matter Mater. Phys. 2006, 73, 024108. [Google Scholar] [CrossRef]
- Vashishta, P.; Kalia, R.K.; Nakano, A. Multimillion atom simulations of dynamics of oxidation of an aluminum nanoparticle and nanoindentation on ceramics. J. Phys. Chem. B 2006, 110, 3727–3733. [Google Scholar] [CrossRef]
- Williamson, R.L.; Hales, J.D.; Novascone, S.R.; Tonks, M.R.; Gaston, D.R.; Permann, C.J.; Andrs, D.; Martineau, R.C. Multidimensional multiphysics simulation of nuclear fuel behavior. J. Nucl. Mater. 2012, 423, 149–163. [Google Scholar] [CrossRef]
- Tonks, M.R.; Millett, P.C.; Nerikar, P.; Du, S.; Andersson, D.; Stanek, C.R.; Gaston, D.; Andrs, D.; Williamson, R. Multiscale development of a fission gas thermal conductivity model: Coupling atomic, meso and continuum level simulations. J. Nucl. Mater. 2013, 440, 193–200. [Google Scholar] [CrossRef]
- Wagner, G.J.; Liu, W.K. Coupling of atomistic and continuum simulations using a bridging scale decomposition. J. Comput. Phys. 2003, 190, 249–274. [Google Scholar] [CrossRef]
- Tadmor, E.B.; Ortiz, M.; Phillips, R. Quasicontinuum analysis of defects in solids. Philos. Mag. A 1996, 73, 1529–1563. [Google Scholar] [CrossRef]
- Xiao, S.P.; Belytschko, T. A bridging domain method for coupling continua with molecular dynamics. Comput. Methods Appl. Mech. Eng. 2004, 193, 1645–1669. [Google Scholar] [CrossRef]
- Tadmor, E.B.; Miller, R.E. Modeling Materials. Continuum, Atomistic and Multiscale Techniques; Cambridge University Press: Cambridge, UK, 2011; Volume 1, ISBN 9780521856980. [Google Scholar]
- Weinan, E.; Engquist, B.; Li, X.; Ren, W.; Vanden-Eijnden, E. Heterogeneous multiscale methods: A review. Commun. Comput. Phys. 2007, 2, 367–450. [Google Scholar]
- Verlet, L. Computer “Experiments” on Classical Fluids. I. Thermodynamical Properties of Lennard-Jones Molecules. Phys. Rev. 1967, 159, 98. [Google Scholar] [CrossRef]
- Gear, C.W. Numerical Initial Value Problems in Ordinary Differential Equations; Prentice Hall: Hoboken, NJ, USA, 1971. [Google Scholar]
- Tuckerman, M.; Martyna, G.J.; Berne, B.J. Reversible multiple time scale molecular dynamics. J. Chem. Phys. 1992, 97, 1990–2001. [Google Scholar] [CrossRef]
- Liu, W.K.; Karpov, E.G.; Zhang, S.; Park, H.S. An Introduction to Computational Nanomechanics and Materials. Comput. Methods Appl. Mech. Eng. 2004, 193, 1529–1578. [Google Scholar] [CrossRef]
- Clausius, R. On a mechanical law applicable to heat. Poggendorffs Ann. 1870, 141, 124–130. [Google Scholar] [CrossRef]
- Baker, W.F.; Beghini, L.L.; Mazurek, A.; Carrion, J.; Beghini, A. Maxwell’s reciprocal diagrams and discrete Michell frames. Struct. Multidiscip. Optim. 2013, 48, 267–277. [Google Scholar] [CrossRef]
- Tsai, D.H. The virial theorem and stress calculation in molecular dynamics. J. Chem. Phys. 1979, 70, 1375. [Google Scholar] [CrossRef]
- Cheung, K.S.; Yip, S. Atomic-level stress in an inhomogeneous system. J. Appl. Phys. 1991, 70, 5688–5690. [Google Scholar] [CrossRef]
- Webb, E.B.; Zimmerman, J.A.; Seel, S.C. Reconsideration of Continuum Thermomechanical Quantities in Atomic Scale Simulations. Math. Mech. Solids 2008, 13, 221–266. [Google Scholar] [CrossRef]
- Irving, J.H.; Kirkwood, J.G. The Statistical Mechanical Theory of Transport Processes. IV. The Equations of Hydrodynamics. J. Chem. Phys. 1950, 18, 817–829. [Google Scholar] [CrossRef]
- Admal, N.C.; Tadmor, E.B. A unified interpretation of stress in molecular systems. J. Elast. 2010, 100, 63–143. [Google Scholar] [CrossRef]
- Lehoucq, R.B.; Von Lilienfeld-Toal, A. Translation of Walter Noll’s “derivation of the fundamental equations of continuum thermodynamics from statistical mechanics. ” J. Elast. 2010, 100, 5–24. [Google Scholar] [CrossRef]
- Admal, N.C.; Tadmor, E.B. Stress and heat flux for arbitrary multibody potentials: A unified framework. J. Chem. Phys. 2011, 134, 184106. [Google Scholar] [CrossRef] [PubMed]
- Hardy, R.J. Formulas for determining local properties in molecular-dynamics simulations: Shock waves. J. Chem. Phys. 1982, 76, 622. [Google Scholar] [CrossRef]
- Hardy, R.J.; Root, S.; Swanson, D.R. Continuum properties from molecular simulations. In Proceedings of the 12th International Conference of the American-Physical-Society-Topical-Group on Shock Compression of Condensed Matter, Part 1 of AIP Conference Proceedings, Anaheim, CA, USA, 10–15 July 2022; Volume 620, pp. 7–10. [Google Scholar]
- Root, S.; Hardy, R.J.; Swanson, D.R. Continuum predictions from molecular dynamics simulations: Shock waves. J. Chem. Phys. 2003, 118, 3161–3165. [Google Scholar] [CrossRef]
- Zimmerman, J.A.; WebbIII, E.B.; Hoyt, J.J.; Jones, R.E.; Klein, P.A.; Bammann, D.J. Calculation of stress in atomistic simulation. Model. Simul. Mater. Sci. Eng. 2004, 12, S319–S332. [Google Scholar] [CrossRef]
- Zimmerman, J.A.; Jones, R.E.; Templeton, J.A. A material frame approach for evaluating continuum variables in atomistic simulations. J. Comput. Phys. 2010, 229, 2364–2389. [Google Scholar] [CrossRef]
- Powles, J.G.; Rickayzen, G.; Heyes, D.M. Temperatures: Old, new and middle aged. Mol. Phys. 2005, 103, 1361–1373. [Google Scholar] [CrossRef]
- Zhou, M.; McDowell, D.L. Equivalent continuum for dynamically deforming atomistic particle systems. Philos. Mag. A 2002, 82, 2547–2574. [Google Scholar] [CrossRef]
- Schneider, T.; Stoll, E. Molecular-dynamics study of structural-phase transitions. I. One-component displacement models. Phys. Rev. B 1976, 13, 1216. [Google Scholar] [CrossRef]
- Schneider, T.; Stoll, E. Molecular-dynamics study of a three-dimensional one-component model for distortive phase transitions. Phys. Rev. B 1978, 17, 1302–1322. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Evans, D.J.; Hoover, W.G.; Failor, B.H.; Moran, B.; Ladd, A.J.C. Non-Equilibrium Molecular-Dynamics via Gauss Principle of Least Constraint. Phys. Rev. A 1983, 28, 1016–1021. [Google Scholar] [CrossRef]
- Zhang, T.; Sun, S.; Bai, H. Thermodynamically-consistent flash calculation in energy industry: From iterative schemes to a unified thermodynamics-informed neural network. Int. J. Energy Res. 2022, 46, 15332–15346. [Google Scholar] [CrossRef]
- Zhang, T.; Sun, S. Thermodynamics-Informed Neural Network (TINN) for Phase Equilibrium Calculations Considering Capillary Pressure. Energies 2021, 14, 7724. [Google Scholar] [CrossRef]
- Tadmor, E.; Phillips, R.; Ortiz, M. Mixed atomistic and continuum models of deformations in solids. J. ACM 1996, 7463, 4529–4534. [Google Scholar] [CrossRef]
- Knap, J.; Ortiz, M. An analysis of the quasicontinuum method. J. Mech. Phys. Solids 2001, 49, 1899–1923. [Google Scholar] [CrossRef]
- Miller, R.E.; Tadmor, E.B. The Quasicontinuum Method: Overview, applications and current directions. J. Comput. Mater. Des. 2002, 9, 203–239. [Google Scholar] [CrossRef]
- Dupuy, L.M.; Tadmor, E.B.; Miller, R.E.; Phillips, R. Finite-temperature quasicontinuum: Molecular dynamics without all the atoms. Phys. Rev. Lett. 2005, 95, 060202. [Google Scholar] [CrossRef]
- To, A.C.; Liu, W.K.; Kopacz, A. A finite temperature continuum theory based on interatomic potential in crystalline solids. Comput. Mech. 2008, 42, 531–541. [Google Scholar] [CrossRef]
- Tadmor, E.B.; Legoll, F.; Kim, W.K.; Dupuy, L.M.; Miller, R.E. Finite-Temperature Quasi-Continuum. Appl. Mech. Rev. 2013, 65, 010803. [Google Scholar] [CrossRef]
- Cai, W.; De Koning, M.; Bulatov, V.V.; Yip, S. Minimizing boundary reflections in coupled-domain simulations. Phys. Rev. Lett. 2000, 85, 3213–3216. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.J.; Karpov, E.G.; Liu, W.K. Molecular dynamics boundary conditions for regular crystal lattices. Comput. Methods Appl. Mech. Eng. 2004, 193, 1579–1601. [Google Scholar] [CrossRef]
- Weinan, E.; Huang, Z. Matching conditions in atomistic-continuum modeling of materials. Phys. Rev. Lett. 2001, 87, 135501. [Google Scholar] [CrossRef] [PubMed]
- Mathew, N.; Picu, R.C.; Bloomfield, M. Concurrent coupling of atomistic and continuum models at finite temperature. Comput. Methods Appl. Mech. Eng. 2011, 200, 765–773. [Google Scholar] [CrossRef]
- Park, H.S.; Liu, W.K. An introduction and tutorial on multiple-scale analysis in solids. Comput. Methods Appl. Mech. Eng. 2004, 193, 1733–1772. [Google Scholar] [CrossRef]
- Karpov, E.G.; Wagner, G.J.; Liu, W.K. A Green’s function approach to deriving non-reflecting boundary conditions in molecular dynamics simulations. Int. J. Numer. Methods Eng. 2005, 62, 1250–1262. [Google Scholar] [CrossRef]
- Gao, H.; Klein, P. Numerical simulation of crack growth in an isotropic solid with randomized internal cohesive bonds. J. Mech. Phys. Solids 1998, 46, 187–218. [Google Scholar] [CrossRef]
- Park, H.S.; Karpov, E.G.; Liu, W.K. A temperature equation for coupled atomistic/continuum simulations. Comput. Methods Appl. Mech. Eng. 2004, 193, 1713–1732. [Google Scholar] [CrossRef]
- Wagner, G.; Jones, R. An atomistic-to-continuum coupling method for heat transfer in solids. Comput. Method 2008, 197, 3351–3365. [Google Scholar] [CrossRef]
- Li, X.; Weinan, E. W E. Variational boundary conditions for molecular dynamics simulations of solids at low temperature. Commun. Comput. Phys. 2006, 1, 135–175. [Google Scholar]
- Zhou, M. Thermomechanical continuum representation of atomistic deformation at arbitrary size scales. Proc. R. Soc. A Math. Phys. Eng. Sci. 2005, 461, 3437–3472. [Google Scholar] [CrossRef]
- Born, M.; Huang, K. Dynamical Theory of Crystal Lattices; Clarendon Press: Oxford, UK, 1954; ISBN 0198503695. [Google Scholar]
- Coohen, L. Time-Frequency Analysis: Theory and Applications; Prentice-Hall: Englewood Cliffs, NJ, USA, 1995. [Google Scholar]
- Xiang, M.Z.; Cui, J.Z.; Li, B.W.; Tian, X. Atom-continuum coupled model for thermo-mechanical behavior of materials in micro-nano scales. Sci. China Phys. Mech. Astron. 2012, 55, 1125–1137. [Google Scholar] [CrossRef]
- To, A.C.; Fu, Y.; Kam Liu, W. Denoising methods for thermomechanical decomposition for quasi-equilibrium molecular dynamics simulations. Comput. Methods Appl. Mech. Eng. 2011, 200, 1979–1992. [Google Scholar] [CrossRef]
- Ramisetti, S.B.; Anciaux, G.; Molinari, J.F. A concurrent atomistic and continuum coupling method with applications to thermo-mechanical problems. Int. J. Numer. Methods Eng. 2014, 97, 707–738. [Google Scholar] [CrossRef]
- Chen, Y.; Shabanov, S.; McDowell, D.L. Concurrent atomistic-continuum modeling of crystalline materials. J. Appl. Phys. 2019, 126, 101101. [Google Scholar] [CrossRef]
- Curtin, W.A.; Miller, R.E. Atomistic / continuum coupling in computational materials science. Model. Simul. Mater. Sci. Eng. 2003, 11, R33–R68. [Google Scholar] [CrossRef]
- Buehler, M.J. Atomistic Modeling of Materials Failure; 2008; ISBN 9780387764252. [Google Scholar]
- Miller, R.E.; Tadmor, E.B. A unified framework and performance benchmark of fourteen multiscale atomistic/continuum coupling methods. Model. Simul. Mater. Sci. Eng. 2009, 17, 053001. [Google Scholar] [CrossRef]
- Wernik, J.M.; Meguid, S.A. Coupling atomistics and continuum in solids: Status, prospects, and challenges. Int. J. Mech. Mater. Des. 2009, 5, 79–110. [Google Scholar] [CrossRef]
- Wang, L. Mechanics of Asphalt Microstructure and Micromechanics; Springer: Berlin/Heidelberg, Germany, 2011; ISBN 9780071640978. [Google Scholar]
- De Pablo, J.J.; Curtin, W.A. Multiscale Modeling in Advanced Materials Research: Challenges, Novel Methods, and Emerging Applications. MRS Bull. 2011, 32, 905–911. [Google Scholar] [CrossRef]
- Panchal, J.H.; Kalidindi, S.R.; McDowell, D.L. Key computational modeling issues in Integrated Computational Materials Engineering. Comput. Des. 2013, 45, 4–25. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, Y.; Thomas, S.; Zhang, T.J. Concurrent AtC Multiscale Modeling of Material Coupled Thermo-Mechanical Behaviors: A Review. CivilEng 2022, 3, 1013-1038. https://doi.org/10.3390/civileng3040057
Lu Y, Thomas S, Zhang TJ. Concurrent AtC Multiscale Modeling of Material Coupled Thermo-Mechanical Behaviors: A Review. CivilEng. 2022; 3(4):1013-1038. https://doi.org/10.3390/civileng3040057
Chicago/Turabian StyleLu, Yang, Stephen Thomas, and Tian Jie Zhang. 2022. "Concurrent AtC Multiscale Modeling of Material Coupled Thermo-Mechanical Behaviors: A Review" CivilEng 3, no. 4: 1013-1038. https://doi.org/10.3390/civileng3040057
APA StyleLu, Y., Thomas, S., & Zhang, T. J. (2022). Concurrent AtC Multiscale Modeling of Material Coupled Thermo-Mechanical Behaviors: A Review. CivilEng, 3(4), 1013-1038. https://doi.org/10.3390/civileng3040057