
Meta-Analysis of Gene Expression in Bulk-Processed Post-Mortem Spinal Cord from ALS Patients and Normal Controls

Abstract

1. Introduction
2. Materials and Methods
2.1. Study Selection
2.2. GSE137810 Sample Filtering
2.3. GSE137810 Read Mapping
2.4. GSE137810 Covariates
2.5. GSE137810-NovaSeq Differential Expression Analysis
2.6. GSE137810-HiSeq Differential Expression Analysis
2.7. GSE255683 Analysis
2.8. SRP064478 Analysis
2.9. GSE26927 Analysis
2.10. E-MTAB-8635 Analysis
2.11. Meta-Analysis
2.12. Single-Nucleus Transcriptomics of Normal Human Spinal Cord (GSE190442)
2.13. Spatial Transcriptomics of the Normal Spinal Cord (GSE222322)
2.14. Spatial Transcriptomics of ALS Spinal Cord
3. Results
3.1. Dataset Comparison
3.2. Differential Expression Bias (mRNA Abundance, Gene Length, GC Content)
3.3. Meta-Analysis Moderates Differential Expression Bias
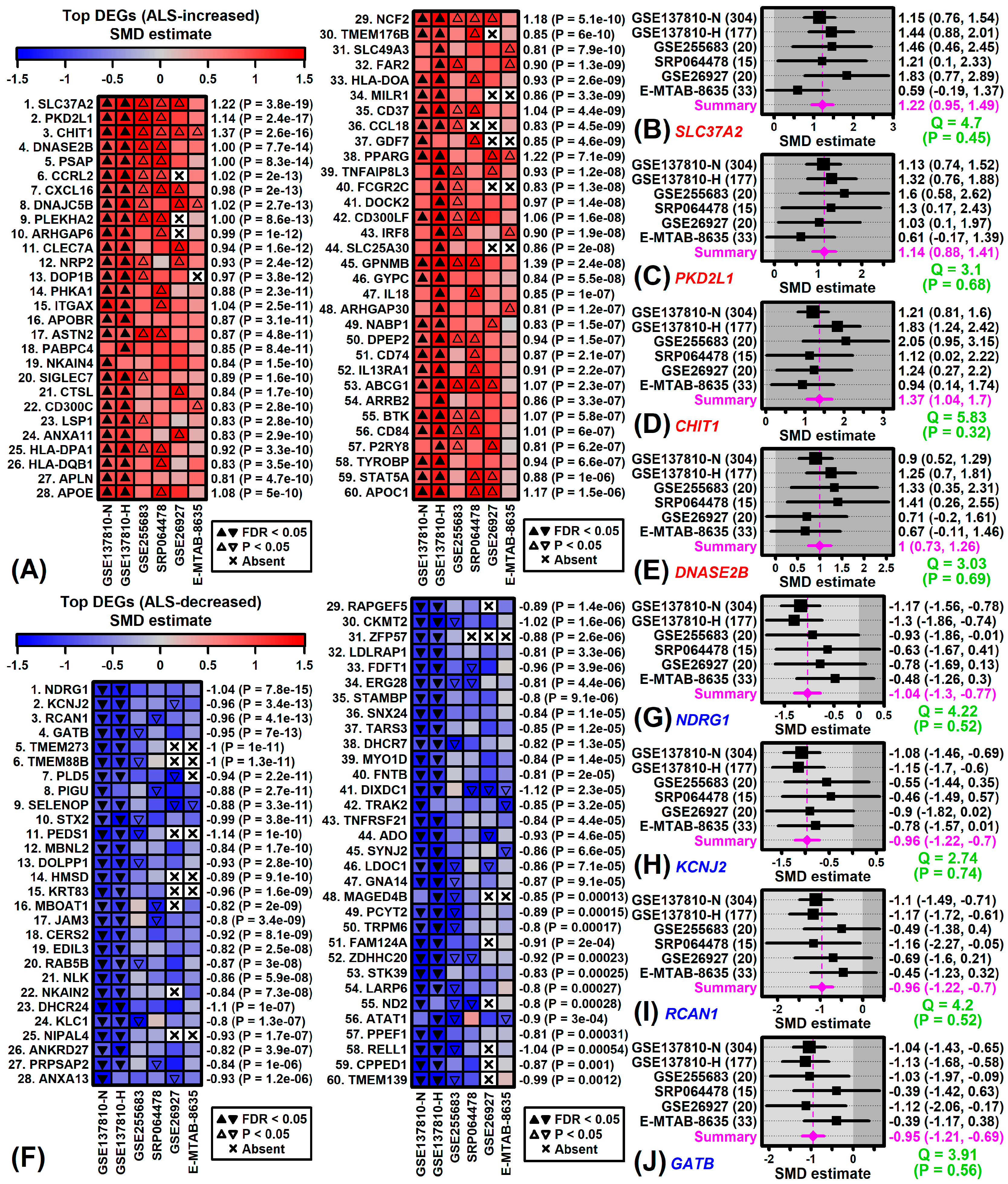
3.4. Genes with Expression Consistently Altered in ALS Spinal Cord
3.5. Spinal Cord DEGs Overlap Significantly with Those Identified in LCM-Dissected Motor Neurons and mRNAs Associated with ALS-Dysregulated Proteins
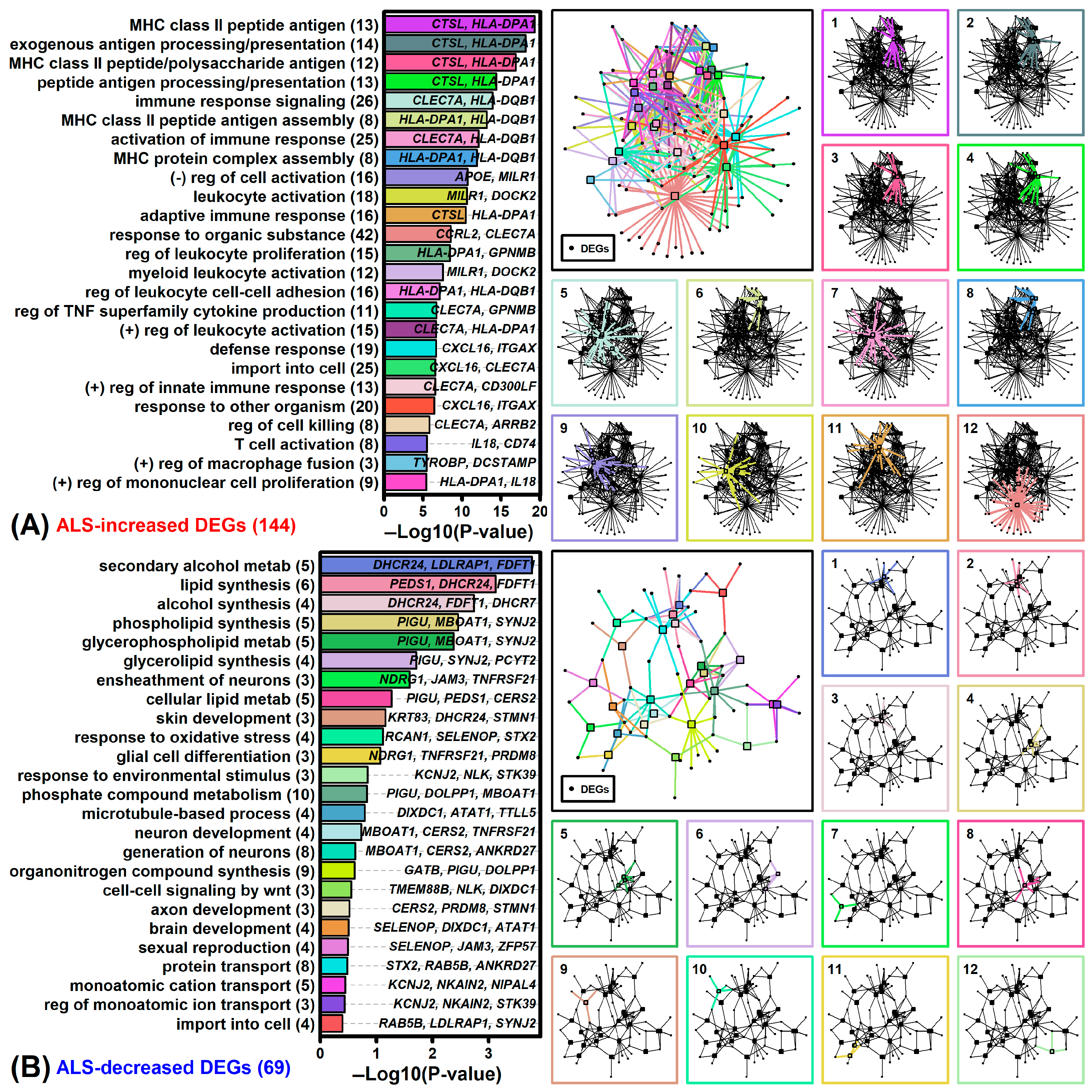
3.6. ALS DEGs and Genes near ALS Risk Loci Are Associated with Plasma Membrane and Sterol Metabolism
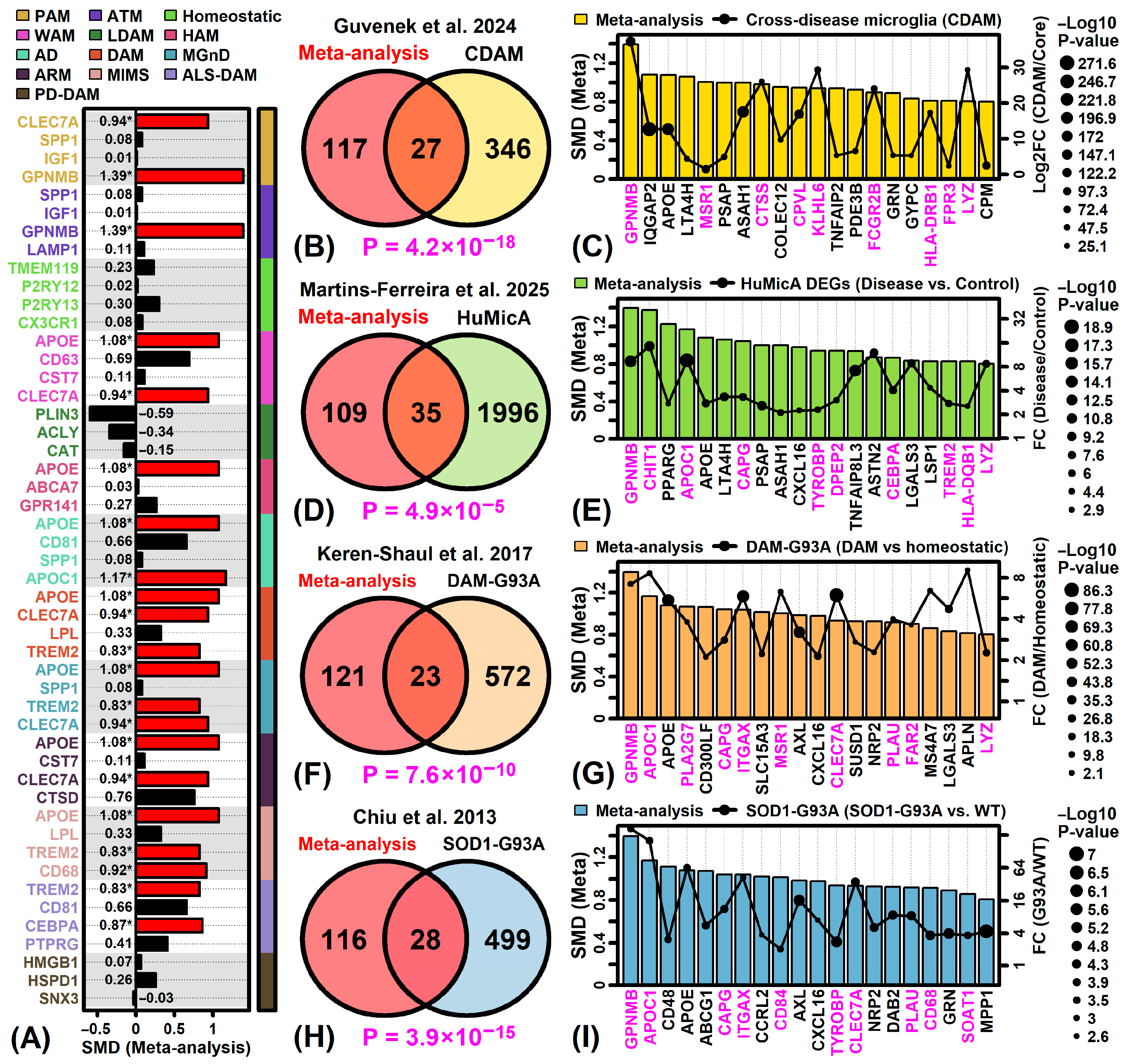
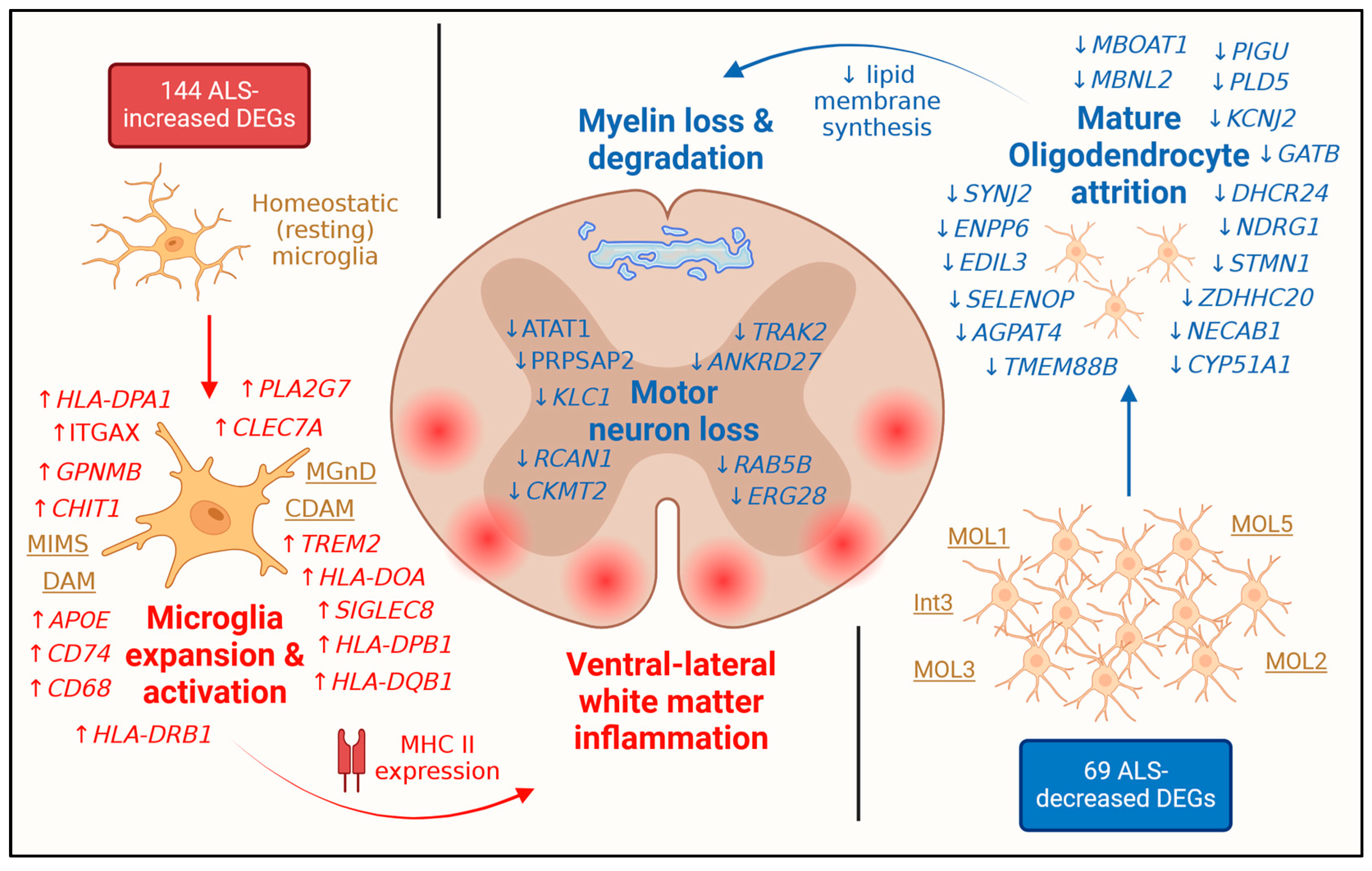
3.7. ALS-Increased DEGs Are Most Strongly Expressed by Microglia
3.8. ALS-Decreased DEGs Are Most Strongly Expressed by Mature Oligodendrocyte Phenotypes
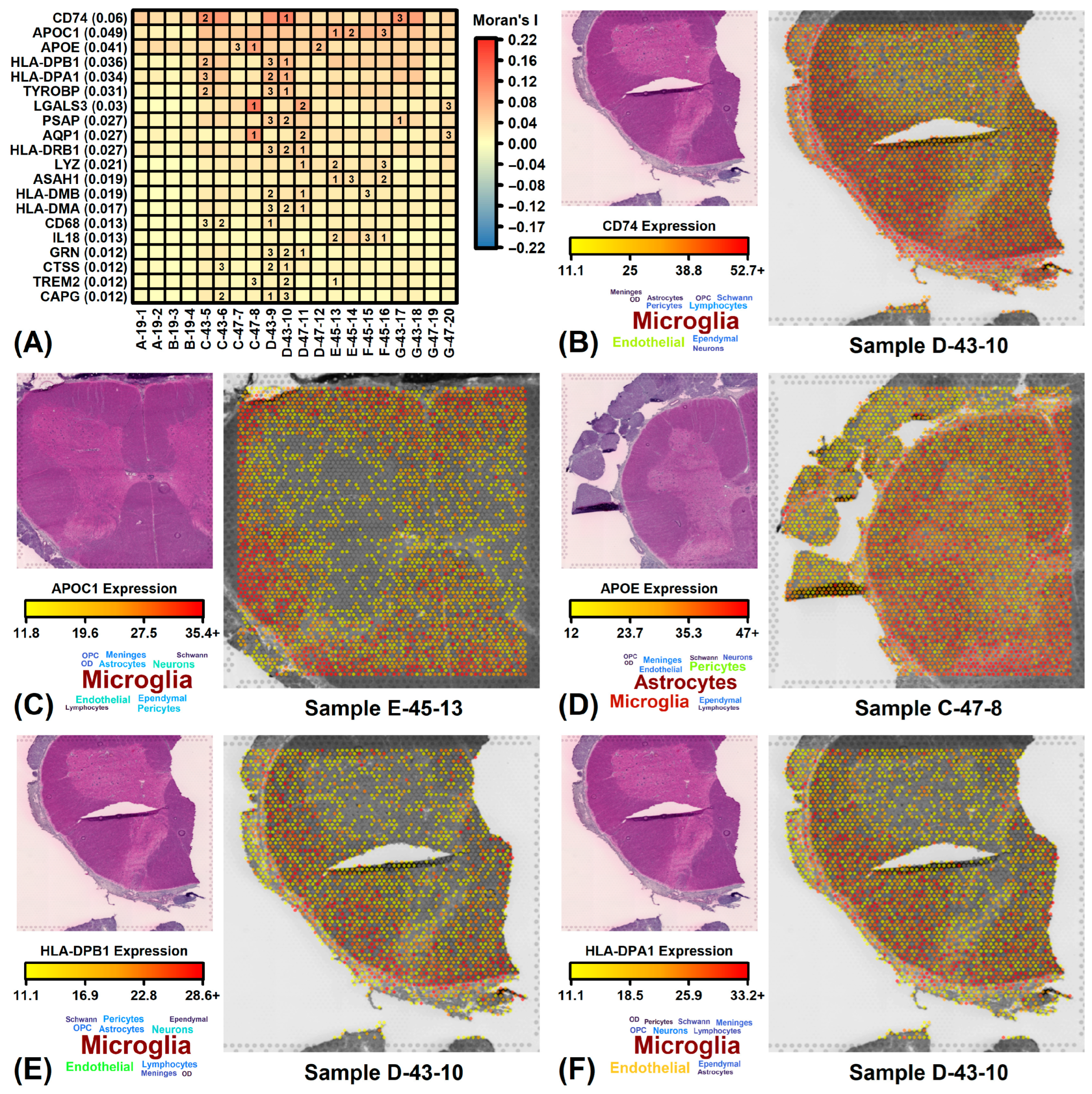
3.9. ALS-Increased DEGs Are Expressed in Dorsal/Lateral White Matter Whereas Decreased DEGs Are Expressed in Ventral/Lateral White Matter
3.10. ALS-Increased Genes Are Weakly Expressed in ALS Cord but Are Most Prominent in Ventral/Lateral White Matter
4. Discussion
5. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATM | axon tract associated microglia |
| ARM | activated response microglia |
| BP | biological process |
| CDAM | cross-disease-associated microglia module |
| CDF | cumulative distribution function |
| CTL | control |
| CPM | count per million |
| DAM | disease-associated microglia |
| DEG | differential expressed gene |
| FDR | false discovery rate |
| FPKM | fragments per kilobase of transcript per million mapped reads |
| GO | gene ontology |
| GWAS | genome-wide association study |
| HAM | human AD microglia |
| HuMicA | human microglia atlas |
| ImOLs | immune oligodendrocytes |
| LCM | laser capture microdissection |
| LCM-MN | laser capture microdissected motor neuron |
| LDAM | lipid droplet accumulating microglia |
| LRT | likelihood ratio test |
| M1 | homeostatic/surveillant microglia |
| M2 | protective microglia |
| MGnD | microglia neurodegenerative phenotype |
| MIMS | microglia inflamed in MS |
| NYGC | New York genome center |
| OD | oligodendrocyte |
| OPC | oligodendrocyte precursor cells |
| PAM | proliferative associated microglia |
| PC | principal component |
| PD-DAM | disease associated microglia in Parkinson’s disease |
| RIN | RNA integrity number |
| SMD | standardized mean difference |
| SOM | self-organizing map |
| snRNA-seq | single-nucleus RNA-seq |
| WAM | white matter associated microglia |
References
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Ye, S.; Liu, X.Y.; Xu, Y.S.; Fan, D.S. The burden of upper motor neuron involvement is correlated with the bilateral limb involvement interval in patients with amyotrophic lateral sclerosis: A retrospective observational study. Neural Regen. Res. 2025, 20, 1505–1512. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Yang, B.; Li, Q.; Zhu, X.; Song, E.; Liu, C.; Song, Y.; Jiang, G. Polystyrene nanoparticles trigger aberrant condensation of TDP-43 and amyotrophic lateral sclerosis-like symptoms. Nat. Nanotechnol. 2024, 19, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Garcidueñas, L.; Stommel, E.W.; Torres-Jardón, R.; Hernández-Luna, J.; Aiello-Mora, M.; González-Maciel, A.; Reynoso-Robles, R.; Pérez-Guillé, B.; Silva-Pereyra, H.G.; Tehuacanero-Cuapa, S.; et al. Alzheimer and Parkinson diseases, frontotemporal lobar degeneration and amyotrophic lateral sclerosis overlapping neuropathology start in the first two decades of life in pollution exposed urbanites and brain ultrafine particulate matter and industrial nanoparticles, including Fe, Ti, Al, V, Ni, Hg, Co, Cu, Zn, Ag, Pt, Ce, La, Pr and W are key players. Metropolitan Mexico City health crisis is in progress. Front. Hum. Neurosci. 2024, 17, 1297467. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Bano, N.; Ahamad, S.; John, U.; Dar, N.J.; Bhat, S.A. Excitotoxicity, Oxytosis/Ferroptosis, and Neurodegeneration: Emerging Insights into Mitochondrial Mechanisms. Aging Dis. 2025, 15, 125–144. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Wei, Y.; Zhang, J.; Gal, J.; Zhu, H. Mitochondrial dysfunction is a converging point of multiple pathological pathways in amyotrophic lateral sclerosis. J. Alzheimer’s Dis. JAD 2010, 20 (Suppl. 2), S311–S324. [Google Scholar]
- Abyadeh, M.; Gupta, V.; Paulo, J.A.; Mahmoudabad, A.G.; Shadfar, S.; Mirshahvaladi, S.; Gupta, V.; Nguyen, C.T.O.; Finkelstein, D.I.; You, Y.Y.; et al. Amyloid-beta and tau protein beyond Alzheimer’s disease. Neural Regen. Res. 2024, 19, 1262–1276. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.Y.; Kotha, S.; Fu, M.; Yang, Q.; Wang, H.; He, W.W.; Mao, X.B. Nanozyme enabled protective therapy for neurological diseases. Nano Today 2024, 54, 102142. [Google Scholar] [CrossRef]
- Zhang, H.; Guo, H.Z.; Li, D.N.; Zhang, Y.L.; Zhang, S.N.; Kang, W.Y.; Liu, C.; Le, W.D.; Wang, L.; Li, D.; et al. Halogen doped graphene quantum dots modulate TDP-43 phase separation and aggregation in the nucleus. Nat. Commun. 2024, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.H.; Zhang, H.; Lu, K.L.; Chen, L.; Zhang, Y.Q.; Xu, Z.W.; Zhou, H.S.; Sun, J.F.; Xu, M.Y.; Ouyang, Q.; et al. Low-intensity pulsed ultrasound modulates disease progression in the SOD1G93A mouse G93A mouse model of amyotrophic lateral sclerosis. Cell Rep. 2024, 43, 114660. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.G.; Xia, Y.L.; Sun, J. Probiotics and microbial metabolites maintain barrier and neuromuscular functions and clean protein aggregation to delay disease progression in TDP43 mutation mice. Gut Microbes 2024, 16, 2363880. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.S.; Chen, H.X.; Zhang, Y.; Peng, L.H.; Wang, Z.K.; Lan, X.Y.; Yu, S.Y.; Yang, Y.S. Fecal microbiota transplantation significantly improved respiratory failure of amyotrophic lateral sclerosis. Gut Microbes 2024, 16, 2353396. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.L.; Fu, Y.L.; Wang, A.T.; Shi, K.X.; Peng, Y.D.; Yi, Y.; Yu, R.H.; Gao, J.C.; Feng, J.F.; Jiang, G.; et al. Brain Delivery of Protein Therapeutics by Cell Matrix-Inspired Biomimetic Nanocarrier. Adv. Mater. 2024, 36, e2405323. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Sneha, N.P.; Dharshini, S.A.P.; Taguchi, Y.H.; Gromiha, M.M. Tracing ALS Degeneration: Insights from Spinal Cord and Cortex Transcriptomes. Genes 2024, 15, 1431. [Google Scholar] [CrossRef] [PubMed]
- Rizzuti, M.; Sali, L.; Melzi, V.; Scarcella, S.; Costamagna, G.; Ottoboni, L.; Quetti, L.; Brambilla, L.; Papadimitriou, D.; Verde, F.; et al. Genomic and transcriptomic advances in amyotrophic lateral sclerosis. Ageing Res. Rev. 2023, 92, 102126. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, J.; Venkatesh, S.; Hasan, R.; Herb, J.T.; de Paiva Lopes, K.; Küçükali, F.; Byrska-Bishop, M.; Evani, U.S.; Narzisi, G.; Fagegaltier, D.; et al. Integrative transcriptomic analysis of the amyotrophic lateral sclerosis spinal cord implicates glial activation and suggests new risk genes. Nat. Neurosci. 2023, 26, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Durrenberger, P.F.; Fernando, F.S.; Kashefi, S.N.; Bonnert, T.P.; Seilhean, D.; Nait-Oumesmar, B.; Schmitt, A.; Gebicke-Haerter, P.J.; Falkai, P.; Grünblatt, E.; et al. Common mechanisms in neurodegeneration and neuroinflammation: A BrainNet Europe gene expression microarray study. J. Neural Transm. 2015, 122, 1055–1068. [Google Scholar] [CrossRef] [PubMed]
- Durrenberger, P.F.; Fernando, F.S.; Magliozzi, R.; Kashefi, S.N.; Bonnert, T.P.; Ferrer, I.; Seilhean, D.; Nait-Oumesmar, B.; Schmitt, A.; Gebicke-Haerter, P.J.; et al. Selection of novel reference genes for use in the human central nervous system: A BrainNet Europe Study. Acta Neuropathol. 2012, 124, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Newton, D.F.; Yang, R.; Gutierrez, J.; Hofmann, J.W.; Yeh, F.L.; Biever, A.; Friedman, B.A. TDP43 proteinopathy exhibits disease, tissue, and context-specific cryptic splicing signatures. bioRxiv 2024, 2024, 29. [Google Scholar] [CrossRef]
- Brohawn, D.G.; O’Brien, L.C.; Bennett, J.P., Jr. RNAseq Analyses Identify Tumor Necrosis Factor-Mediated Inflammation as a Major Abnormality in ALS Spinal Cord. PLoS ONE 2016, 11, e0160520. [Google Scholar] [CrossRef] [PubMed]
- La Cognata, V.; Gentile, G.; Aronica, E.; Cavallaro, S. Splicing Players Are Differently Expressed in Sporadic Amyotrophic Lateral Sclerosis Molecular Clusters and Brain Regions. Cells 2020, 9, 159. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.E.; Ferraiuolo, L.; Goodall, E.F.; Heath, P.R.; Higginbottom, A.; Mortiboys, H.; Hollinger, H.C.; Hartley, J.A.; Brockington, A.; Burness, C.E.; et al. Mutations in CHMP2B in lower motor neuron predominant amyotrophic lateral sclerosis (ALS). PLoS ONE 2010, 5, e9872. [Google Scholar] [CrossRef] [PubMed]
- Rabin, S.J.; Kim, J.M.; Baughn, M.; Libby, R.T.; Kim, Y.J.; Fan, Y.; Libby, R.T.; La Spada, A.; Stone, B.; Ravits, J. Sporadic ALS has compartment-specific aberrant exon splicing and altered cell-matrix adhesion biology. Hum. Mol. Genet. 2010, 19, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Kirby, J.; Ning, K.; Ferraiuolo, L.; Heath, P.R.; Ismail, A.; Kuo, S.W.; Valori, C.F.; Cox, L.; Sharrack, B.; Wharton, S.B.; et al. Phosphatase and tensin homologue/protein kinase B pathway linked to motor neuron survival in human superoxide dismutase 1-related amyotrophic lateral sclerosis. Brain A J. Neurol. 2011, 134 Pt 2, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Highley, J.R.; Kirby, J.; Jansweijer, J.A.; Webb, P.S.; Hewamadduma, C.A.; Heath, P.R.; Higginbottom, A.; Raman, R.; Ferraiuolo, L.; Cooper-Knock, J.; et al. Loss of nuclear TDP-43 in amyotrophic lateral sclerosis (ALS) causes altered expression of splicing machinery and widespread dysregulation of RNA splicing in motor neurones. Neuropathol. Appl. Neurobiol. 2014, 40, 670–685. [Google Scholar] [CrossRef] [PubMed]
- Cooper-Knock, J.; Bury, J.J.; Heath, P.R.; Wyles, M.; Higginbottom, A.; Gelsthorpe, C.; Highley, J.R.; Hautbergue, G.; Rattray, M.; Kirby, J.; et al. C9ORF72 GGGGCC Expanded Repeats Produce Splicing Dysregulation which Correlates with Disease Severity in Amyotrophic Lateral Sclerosis. PLoS ONE 2015, 10, e0127376. [Google Scholar] [CrossRef] [PubMed]
- Krach, F.; Batra, R.; Wheeler, E.C.; Vu, A.Q.; Wang, R.; Hutt, K.; Rabin, S.J.; Baughn, M.W.; Libby, R.T.; Diaz-Garcia, S.; et al. Transcriptome-pathology correlation identifies interplay between TDP-43 and the expression of its kinase CK1E in sporadic ALS. Acta Neuropathol. 2018, 136, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Nizzardo, M.; Taiana, M.; Rizzo, F.; Aguila Benitez, J.; Nijssen, J.; Allodi, I.; Melzi, V.; Bresolin, N.; Comi, G.P.; Hedlund, E.; et al. Synaptotagmin 13 is neuroprotective across motor neuron diseases. Acta Neuropathol. 2020, 139, 837–853. [Google Scholar] [CrossRef] [PubMed]
- Pineda, S.S.; Lee, H.; Ulloa-Navas, M.J.; Linville, R.M.; Garcia, F.J.; Galani, K.; Engelberg-Cook, E.; Castanedes, M.C.; Fitzwalter, B.E.; Pregent, L.J.; et al. Single-cell dissection of the human motor and prefrontal cortices in ALS and FTLD. Cell 2024, 187, 1971–1989.e16. [Google Scholar] [CrossRef] [PubMed]
- Limone, F.; Mordes, D.A.; Couto, A.; Joseph, B.J.; Mitchell, J.M.; Therrien, M.; Ghosh, S.D.; Meyer, D.; Zhang, Y.; Goldman, M.; et al. Single-nucleus sequencing reveals enriched expression of genetic risk factors in extratelencephalic neurons sensitive to degeneration in ALS. Nat. Aging 2024, 4, 984–997. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jaiswal, M.K.; Chien, J.F.; Kozlenkov, A.; Jung, J.; Zhou, P.; Gardashli, M.; Pregent, L.J.; Engelberg-Cook, E.; Dickson, D.W.; et al. Divergent single cell transcriptome and epigenome alterations in ALS and FTD patients with C9orf72 mutation. Nat. Commun. 2023, 14, 5714. [Google Scholar] [CrossRef] [PubMed]
- Swindell, W.R. Meta-analysis of differential gene expression in lower motor neurons isolated by laser capture microdissection from post-mortem ALS spinal cords. Front. Genet. 2024, 15, 1385114. [Google Scholar] [CrossRef] [PubMed]
- Toro-Domínguez, D.; Villatoro-García, J.A.; Martorell-Marugán, J.; Román-Montoya, Y.; Alarcón-Riquelme, M.E.; Carmona-Sáez, P. A survey of gene expression meta-analysis: Methods and applications. Brief. Bioinform. 2021, 22, 1694–1705. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, A.; Mondry, A.; Holmes, C.C.; Altman, D.G. Key issues in conducting a meta-analysis of gene expression microarray datasets. PLoS Med. 2008, 5, e184. [Google Scholar] [CrossRef] [PubMed]
- Campain, A.; Yang, Y.H. Comparison study of microarray meta-analysis methods. BMC Bioinform. 2010, 11, 408. [Google Scholar] [CrossRef] [PubMed]
- Grad, L.I.; Rouleau, G.A.; Ravits, J.; Cashman, N.R. Clinical Spectrum of Amyotrophic Lateral Sclerosis (ALS). Cold Spring Harb. Perspect. Med. 2017, 7, a024117. [Google Scholar] [CrossRef] [PubMed]
- Goyal, N.A.; Berry, J.D.; Windebank, A.; Staff, N.P.; Maragakis, N.J.; van den Berg, L.H.; Genge, A.; Miller, R.; Baloh, R.H.; Kern, R.; et al. Addressing heterogeneity in amyotrophic lateral sclerosis CLINICAL TRIALS. Muscle Nerve 2020, 62, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Su, X.W.; Broach, J.R.; Connor, J.R.; Gerhard, G.S.; Simmons, Z. Genetic heterogeneity of amyotrophic lateral sclerosis: Implications for clinical practice and research. Muscle Nerve 2014, 49, 786–803. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, C.Y. From bulk, single-cell to spatial RNA sequencing. Int. J. Oral Sci. 2021, 13, 36. [Google Scholar] [CrossRef] [PubMed]
- Law, C.W.; Chen, Y.; Shi, W.; Smyth, G.K. Voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014, 15, R29. [Google Scholar] [CrossRef] [PubMed]
- Vestal, B.E.; Wynn, E.; Moore, C.M. LmerSeq: An R package for analyzing transformed RNA-Seq data with linear mixed effects models. BMC Bioinform. 2022, 23, 489. [Google Scholar] [CrossRef] [PubMed]
- Yadav, A.; Matson, K.J.E.; Li, L.; Hua, I.; Petrescu, J.; Kang, K.; Alkaslasi, M.R.; Lee, D.I.; Hasan, S.; Galuta, A.; et al. A cellular taxonomy of the adult human spinal cord. Neuron 2023, 111, 328–344.e7. [Google Scholar] [CrossRef] [PubMed]
- Maniatis, S.; Äijö, T.; Vickovic, S.; Braine, C.; Kang, K.; Mollbrink, A.; Fagegaltier, D.; Andrusivová, Ž.; Saarenpää, S.; Saiz-Castro, G.; et al. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science (New York NY) 2019, 364, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Sollis, E.; Mosaku, A.; Abid, A.; Buniello, A.; Cerezo, M.; Gil, L.; Groza, T.; Güneş, O.; Hall, P.; Hayhurst, J.; et al. The NHGRI-EBI GWAS Catalog: Knowledgebase and deposition resource. Nucleic Acids Res. 2023, 51, D977–D985. [Google Scholar] [CrossRef] [PubMed]
- Malaspina, A.; Kaushik, N.; de Belleroche, J. Differential expression of 14 genes in amyotrophic lateral sclerosis spinal cord detected using gridded cDNA arrays. J. Neurochem. 2001, 77, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Offen, D.; Barhum, Y.; Melamed, E.; Embacher, N.; Schindler, C.; Ransmayr, G. Spinal cord mRNA profile in patients with ALS: Comparison with transgenic mice expressing the human SOD-1 mutant. J. Mol. Neurosci. MN 2009, 38, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Andrés-Benito, P.; Moreno, J.; Aso, E.; Povedano, M.; Ferrer, I. Amyotrophic lateral sclerosis, gene deregulation in the anterior horn of the spinal cord and frontal cortex area 8: Implications in frontotemporal lobar degeneration. Aging 2017, 9, 823–851. [Google Scholar] [CrossRef] [PubMed]
- D’Erchia, A.M.; Gallo, A.; Manzari, C.; Raho, S.; Horner, D.S.; Chiara, M.; Valletti, A.; Aiello, I.; Mastropasqua, F.; Ciaccia, L.; et al. Massive transcriptome sequencing of human spinal cord tissues provides new insights into motor neuron degeneration in ALS. Sci. Rep. 2017, 7, 10046. [Google Scholar] [CrossRef] [PubMed]
- Dangond, F.; Hwang, D.; Camelo, S.; Pasinelli, P.; Frosch, M.P.; Stephanopoulos, G.; Stephanopoulos, G.; Brown, R.H., Jr.; Gullans, S.R. Molecular signature of late-stage human ALS revealed by expression profiling of postmortem spinal cord gray matter. Physiol. Genom. 2004, 16, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Butovsky, O.; Jedrychowski, M.P.; Cialic, R.; Krasemann, S.; Murugaiyan, G.; Fanek, Z.; Greco, D.J.; Wu, P.M.; Doykan, C.E.; Kiner, O.; et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann. Neurol. 2015, 77, 75–99. [Google Scholar] [CrossRef] [PubMed]
- Kodama, Y.; Shumway, M.; Leinonen, R. The Sequence Read Archive: Explosive growth of sequencing data. Nucleic Acids Res. 2012, 40, D54–D56. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Pirrung, M.; McCue, L.A. FQC Dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 2017, 33, 3137–3139. [Google Scholar] [CrossRef] [PubMed]
- TrimGalore. Available online: https://github.com/FelixKrueger/TrimGalore (accessed on 13 April 2025).
- BBMap Short Read Aligner, and Other Bioinformatic Tools. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 13 April 2025).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Ramsköld, D.; Wang, E.T.; Burge, C.B.; Sandberg, R. An abundance of ubiquitously expressed genes revealed by tissue transcriptome sequence data. PLoS Comput. Biol. 2009, 5, e1000598. [Google Scholar] [CrossRef] [PubMed]
- Hart, T.; Komori, H.K.; LaMere, S.; Podshivalova, K.; Salomon, D.R. Finding the active genes in deep RNA-seq gene expression studies. BMC Genom. 2013, 14, 778. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.E.; Silver, J.; Oshlack, A.; Holmes, M.; Diyagama, D.; Holloway, A.; Smyth, G.K. A comparison of background correction methods for two-colour microarrays. Bioinformatics 2007, 23, 2700–2707. [Google Scholar] [CrossRef] [PubMed]
- Bolstad, B.M.; Irizarry, R.A.; Astrand, M.; Speed, T.P. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 2003, 19, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, D.; Cook, M. A statistical framework for consolidating “sibling” probe sets for Affymetrix GeneChip data. BMC Genom. 2008, 9, 188. [Google Scholar] [CrossRef] [PubMed]
- Weng, L.; Dai, H.; Zhan, Y.; He, Y.; Stepaniants, S.B.; Bassett, D.E. Rosetta error model for gene expression analysis. Bioinformatics 2006, 22, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Sarkans, U.; Füllgrabe, A.; Ali, A.; Athar, A.; Behrangi, E.; Diaz, N.; Fexova, S.; George, N.; Iqbal, H.; Kurri, S.; et al. From ArrayExpress to BioStudies. Nucleic Acids Res. 2021, 49, D1502–D1506. [Google Scholar] [CrossRef] [PubMed]
- Workman, C.; Jensen, L.J.; Jarmer, H.; Berka, R.; Gautier, L.; Nielser, H.B.; Saxild, H.H.; Nielsen, C.; Brunak, S.; Knudsen, S. A new non-linear normalization method for reducing variability in DNA microarray experiments. Genome Biol. 2002, 3, research0048. [Google Scholar] [CrossRef] [PubMed]
- Hedges, L.V. Distribution theory for Glass’s estimator of effect size and related estimators. J. Educ. Stat. 1981, 6, 107–128. [Google Scholar] [CrossRef]
- Hedges, L.V.; Olkin, I. Statistical Methods for Meta-Analysis; Academic Press: Cambridge, MA, USA, 2014. [Google Scholar]
- Lipsey, M.W.; Wilson, D.B. Practical Meta-Analysis; Sage Publications: Thousand Oaks, CA, USA, 2001. [Google Scholar]
- Schwarzer, G.; Carpenter, J.R.; Rücker, G. Meta-Analysis with R; Springer: Berlin/Heidelberg, Germany, 2015; Volume 4784. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A powerful and practical approach to multiple testing. J. R. Stat. Soc. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Hafemeister, C.; Satija, R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019, 20, 296. [Google Scholar] [CrossRef] [PubMed]
- Lorenzi, L.; Chiu, H.S.; Avila Cobos, F.; Gross, S.; Volders, P.J.; Cannoodt, R.; Nuytens, J.; Vanderheyden, K.; Anckaert, J.; Lefever, S.; et al. The RNA Atlas expands the catalog of human non-coding RNAs. Nat. Biotechnol. 2021, 39, 1453–1465. [Google Scholar] [CrossRef] [PubMed]
- Oeckl, P.; Weydt, P.; Thal, D.R.; Weishaupt, J.H.; Ludolph, A.C.; Otto, M. Proteomics in cerebrospinal fluid and spinal cord suggests UCHL1, MAP2 and GPNMB as biomarkers and underpins importance of transcriptional pathways in amyotrophic lateral sclerosis. Acta Neuropathol. 2020, 139, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Zaldívar, H.M.; Polakovicova, I.; Salas-Huenuleo, E.; Corvalán, A.H.; Kogan, M.J.; Yefi, C.P.; Andia, M.E. Extracellular vesicles through the blood-brain barrier: A review. Fluids Barriers CNS 2022, 19, 60. [Google Scholar] [CrossRef] [PubMed]
- Swindell, W.R.; Kruse, C.P.S.; List, E.O.; Berryman, D.E.; Kopchick, J.J. ALS blood expression profiling identifies new biomarkers, patient subgroups, and evidence for neutrophilia and hypoxia. J. Transl. Med. 2019, 17, 170. [Google Scholar] [CrossRef] [PubMed]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Swindell, W.R.; Johnston, A.; Voorhees, J.J.; Elder, J.T.; Gudjonsson, J.E. Dissecting the psoriasis transcriptome: Inflammatory- and cytokine-driven gene expression in lesions from 163 patients. BMC Genom. 2013, 14, 527. [Google Scholar] [CrossRef] [PubMed]
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef] [PubMed]
- Guvenek, A.; Parikshak, N.; Zamolodchikov, D.; Gelfman, S.; Moscati, A.; Dobbyn, L.; Stahl, E.; Shuldiner, A.; Coppola, G. Transcriptional profiling in microglia across physiological and pathological states identifies a transcriptional module associated with neurodegeneration. Commun. Biol. 2024, 7, 1168. [Google Scholar] [CrossRef] [PubMed]
- Martins-Ferreira, R.; Calafell-Segura, J.; Leal, B.; Rodríguez-Ubreva, J.; Martínez-Saez, E.; Mereu, E.; Pinho, E.C.P.; Laguna, A.; Ballestar, E. The Human Microglia Atlas (HuMicA) unravels changes in disease-associated microglia subsets across neurodegenerative conditions. Nat. Commun. 2025, 16, 739. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef] [PubMed]
- Chiu, I.M.; Morimoto, E.T.; Goodarzi, H.; Liao, J.T.; O’Keeffe, S.; Phatnani, H.P.; Muratet, M.; Carroll, M.C.; Levy, S.; Tavazoie, S.; et al. A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 2013, 4, 385–401. [Google Scholar] [CrossRef] [PubMed]
- Valihrach, L.; Matusova, Z.; Zucha, D.; Klassen, R.; Benesova, S.; Abaffy, P.; Kubista, M.; Anderova, M. Recent advances in deciphering oligodendrocyte heterogeneity with single-cell transcriptomics. Front. Cell. Neurosci. 2022, 16, 1025012. [Google Scholar] [CrossRef] [PubMed]
- Marques, S.; Zeisel, A.; Codeluppi, S.; van Bruggen, D.; Mendanha Falcão, A.; Xiao, L.; Li, H.; Häring, M.; Hochgerner, H.; Romanov, R.A.; et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 2016, 352, 1326–1329. [Google Scholar] [CrossRef] [PubMed]
- Sadick, J.S.; O’Dea, M.R.; Hasel, P.; Dykstra, T.; Faustin, A.; Liddelow, S.A. Astrocytes and oligodendrocytes undergo subtype-specific transcriptional changes in Alzheimer’s disease. Neuron 2022, 110, 1788–1805.e10. [Google Scholar] [CrossRef] [PubMed]
- Jäkel, S.; Agirre, E.; Mendanha Falcão, A.; van Bruggen, D.; Lee, K.W.; Knuesel, I.; Malhotra, D.; Ffrench-Constant, C.; Williams, A.; Castelo-Branco, G. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 2019, 566, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Swindell, W.R.; Xing, X.; Voorhees, J.J.; Elder, J.T.; Johnston, A.; Gudjonsson, J.E. Integrative RNA-seq and microarray data analysis reveals GC content and gene length biases in the psoriasis transcriptome. Physiol. Genom. 2014, 46, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H. Multicollinearity and misleading statistical results. Korean J. Anesthesiol. 2019, 72, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Jiang, S.; Land, K.C. Multicollinearity in hierarchical linear models. Soc. Sci. Res. 2015, 53, 118–136. [Google Scholar] [CrossRef] [PubMed]
- Yoo, W.; Mayberry, R.; Bae, S.; Singh, K.; He, Q.P.; Lillard, J.W., Jr. A Study of Effects of MultiCollinearity in the Multivariable Analysis. Int. J. Appl. Sci. Technol. 2014, 4, 9–19. [Google Scholar] [PubMed]
- Ioannidis, J.P. Why most published research findings are false. PLoS Med. 2005, 2, e124. [Google Scholar] [CrossRef] [PubMed]
- Yu, L. RNA-Seq Reproducibility Assessment of the Sequencing Quality Control Project. Cancer Inform. 2020, 19, 1176935120922498. [Google Scholar] [CrossRef] [PubMed]
- Lemon, R.N. Descending pathways in motor control. Annu. Rev. Neurosci. 2008, 31, 195–218. [Google Scholar] [CrossRef] [PubMed]
- Lemon, R. The Corticospinal System and Amyotrophic Lateral Sclerosis: IFCN handbook chapter. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2024, 160, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Ahmad, T.K.; Gozda, K.; Truong, J.; Kong, J.; Namaka, M. Implications of white matter damage in amyotrophic lateral sclerosis. Mol. Med. Rep. 2017, 16, 4379–4392. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004, 185, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Toko, M.; Ohshita, T.; Nakamori, M.; Ueno, H.; Akiyama, Y.; Maruyama, H. Myelin measurement in amyotrophic lateral sclerosis with synthetic MRI: A potential diagnostic and predictive method. J. Neurol. Sci. 2025, 468, 123337. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.J.; Zhan, C.; Cai, L.M.; Lin, J.H.; Zhou, M.X.; Zou, Z.Y.; Yao, X.F.; Lin, Y.J. White matter microstructural impairments in amyotrophic lateral sclerosis: A mean apparent propagator MRI study. NeuroImage Clin. 2021, 32, 102863. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, V.; Pioro, E.P. Graph theory network analysis reveals widespread white matter damage in brains of patients with classic ALS. Amyotroph. Lateral Scler. Front. Degener. 2025, 26, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Desai, A.B.; Agarwal, A.; Mohamed, A.S.; Mohamed, K.H.; Middlebrooks, E.H.; Bhatt, A.A.; Gupta, V.; Kumar, N.; Sechi, E.; Flanagan, E.P.; et al. Motor Neuron Diseases and Central Nervous System Tractopathies: Clinical-Radiologic Correlation and Diagnostic Approach. Radiogr. A Rev. Publ. Radiol. Soc. N. Am. Inc. 2025, 45, e240067. [Google Scholar] [CrossRef] [PubMed]
- Almgren, H.; Mahoney, C.J.; Huynh, W.; D’Souza, A.; Berte, S.; Lv, J.; Wang, C.; Kiernan, M.C.; Calamante, F.; Tu, S. Quantifying neurodegeneration within subdivisions of core motor pathways in amyotrophic lateral sclerosis using diffusion MRI. J. Neurol. 2025, 272, 215. [Google Scholar] [CrossRef] [PubMed]
- Wendebourg, M.J.; Kesenheimer, E.; Sander, L.; Weigel, M.; Weidensteiner, C.; Haas, T.; Madoerin, P.; Diebold, M.; Deigendesch, N.; Neuhaus, D.; et al. The Lateral Corticospinal Tract Sign: An MRI Marker for Amyotrophic Lateral Sclerosis. Radiology 2024, 312, e231630. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; Toledo, J.B.; Van Deerlin, V.M.; Elman, L.; McCluskey, L.; Lee, V.M.; Trojanowski, J.Q. Microglial activation correlates with disease progression and upper motor neuron clinical symptoms in amyotrophic lateral sclerosis. PLoS ONE 2012, 7, e39216. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.Y.; Liu, L.; Yang, Q.W. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog. Neurobiol. 2016, 142, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Perlmutter, L.S.; Scott, S.A.; Barrón, E.; Chui, H.C. MHC class II-positive microglia in human brain: Association with Alzheimer lesions. J. Neurosci. Res. 1992, 33, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Mathys, H.; Adaikkan, C.; Gao, F.; Young, J.Z.; Manet, E.; Hemberg, M.; De Jager, P.L.; Ransohoff, R.M.; Regev, A.; Tsai, L.H. Temporal Tracking of Microglia Activation in Neurodegeneration at Single-Cell Resolution. Cell Rep. 2017, 21, 366–380. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; Kawamata, T.; Walker, D.G.; Akiyama, H.; Tooyama, I.; McGeer, E.G. Microglia in degenerative neurological disease. Glia 1993, 7, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Dachet, F.; Liu, J.; Ravits, J.; Song, F. Predicting disease specific spinal motor neurons and glia in sporadic ALS. Neurobiol. Dis. 2019, 130, 104523. [Google Scholar] [CrossRef] [PubMed]
- Imamura, K.; Hishikawa, N.; Sawada, M.; Nagatsu, T.; Yoshida, M.; Hashizume, Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol. 2003, 106, 518–526. [Google Scholar] [CrossRef] [PubMed]
- Hayes, G.M.; Woodroofe, M.N.; Cuzner, M.L. Microglia are the major cell type expressing MHC class II in human white matter. J. Neurol. Sci. 1987, 80, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Weterman, M.A.; Ajubi, N.; van Dinter, I.M.; Degen, W.G.; van Muijen, G.N.; Ruitter, D.J.; Bloemers, H.P. NMB, a novel gene, is expressed in low-metastatic human melanoma cell lines and xenografts. Int. J. Cancer 1995, 60, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Barreto-Núñez, R.; Béland, L.C.; Boutej, H.; Picher-Martel, V.; Dupré, N.; Barbeito, L.; Kriz, J. Chronically activated microglia in ALS gradually lose their immune functions and develop unconventional proteome. Glia 2024, 72, 1319–1339. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Shimazawa, M.; Kimura, M.; Takata, M.; Tsuruma, K.; Yamada, M.; Takahashi, H.; Hozumi, I.; Niwa, J.; Iguchi, Y.; et al. The potential of GPNMB as novel neuroprotective factor in amyotrophic lateral sclerosis. Sci. Rep. 2012, 2, 573. [Google Scholar] [CrossRef] [PubMed]
- Budge, K.M.; Neal, M.L.; Richardson, J.R.; Safadi, F.F. Glycoprotein NMB: An Emerging Role in Neurodegenerative Disease. Mol. Neurobiol. 2018, 55, 5167–5176. [Google Scholar] [CrossRef] [PubMed]
- Budge, K.M.; Neal, M.L.; Richardson, J.R.; Safadi, F.F. Transgenic Overexpression of GPNMB Protects Against MPTP-Induced Neurodegeneration. Mol. Neurobiol. 2020, 57, 2920–2933. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Wuolikainen, A.; Wu, J.; Öhman, A.; Wingsle, G.; Moritz, T.; Andersen, P.M.; Forsgren, L.; Trupp, M. Targeted Multiple Reaction Monitoring Analysis of CSF Identifies UCHL1 and GPNMB as Candidate Biomarkers for ALS. J. Mol. Neurosci. MN 2019, 69, 643–657. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.; Barres, B. SnapShot: Astrocytes in Health and Disease. Cell 2015, 162, 1170.e1. [Google Scholar] [CrossRef] [PubMed]
- Izrael, M.; Slutsky, S.G.; Revel, M. Rising Stars: Astrocytes as a Therapeutic Target for ALS Disease. Front. Neurosci. 2020, 14, 824. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Nave, K.A. Oligodendrocytes: Myelination and Axonal Support. Cold Spring Harb. Perspect. Biol. 2015, 8, a020479. [Google Scholar] [CrossRef] [PubMed]
- Stadelmann, C.; Timmler, S.; Barrantes-Freer, A.; Simons, M. Myelin in the Central Nervous System: Structure, Function, and Pathology. Physiol. Rev. 2019, 99, 1381–1431. [Google Scholar] [CrossRef] [PubMed]
- Festa, L.K.; Grinspan, J.B.; Jordan-Sciutto, K.L. White matter injury across neurodegenerative disease. Trends Neurosci. 2024, 47, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Barton, S.K.; Gregory, J.M.; Selvaraj, B.T.; McDade, K.; Henstridge, C.M.; Spires-Jones, T.L.; James, O.G.; Mehta, A.R.; Story, D.; Burr, K.; et al. Dysregulation in Subcellular Localization of Myelin Basic Protein mRNA Does Not Result in Altered Myelination in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2021, 15, 705306. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.H.; Li, Y.; Fukaya, M.; Lorenzini, I.; Cleveland, D.W.; Ostrow, L.W.; Rothstein, J.D.; Bergles, D.E. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat. Neurosci. 2013, 16, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Bento-Abreu, A.; Nonneman, A.; Haeck, W.; Staats, K.; Geelen, V.; Hersmus, N.; Küsters, B.; Van Den Bosch, L.; Van Damme, P.; et al. Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain A J. Neurol. 2013, 136 Pt 2, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Pons, A.L.; Higginbottom, A.; Cooper-Knock, J.; Alrafiah, A.; Alofi, E.; Kirby, J.; Shaw, P.J.; Wood, J.D.; Highley, J.R. Oligodendrocyte pathology exceeds axonal pathology in white matter in human amyotrophic lateral sclerosis. J. Pathol. 2020, 251, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Sadler, G.L.; Lewis, K.N.; Narayana, V.K.; De Souza, D.P.; Mason, J.; McLean, C.; Gonsalvez, D.G.; Turner, B.J.; Barton, S.K. Lipid Metabolism Is Dysregulated in the Motor Cortex White Matter in Amyotrophic Lateral Sclerosis. Metabolites 2022, 12, 554. [Google Scholar] [PubMed]
- Sirisi, S.; Querol-Vilaseca, M.; Dols-Icardo, O.; Pegueroles, J.; Montal, V.; Muñoz, L.; Torres, S.; Ferrer-Raventós, P.; Iulita, M.F.; Sánchez-Aced, É.; et al. Myelin loss in C9orf72 hexanucleotide expansion carriers. J. Neurosci. Res. 2022, 100, 1862–1875. [Google Scholar] [CrossRef] [PubMed]
- Floriddia, E.M.; Lourenço, T.; Zhang, S.; van Bruggen, D.; Hilscher, M.M.; Kukanja, P.; Dos Santos, J.P.G.; Altınkök, M.; Yokota, C.; Llorens-Bobadilla, E.; et al. Distinct oligodendrocyte populations have spatial preference and different responses to spinal cord injury. Nat. Commun. 2020, 11, 5860. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, A.; Hochgerner, H.; Lönnerberg, P.; Johnsson, A.; Memic, F.; van der Zwan, J.; Häring, M.; Braun, E.; Borm, L.E.; La Manno, G.; et al. Molecular Architecture of the Mouse Nervous System. Cell 2018, 174, 999–1014.e22. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identifier | n (ALS) 1 | n (CTL) 1 | Gene Count 2 | ALS-Increased 3 | ALS-Decreased 4 |
|---|---|---|---|---|---|
| GSE137810-N a | 253 (137) | 51 (35) | 16,044 | 1000 | 810 |
| GSE137810-H b | 150 (75) | 27 (17) | 15,936 | 291 | 93 |
| GSE255683 c | 10 (10) | 10 (10) | 15,204 | 0 | 0 |
| SRP064478 d | 7 (7) | 8 (8) | 14,791 | 0 | 0 |
| GSE26927 e | 10 (9) | 10 (7) | 13,634 | 0 | 0 |
| E-MTAB-8635 f | 24 (24) | 9 (9) | 16,921 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swindell, W.R. Meta-Analysis of Gene Expression in Bulk-Processed Post-Mortem Spinal Cord from ALS Patients and Normal Controls. NeuroSci 2025, 6, 65. https://doi.org/10.3390/neurosci6030065
Swindell WR. Meta-Analysis of Gene Expression in Bulk-Processed Post-Mortem Spinal Cord from ALS Patients and Normal Controls. NeuroSci. 2025; 6(3):65. https://doi.org/10.3390/neurosci6030065
Chicago/Turabian StyleSwindell, William R. 2025. "Meta-Analysis of Gene Expression in Bulk-Processed Post-Mortem Spinal Cord from ALS Patients and Normal Controls" NeuroSci 6, no. 3: 65. https://doi.org/10.3390/neurosci6030065
APA StyleSwindell, W. R. (2025). Meta-Analysis of Gene Expression in Bulk-Processed Post-Mortem Spinal Cord from ALS Patients and Normal Controls. NeuroSci, 6(3), 65. https://doi.org/10.3390/neurosci6030065