
In the Brain, It Is Not All about Sugar


{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Brain and Its Fuels
3. Fatty Acid Fueling
4. Fatty Acid Synthesis and Oxidation
4.1. Fatty Acid Synthesis
4.2. Fatty Acid Oxidation
4.2.1. Carnitine Shuttle
4.2.2. Fatty Acid β-Oxidation
5. Ketone Body Metabolism
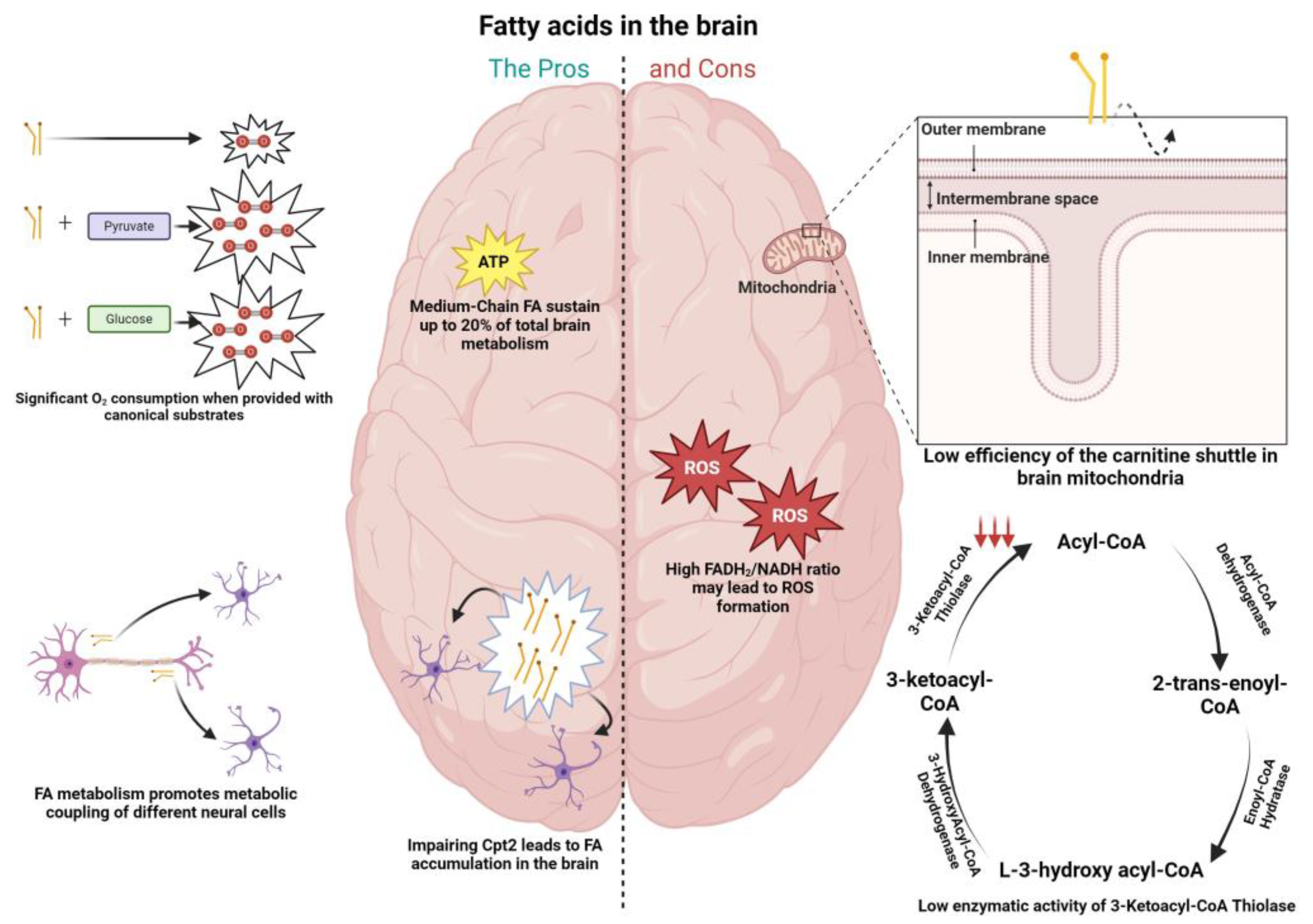
6. Fatty Acid Metabolism in the Brain
6.1. Cons to FA Metabolism in the Brain
6.2. The Pros of FA Metabolism in the Brain
6.3. The Role of Fatty Acids in Brain Pathophysiology
7. Ketone Bodies in the Brain
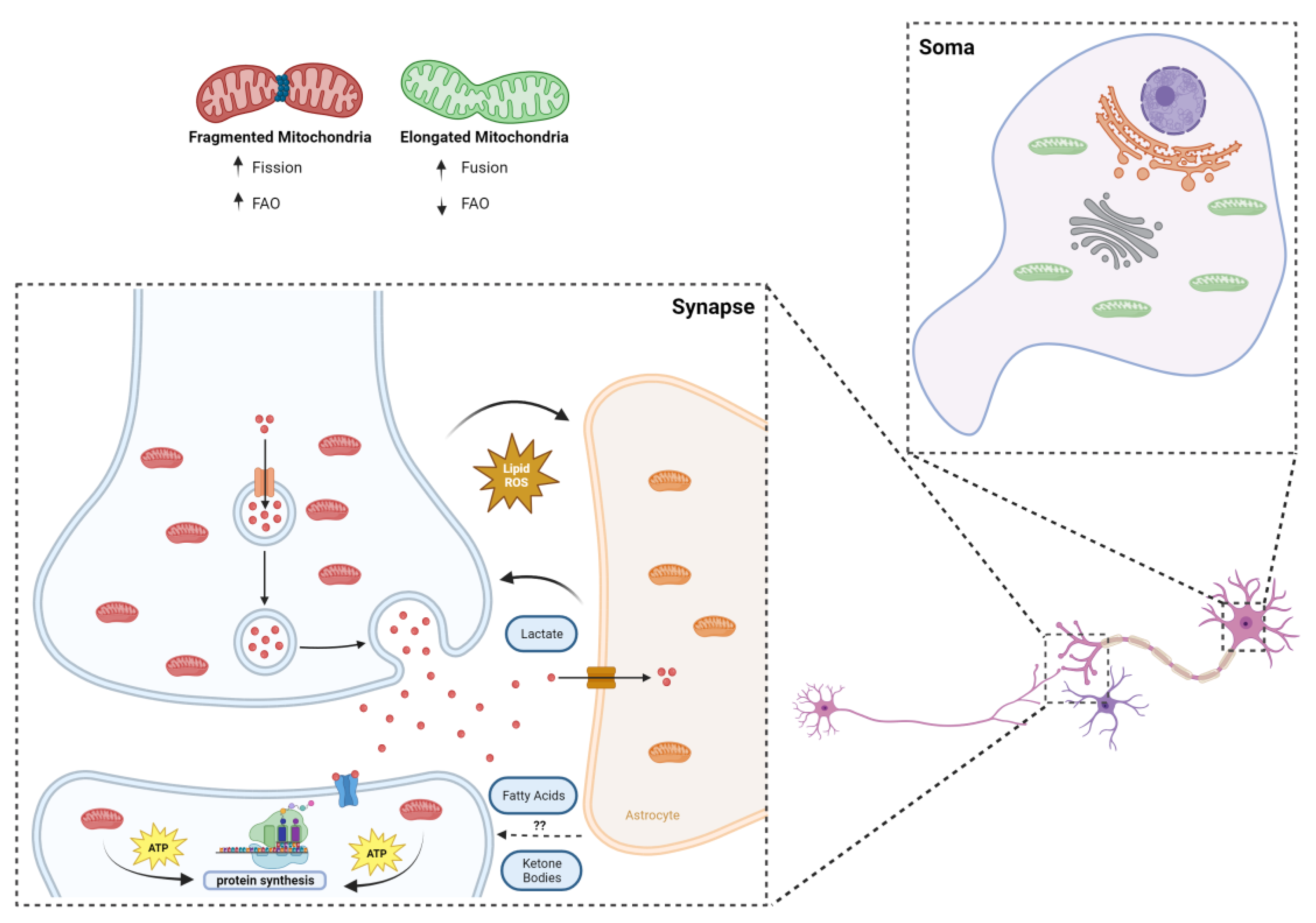
8. Fatty Acids and Synaptic Mitochondria
9. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Spinelli, J.B.; Haigis, M.C. The Multifaceted Contributions of Mitochondria to Cellular Metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Guan, D.; Wang, S.; Chai, L.Y.A.; Xu, S.; Lam, K.P. Glycolysis and Oxidative Phosphorylation Play Critical Roles in Natural Killer Cell Receptor-Mediated Natural Killer Cell Functions. Front. Immunol. 2020, 11, 202. [Google Scholar] [CrossRef] [PubMed]
- Steiner, P. Brain Fuel Utilization in the Developing Brain. Ann. Nutr. Metab. 2020, 75 (Suppl. S1), 8–18. [Google Scholar] [CrossRef]
- Romano, A.; Koczwara, J.B.; Gallelli, C.A.; Vergara, D.; Di Bonaventura, M.V.M.; Gaetani, S.; Giudetti, A.M. Fats for thoughts: An update on brain fatty acid metabolism. Int. J. Biochem. Cell Biol. 2017, 84, 40–45. [Google Scholar] [CrossRef]
- Ebert, D.; Haller, R.G.; Walton, M.E. Energy contribution of octanoate to intact rat brain metabolism measured by 13C nuclear magnetic resonance spectroscopy. J. Neurosci. 2003, 23, 5928–5935. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.G.; Laranjeira, A.; Van Huffel, L.; Gartner, A.; Vilain, S.; Bastianen, J.; Van Veldhoven, P.P.; Dotti, C.G. Glial b-Oxidation regulates Drosophila Energy Metabolism. Sci. Rep. 2015, 5, 7805. [Google Scholar] [CrossRef]
- Panov, A.; Orynbayeva, Z.; Vavilin, V.; Lyakhovich, V. Fatty acids in energy metabolism of the central nervous system. Biomed. Res. Int. 2014, 2014, 472459. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Wu, Y.; Knapp, J.; Park, D.; Gupta, K.; De Camilli, P.; Ryan, T.A. DDHD2 is necessary for activity-driven fatty acid fueling of nerve terminal function. bioRxiv 2023. [Google Scholar] [CrossRef]
- Hamilton, J.A.; Hillard, C.J.; Spector, A.A.; Watkins, P.A. Brain uptake and utilization of fatty acids, lipids and lipoproteins: Application to neurological disorders. J. Mol. Neurosci. 2007, 33, 2–11. [Google Scholar] [CrossRef]
- Tracey, T.J.; Steyn, F.J.; Wolvetang, E.J.; Ngo, S.T. Neuronal lipid metabolism: Multiple pathways driving functional outcomes in health and disease. Front. Mol. Neurosci. 2018, 11, 10. [Google Scholar] [CrossRef]
- Mi, Y.; Qi, G.; Vitali, F.; Shang, Y.; Raikes, A.C.; Wang, T.; Jin, Y.; Brinton, R.D.; Gu, H.; Yin, F. Loss of fatty acid degradation by astrocytic mitochondria triggers neuroinflammation and neurodegeneration. Nat. Metab. 2023, 5, 445–465. [Google Scholar] [CrossRef]
- Schönfeld, P.; Reiser, G. Brain energy metabolism spurns fatty acids as fuel due to their inherent mitotoxicity and potential capacity to unleash neurodegeneration. Neurochem. Int. 2017, 109, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Denker, N.; Harders, A.R.; Arend, C.; Dringen, R. Consumption and Metabolism of Extracellular Pyruvate by Cultured Rat Brain Astrocytes. Neurochem. Res. 2023, 48, 1438–1454. [Google Scholar] [CrossRef] [PubMed]
- Schönfeld, P.; Reiser, G. Why does brain metabolism not favor burning of fatty acids to provide energy-Reflections on disadvantages of the use of free fatty acids as fuel for brain. J. Cereb. Blood Flow Metab. 2013, 33, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A. Brain lactate metabolism: The discoveries and the controversies. J. Cereb. Blood Flow Metab. 2012, 32, 1107–1138. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Miao, Q.Q.; Zhang, Q.; Mao, S.; Li, M.; Xu, X.; Xia, X.; Wei, K.; Fan, Y.; Zheng, X.; et al. Aerobic glycolysis is the predominant means of glucose metabolism in neuronal somata, which protects against oxidative damage. Nat. Neurosci. 2023, 26, 2081–2089. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Myeong, J.; Hashemiaghdam, A.; Zhang, H.; Niu, X. Mitochondrial pyruvate transport regulates presynaptic metabolism and neurotransmission. bioRxiv 2024. [Google Scholar] [CrossRef]
- Divakaruni, A.S.; Wallace, M.; Buren, C.; Martyniuk, K.; Andreyev, A.Y.; Li, E.; Fields, J.A.; Cordes, T.; Reynolds, I.J.; Bloodgood, B.L.; et al. Inhibition of the mitochondrial pyruvate carrier protects from excitotoxic neuronal death. J. Cell Biol. 2017, 216, 1091–1105. [Google Scholar] [CrossRef] [PubMed]
- Glatz, J.F.C.; Luiken, J.J.F.P.; Bonen, A. Membrane fatty acid transporters as regulators of lipid metabolism: Implications for metabolic disease. Physiol. Rev. 2010, 90, 367–417. [Google Scholar] [CrossRef] [PubMed]
- Suburu, J.; Gu, Z.; Chen, H.; Chen, W.; Hao, Z.; Chen, Y.Q. Fatty acid metabolism: Implications for diet, genetic variation, and disease. Food Biosci. 2013, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Diskin, C.; Ryan, T.A.J.; O’Neill, L.A.J. Modification of Proteins by Metabolites in Immunity. Immunity 2021, 54, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Ji, B.; Skup, M. Roles of palmitoylation in structural long-term synaptic plasticity. Mol. Brain 2021, 14, 8. [Google Scholar] [CrossRef] [PubMed]
- Ho, G.P.H.; Wilkie, E.C.; White, A.J.; Selkoe, D.J. Palmitoylation of the Parkinson’s disease-associated protein synaptotagmin-11 links its turnover to α-synuclein homeostasis. Sci. Signal. 2023, 16, eadd7220. [Google Scholar] [CrossRef] [PubMed]
- Sambra, V.; Echeverria, F.; Valenzuela, A.; Chouinard-Watkins, R.; Valenzuela, R. Docosahexaenoic and arachidonic acids as neuroprotective nutrients throughout the life cycle. Nutrients 2021, 13, 986. [Google Scholar] [CrossRef] [PubMed]
- Nowinski, S.M.; Solmonson, A.; Rusin, S.F.; Maschek, J.A.; Bensard, C.L.; Fogarty, S.; Jeong, M.Y.; Lettlova, S.; Berg, J.A.; Morgan, J.T.; et al. Mitochondrial fatty acid synthesis coordinates oxidative metabolism in mammalian mitochondria. eLife 2020, 9, e58041. [Google Scholar] [CrossRef] [PubMed]
- Garcia Corrales, A.V.; Haidar, M.; Bogie, J.F.J.; Hendriks, J.J.A. Fatty acid synthesis in glial cells of the cns. Int. J. Mol. Sci. 2021, 22, 8159. [Google Scholar] [CrossRef] [PubMed]
- El-Gharbawy, A.; Vockley, J. Defects of Fatty Acid Oxidation and the Carnitine Shuttle System. Pediatr. Clin. N. Am. 2018, 65, 317–335. [Google Scholar] [CrossRef] [PubMed]
- Houten, S.M.; Violante, S.; Ventura, F.V.; Wanders, R.J.A. The Biochemistry and Physiology of Mitochondrial Fatty Acid β-Oxidation and Its Genetic Disorders. Annu. Rev. Physiol. 2016, 78, 23–44. [Google Scholar] [CrossRef]
- Houten, S.M.; Wanders, R.J.A. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Yun, C.L.; Zierath, J.R. AMP-activated protein kinase signaling in metabolic regulation. J. Clin. Investig. 2006, 116, 1776–1783. [Google Scholar]
- Puchalska, P.; Crawford, P.A. Multi-dimensional Roles of Ketone Bodies in Fuel Metabolism, Signaling, and Therapeutics. Cell Metab. 2017, 25, 262–284. [Google Scholar] [CrossRef] [PubMed]
- Sierra, A.Y.; Gratacós, E.; Carrasco, P.; Clotet, J.; Ureña, J.; Serra, D.; Asins, G.; Hegardt, F.G.; Casals, N. CPT1c is localized in endoplasmic reticulum of neurons and has carnitine palmitoyltransferase activity. J. Biol. Chem. 2008, 283, 6878–6885. [Google Scholar] [CrossRef]
- Middleton, B. The oxoacyl-coenzyme A thiolases of animal tissues. Biochem. J. 1973, 132, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, M.S.; Jackson, J.; Sheu, S.H.; Chang, C.L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 2019, 177, 1522–1535.e14. [Google Scholar] [CrossRef] [PubMed]
- Thevenet, J.; De Marchi, U.; Domingo, J.S.; Christinat, N.; Bultot, L.; Lefebvre, G.; Sakamoto, K.; Descombes, P.; Masoodi, M.; Wiederkehr, A. Medium-chain fatty acids inhibit mitochondrial metabolism in astrocytes promoting astrocyte-neuron lactate and ketone body shuttle systems. FASEB J. 2016, 30, 1913–1926. [Google Scholar] [CrossRef] [PubMed]
- Adibhatla, R.M.; Hatcher, J.F. Altered lipid metabolism in brain injury and disorders. Subcell. Biochem. 2008, 49, 241–268. [Google Scholar] [PubMed]
- Leng, L.; Yuan, Z.; Pan, R.; Su, X.; Wang, H.; Xue, J.; Zhuang, K.; Gao, J.; Chen, Z.; Lin, H.; et al. Microglial hexokinase 2 deficiency increases ATP generation through lipid metabolism leading to β-amyloid clearance. Nat. Metab. 2022, 4, 1287–1305. [Google Scholar] [CrossRef] [PubMed]
- Drougard, A.; Ma, E.H.; Wegert, V.; Sheldon, R.; Panzeri, I.; Vatsa, N.; Apostle, S.; Fagnocchi, L.; Schaf, J.; Gossens, K.; et al. A rapid microglial metabolic response controls metabolism and improves memory. Elife 2023, 12, RP87120. [Google Scholar]
- Liu, L.; Zhang, K.; Sandoval, H.; Yamamoto, S.; Jaiswal, M.; Sanz, E.; Li, Z.; Hui, J.; Graham, B.H.; Quintana, A.; et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 2015, 160, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Edmond, J.; Robbins, R.A.; Bergstrom, J.D.; Cole, R.A.; de Vellis, J. Capacity for substrate utilization in oxidative metabolism by neurons, astrocytes, and oligodendrocytes from developing brain in primary culture. J. Neurosci. Res. 1987, 18, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Cahill, G.F. Fuel Metabolism in Starvation. Annu. Rev. Nutr. 2006, 26, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.; Kempf, K.; Röhling, M.; Lenzen-Schulte, M.; Schloot, N.C.; Martin, S. Ketone bodies: From enemy to friend and guardian angel. BMC Med. 2021, 19, 313. [Google Scholar] [CrossRef] [PubMed]
- Silva, B.; Mantha, O.L.; Schor, J.; Pascual, A.; Plaçais, P.Y.; Pavlowsky, A.; Preat, T. Glia fuel neurons with locally synthesized ketone bodies to sustain memory under starvation. Nat. Metab. 2022, 4, 213–224. [Google Scholar] [CrossRef]
- Klosinski, L.P.; Yao, J.; Yin, F.; Fonteh, A.N.; Harrington, M.G.; Christensen, T.A.; Trushina, E.; Brinton, R.D. White Matter Lipids as a Ketogenic Fuel Supply in Aging Female Brain: Implications for Alzheimer’s Disease. EBioMedicine 2015, 2, 1888–1904. [Google Scholar] [CrossRef] [PubMed]
- Fedorovich, S.V.; Waseem, T.V.; Puchkova, L.V. Biogenetic and morphofunctional heterogeneity of mitochondria: The case of synaptic mitochondria. Rev. Neurosci. 2017, 28, 363–373. [Google Scholar] [CrossRef] [PubMed]
- Stowers, R.S.; Megeath, L.J.; Górska-Andrzejak, J.; Meinertzhagen, I.A.; Schwarz, T.L. Axonal transport of mitochondria to synapses depends on Milton, a novel Drosophila protein. Neuron 2002, 36, 1063–1077. [Google Scholar] [CrossRef] [PubMed]
- Lanfranchi, M.; Meyer-Dilhet, G.; Dos Reis, R.; Garcia, A.; Blondet, C.; Javin, L.; Amar, A.; Courchet, J. The AMPK-related kinase NUAK1 controls cortical axons branching though a local modulation of mitochondrial metabolic functions. bioRxiv 2020. [Google Scholar] [CrossRef]
- Pathak, D.; Shields, L.Y.; Mendelsohn, B.A.; Haddad, D.; Lin, W.; Gerencser, A.A.; Kim, H.; Brand, M.D.; Edwards, R.H.; Nakamura, K. The role of mitochondrially derived ATP in synaptic vesicle recycling. J. Biol. Chem. 2015, 290, 22325–22336. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.C.; Clark, J.B. Preparation and properties of mitochondria derived from synaptosomes. Biochem. J. 1976, 154, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Lai, J.C.K.; Walsh, J.M.; Dennis, S.C.; Clark, J.B. Synaptic and Non-Synaptic Mitochondria from Rat Brain: Isolation and Characterization. J. Neurochem. 1977, 28, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.F.; Lai, J.C.K.; Lim, L.; Clark, J.B. The Activities of Some Energy-Metabolising Enzymes in Nonsynaptic (Free) and Synaptic Mitochondria Derived from Selected Brain Regions. J. Neurochem. 1984, 42, 1306–1312. [Google Scholar] [CrossRef] [PubMed]
- Dagani, F.; Gorini, A.; Polgatti, M.; Villa, R.F.; Benzi, G. Rat cortex synaptic and nonsynaptic mitochondria: Enzymatic characterization and pharmacological effects of naftidrofuryl. J. Neurosci. Res. 1983, 10, 135–140. [Google Scholar] [CrossRef] [PubMed]
- McKenna, M.C.; Hopkins, I.B.; Lindauer, S.L.; Bamford, P. Aspartate aminotransferase in synaptic and nonsynaptic mitochondria: Differential effect of compounds that influence transient hetero-enzyme complex (metabolon) formation. Neurochem. Int. 2006, 48, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Kiebisch, M.A.; Han, X.; Cheng, H.; Lunceford, A.; Clarke, C.F.; Moon, H.; Chuang, J.H.; Seyfried, T.N. Lipidomic analysis and electron transport chain activities in C57BL/6J mouse brain mitochondria. J. Neurochem. 2008, 106, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Stauch, K.L.; Purnell, P.R.; Fox, H.S. Quantitative proteomics of synaptic and nonsynaptic mitochondria: Insights for synaptic mitochondrial vulnerability. J. Proteome Res. 2014, 13, 2620–2636. [Google Scholar] [CrossRef] [PubMed]
- Völgyi, K.; Gulyássy, P.; Háden, K.; Kis, V.; Badics, K.; Kékesi, K.A.; Simor, A.; Györffy, B.; Tóth, E.A.; Lubec, G.; et al. Synaptic mitochondria: A brain mitochondria cluster with a specific proteome. J. Proteomics 2015, 120, 142–157. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Okamoto, K.I.; Hayashi, Y.; Sheng, M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell 2004, 119, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Rangaraju, V.; Lauterbach, M.; Schuman, E.M. Spatially Stable Mitochondrial Compartments Fuel Local Translation during Plasticity. Cell 2019, 176, 73–84.e15. [Google Scholar] [CrossRef]
- Morant-Ferrando, B.; Jimenez-Blasco, D.; Alonso-Batan, P.; Agulla, J.; Lapresa, R.; Garcia-Rodriguez, D.; Yunta-Sanchez, S.; Lopez-Fabuel, I.; Fernandez, E.; Carmeliet, P.; et al. Fatty acid oxidation organizes mitochondrial supercomplexes to sustain astrocytic ROS and cognition. Nat. Metab. 2023, 5, 1290–1302. [Google Scholar] [CrossRef] [PubMed]
- Dienel, G.A.; Hertz, L. Glucose and lactate metabolism during brain activation. J. Neurosci. Res. 2001, 66, 824–838. [Google Scholar] [CrossRef] [PubMed]
- Ngo, J.; Choi, D.W.; Stanley, I.A.; Stiles, L.; Molina, A.J.A.; Chen, P.; Lako, A.; Sung, I.C.H.; Goswami, R.; Kim, M.; et al. Mitochondrial morphology controls fatty acid utilization by changing CPT1 sensitivity to malonyl-CoA. EMBO J. 2023, 42, e111901. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Antunes, B.C.; Mateus, T.; Morais, V.A. In the Brain, It Is Not All about Sugar. NeuroSci 2024, 5, 209-221. https://doi.org/10.3390/neurosci5020016
Antunes BC, Mateus T, Morais VA. In the Brain, It Is Not All about Sugar. NeuroSci. 2024; 5(2):209-221. https://doi.org/10.3390/neurosci5020016
Chicago/Turabian StyleAntunes, Bernardo C., Tomás Mateus, and Vanessa A. Morais. 2024. "In the Brain, It Is Not All about Sugar" NeuroSci 5, no. 2: 209-221. https://doi.org/10.3390/neurosci5020016
APA StyleAntunes, B. C., Mateus, T., & Morais, V. A. (2024). In the Brain, It Is Not All about Sugar. NeuroSci, 5(2), 209-221. https://doi.org/10.3390/neurosci5020016