
GSK3β Activity in Reward Circuit Functioning and Addiction

 , , ,
, , ,
Abstract
:1. Introduction
1.1. GSK3β Characteristic
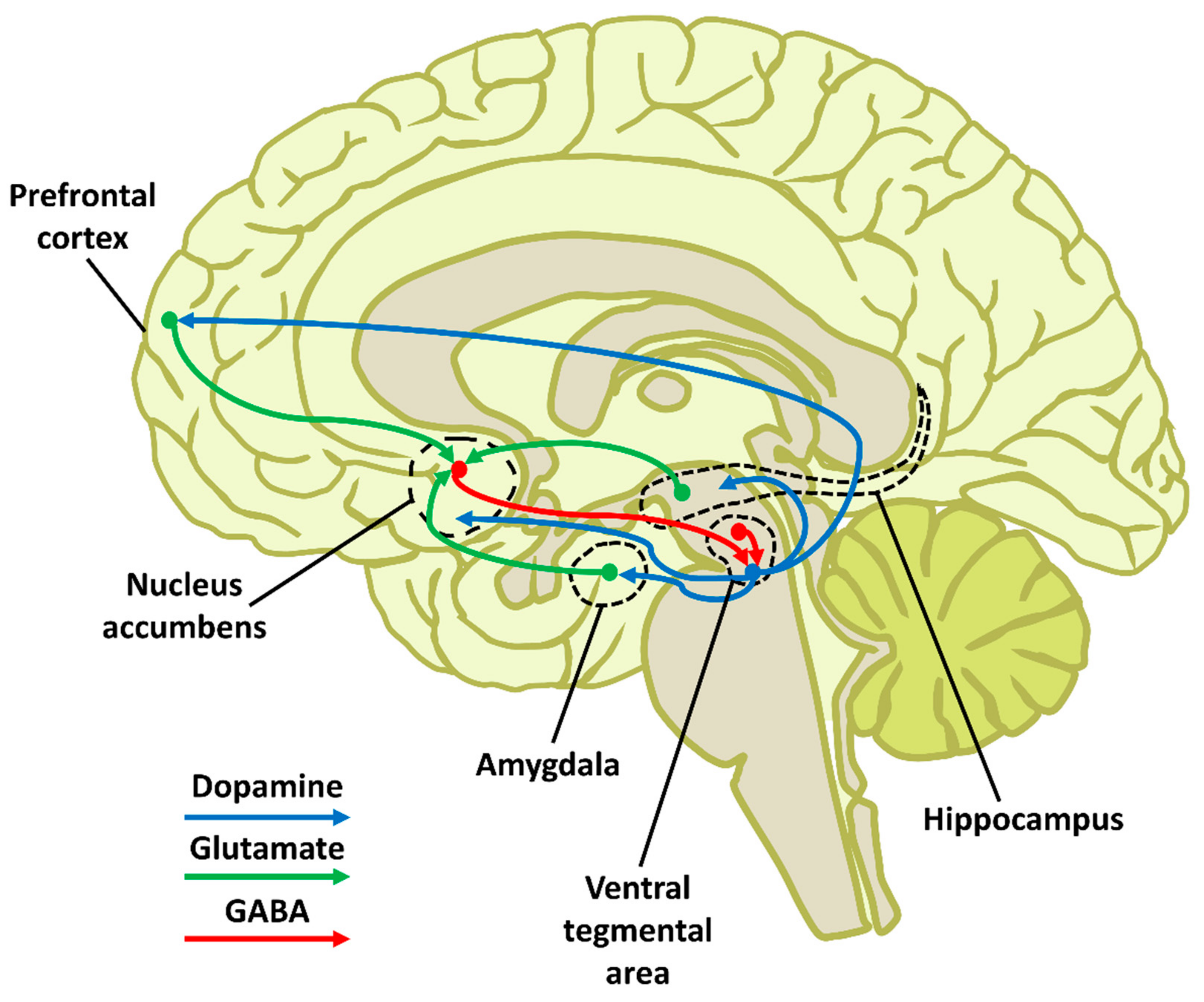
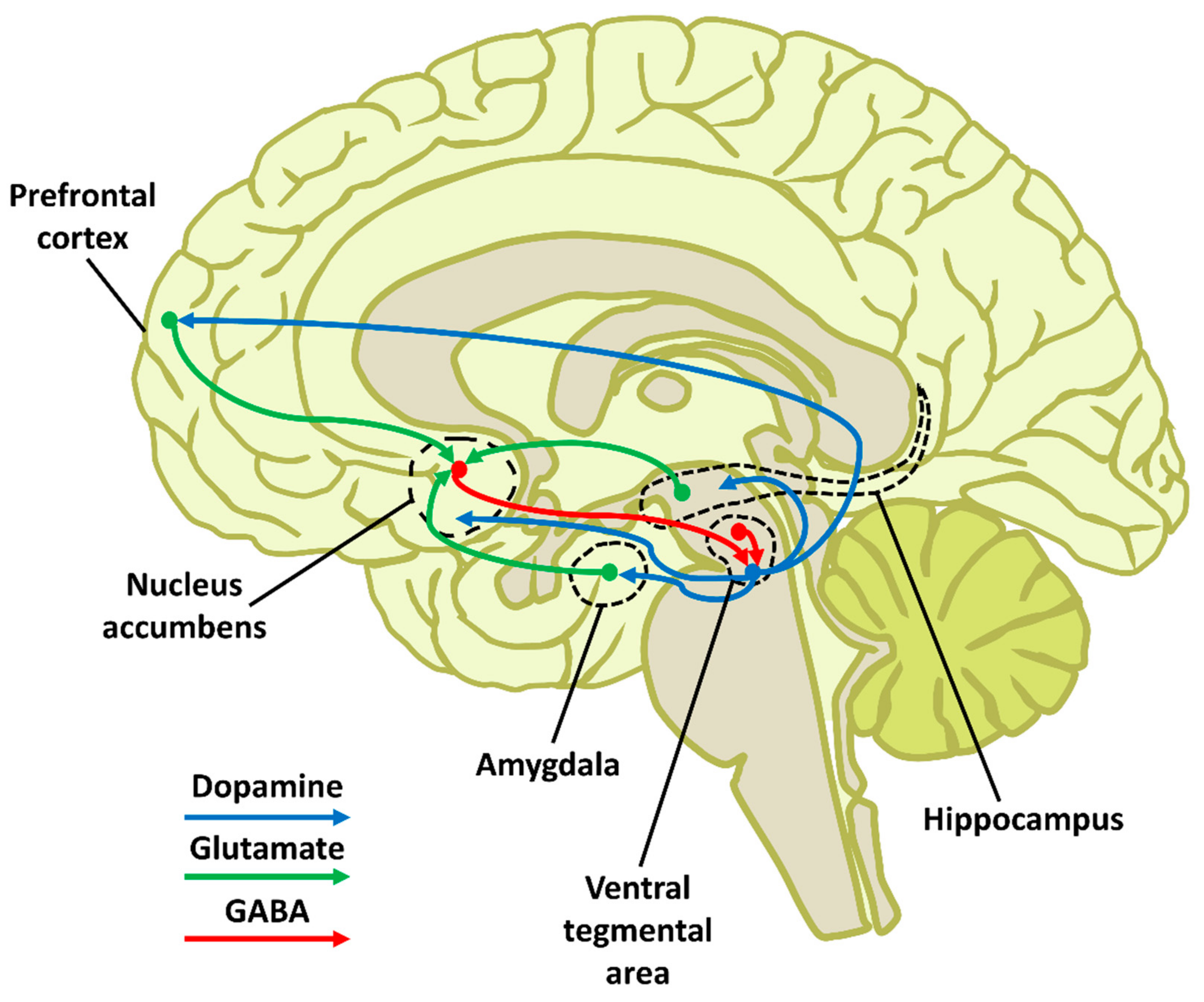
1.2. Reward Circuit Anatomy and Functioning
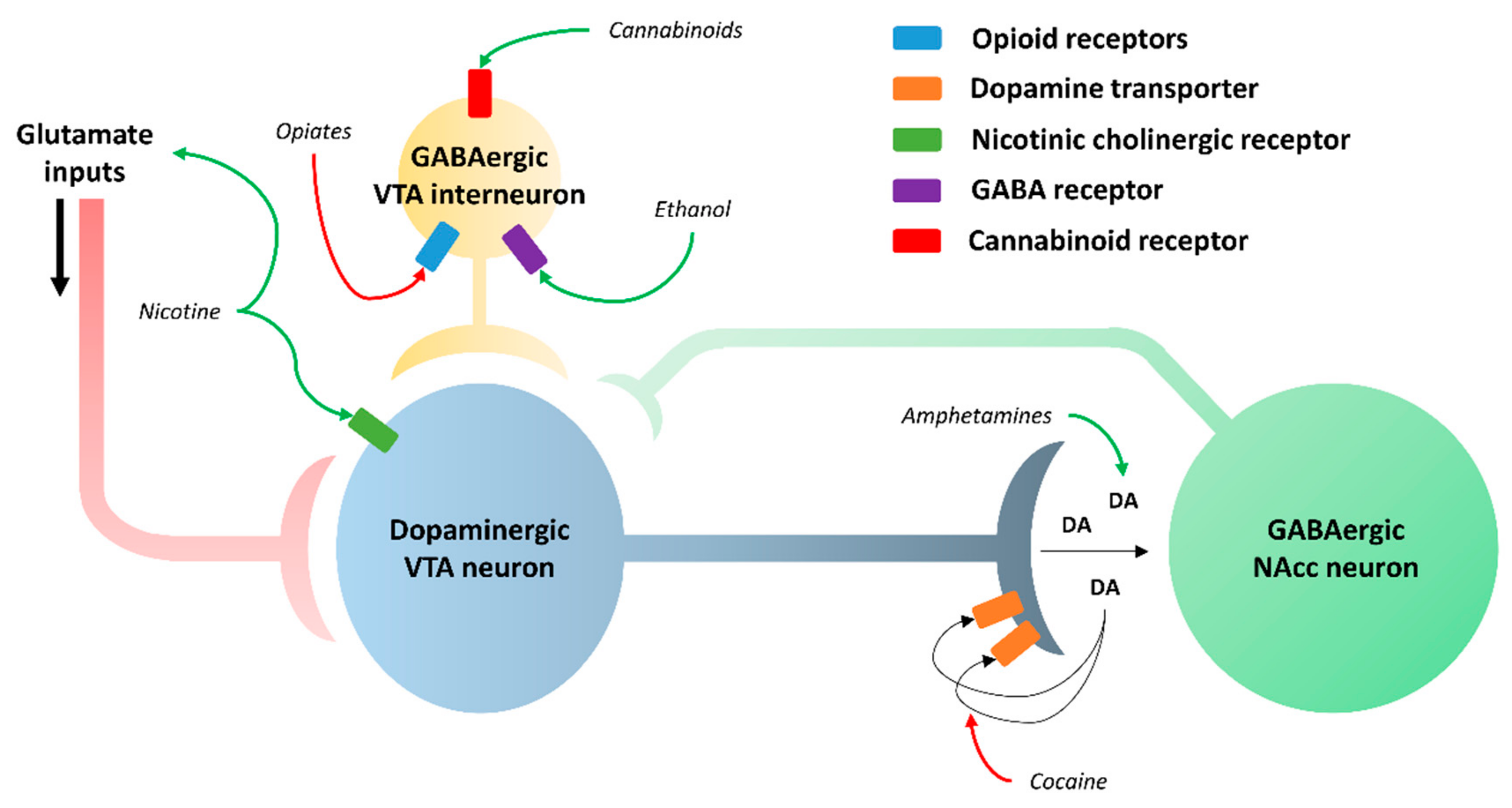
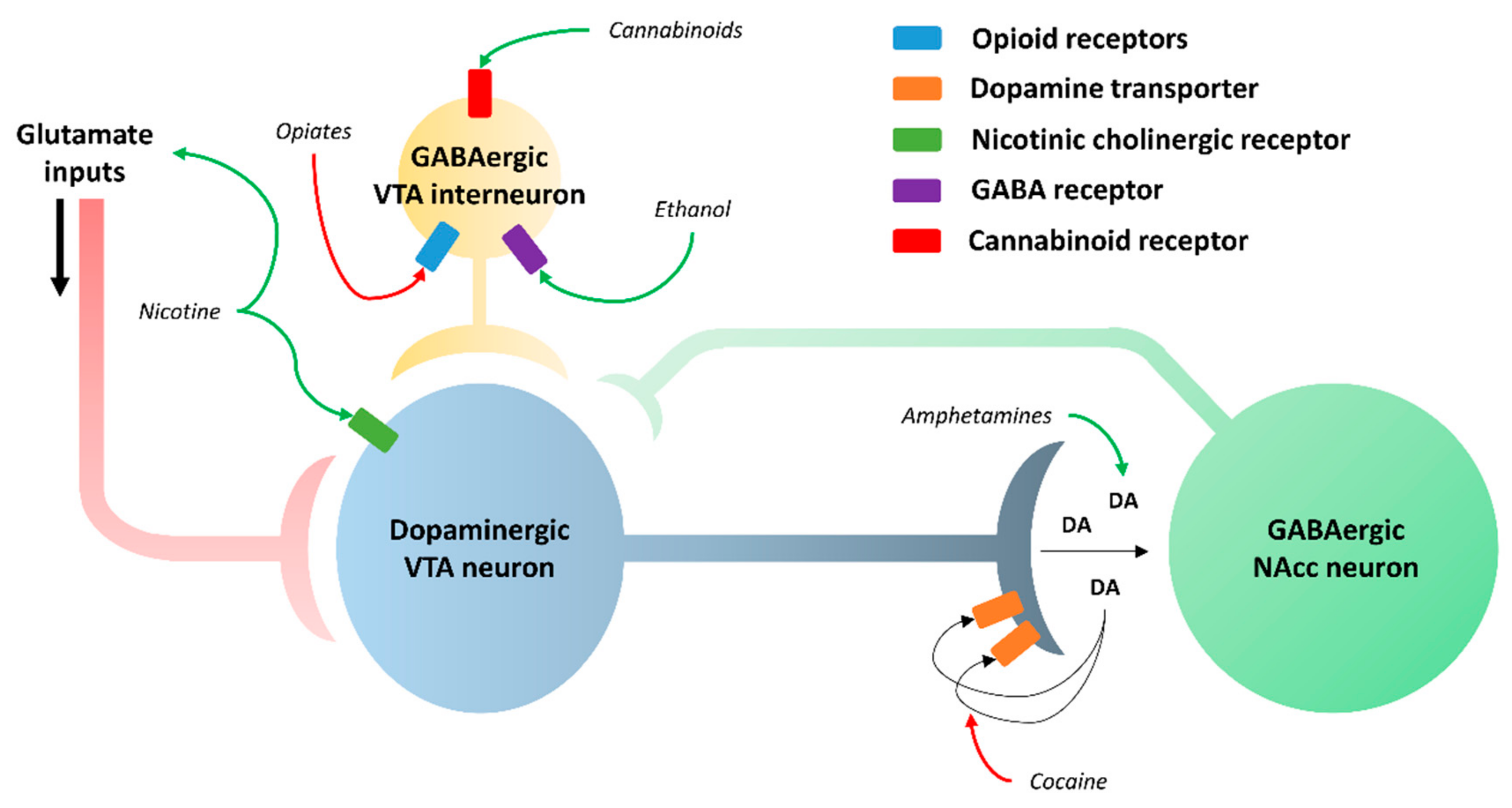
1.3. Addiction Mechanism
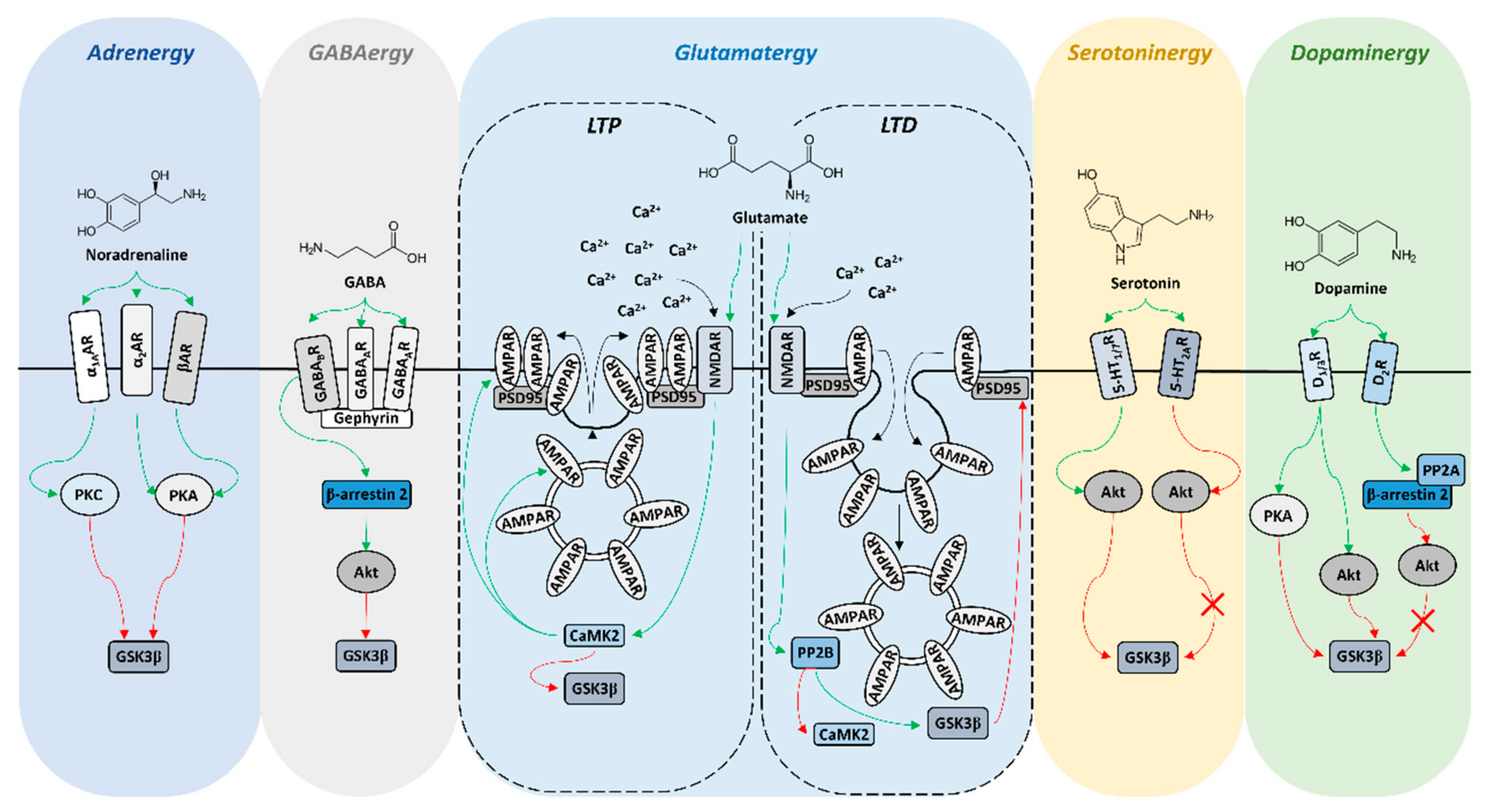
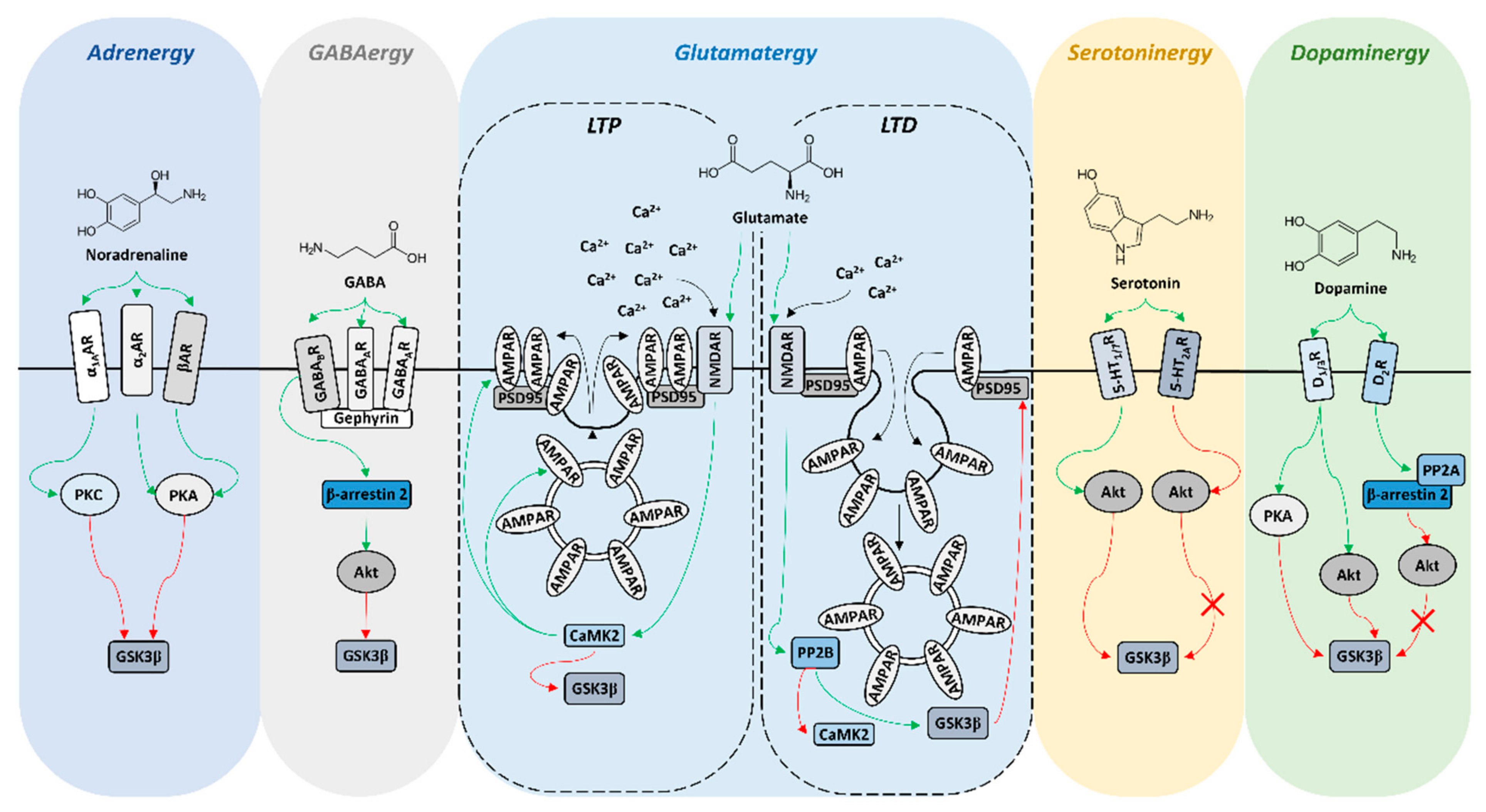
2. GSK3β Expression Profile in the Reward Circuit-Related Structures
3. GSK3β Activity in the Reward Circuit
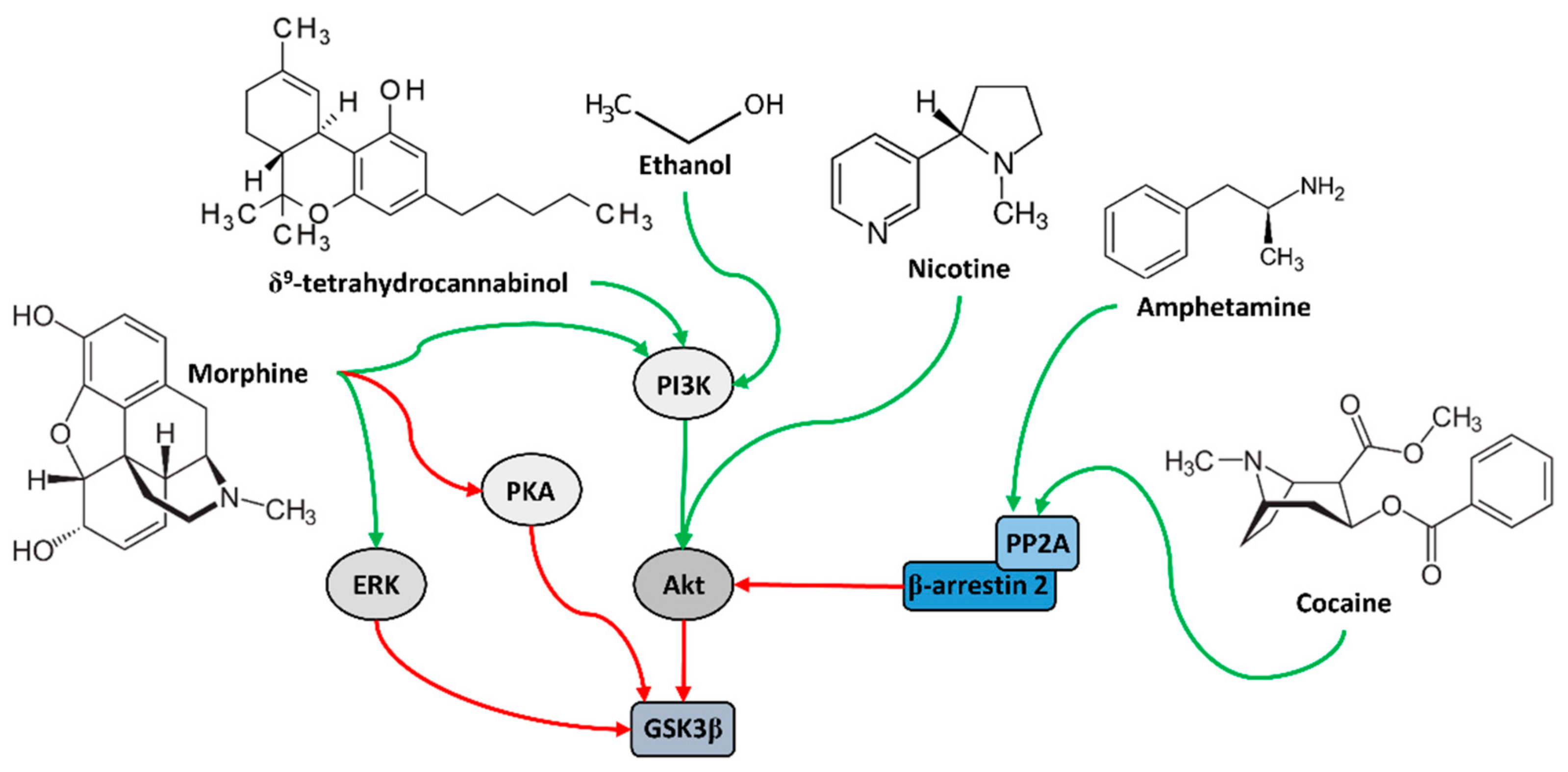
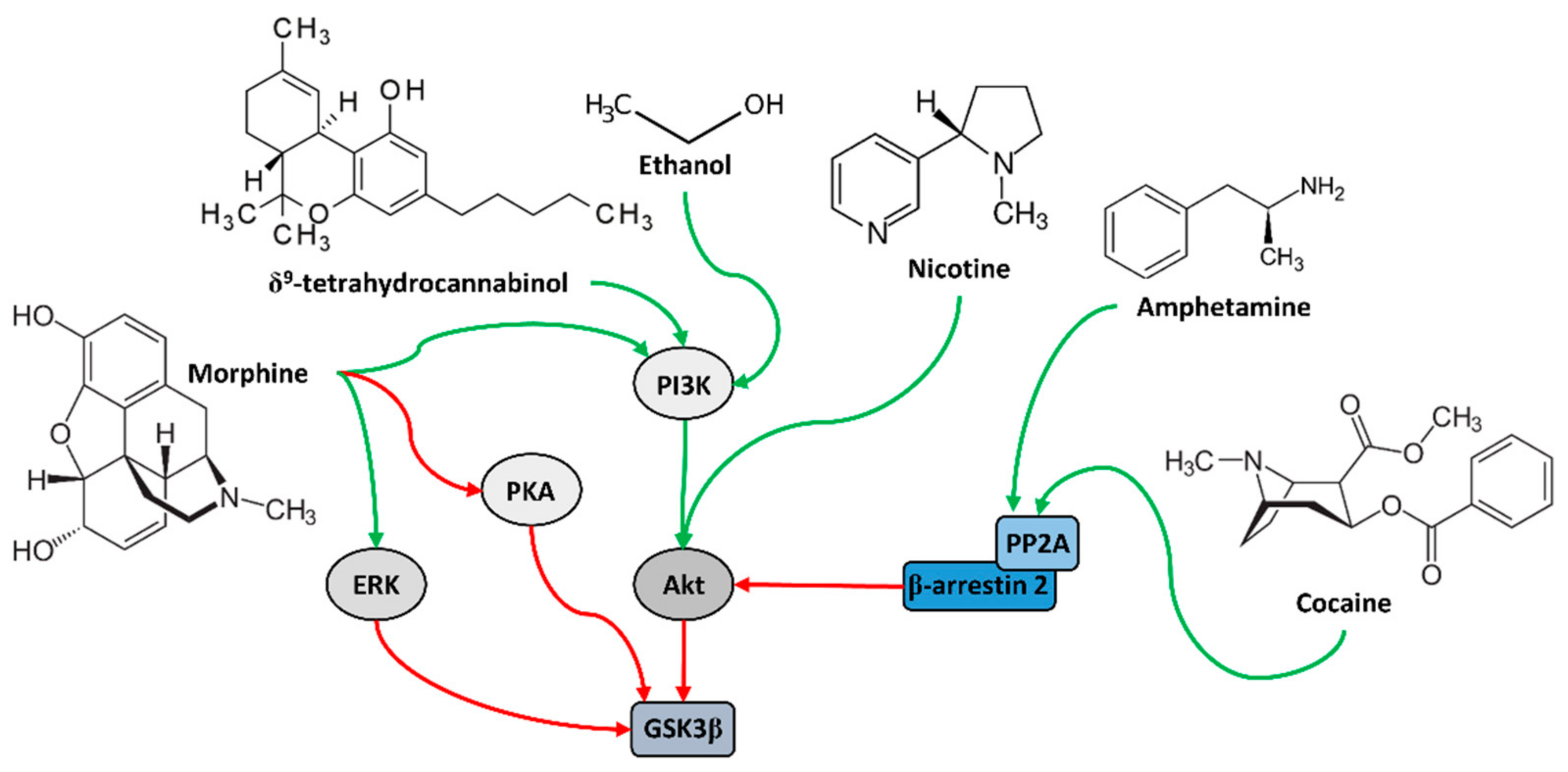
4. Role of GSK3β in Addiction
5. Addiction Treatment and Its Effect on GSK3β
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hughes, K.; Nikolakaki, E.; Plyte, S.E.; Totty, N.F.; Woodgett, J.R. Modulation of the Glycogen Synthase Kinase-3 Family by Tyrosine Phosphorylation. EMBO J. 1993, 12, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, C.; Cohen, P. The α-Isoform of Glycogen Synthase Kinase-3 from Rabbit Skeletal Muscle Is Inactivated by P70 S6 Kinase or MAP Kinase-Activated Protein Kinase-1 in Vitro. FEBS Lett. 1994, 338, 37–42. [Google Scholar] [CrossRef] [Green Version]
- Brady, M.J.; Bourbonais, F.J.; Saltiel, A.R. The Activation of Glycogen Synthase by Insulin Switches from Kinase Inhibition to Phosphatase Activation during Adipogenesis in 3T3-L1 Cells. J. Biol. Chem. 1998, 273, 14063–14066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sutherland, C.; Leighton, I.A.; Cohen, P. Inactivation of Glycogen Synthase Kinase-3β by Phosphorylation: New Kinase Connections in Insulin and Growth-Factor Signalling. Biochem. J. 1993, 296, 15–19. [Google Scholar] [CrossRef]
- Tanji, C.; Yamamoto, H.; Yorioka, N.; Kohno, N.; Kikuchi, K.; Kikuchi, A. A-Kinase Anchoring Protein AKAP220 Binds to Glycogen Synthase Kinase-3β (GSK-3β) and Mediates Protein Kinase A-Dependent Inhibition of GSK-3β. J. Biol. Chem. 2002, 277, 36955–36961. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Wang, X.; Meintzer, M.K.; Laessig, T.; Birnbaum, M.J.; Heidenreich, K.A. Cyclic AMP Promotes Neuronal Survival by Phosphorylation of Glycogen Synthase Kinase 3beta. Mol. Cell. Biol. 2000, 20, 9356–9363. [Google Scholar] [CrossRef] [Green Version]
- Duda, P.; Wiśniewski, J.; Wójtowicz, T.; Wójcicka, O.; Jaśkiewicz, M.; Drulis-Fajdasz, D.; Rakus, D.; McCubrey, J.A.; Gizak, A. Targeting GSK3 Signaling as a Potential Therapy of Neurodegenerative Diseases and Aging. Expert Opin. Ther. Targets 2018, 22, 833–848. [Google Scholar] [CrossRef]
- Salcedo-Tello, P.; Ortiz-Matamoros, A.; Arias, C. GSK3 Function in the Brain during Development, Neuronal Plasticity, and Neurodegeneration. J. Alzheimer’s Dis. 2011, 2011, 189728. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, M.; Tomizawa, K.; Ishiguro, K. Distribution of Tau Protein Kinase I/Glycogen Synthase Kinase-3β, Phosphatases 2A and 2B, and Phosphorylated Tau in the Developing Rat Brain. Brain Res. 2000, 857, 193–206. [Google Scholar] [CrossRef]
- Takahashi, M.; Tomizawa, K.; Kato, R.; Sato, K.; Uchida, T.; Fujita, S.C.; Imahori, K. Localization and Developmental Changes of τ Protein Kinase I/Glycogen Synthase Kinase-3β in Rat Brain. J. Neurochem. 1994, 63, 245–255. [Google Scholar] [CrossRef]
- Mukai, F.; Ishiguro, K.; Sano, Y.; Fujita, S.C. Alternative Splicing Isoform of Tau Protein Kinase I/Glycogen Synthase Kinase 3β. J. Neurochem. 2002, 81, 1073–1083. [Google Scholar] [CrossRef]
- Trivedi, N.; Marsh, P.; Goold, R.G.; Wood-Kaczmar, A.; Gordon-Weeks, P.R. Glycogen Synthase Kinase-3β Phosphorylation of MAP1B at Ser1260 and Thr1265 Is Spatially Restricted to Growing Axons. J. Cell. Sci. 2005, 118, 993–1005. [Google Scholar] [CrossRef] [Green Version]
- Goold, R.G.; Gordon-Weeks, P.R. Glycogen Synthase Kinase 3β and the Regulation of Axon Growth. Biochem. Soc. Trans. 2004, 32, 809–811. [Google Scholar] [CrossRef]
- Castaño, Z.; Gordon-Weeks, P.R.; Kypta, R.M. The Neuron-Specific Isoform of Glycogen Synthase Kinase-3β Is Required for Axon Growth. J. Neurochem. 2010, 113, 117–130. [Google Scholar] [CrossRef]
- Soutar, M.P.M.; Kim, W.Y.; Williamson, R.; Peggie, M.; Hastie, C.J.; McLauchlan, H.; Snider, W.D.; Gordon-Weeks, P.R.; Sutherland, C. Evidence That Glycogen Synthase Kinase-3 Isoforms Have Distinct Substrate Preference in the Brain. J. Neurochem. 2010, 115, 974–983. [Google Scholar] [CrossRef]
- Emamian, E. AKT/GSK3 Signaling Pathway and Schizophrenia. Front. Mol. Neurosci. 2012, 5, 33. [Google Scholar] [CrossRef] [Green Version]
- Denny, C.A.; Heinecke, K.A.; Kim, Y.P.; Baek, R.C.; Loh, K.S.; Butters, T.D.; Bronson, R.T.; Platt, F.M.; Seyfried, T.N. Restricted Ketogenic Diet Enhances the Therapeutic Action of N-Butyldeoxynojirimycin towards Brain GM2 Accumulation in Adult Sandhoff Disease Mice. J. Neurochem. 2010, 113, 1525–1535. [Google Scholar] [CrossRef]
- Hashizume, T.; Kanematsu, S. Effects of Prostaglandins E2, F2α, and D2 on the Release of Growth Hormone, Prolactin, and Luteinizing Hormone from Cultured Bovine Anterior Pituitary Cells. J. Reprod. Dev. 1995, 41, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, J.M.; Sotnikova, T.D.; Gainetdinov, R.R.; Caron, M.G. Paradoxical Striatal Cellular Signaling Responses to Psychostimulants in Hyperactive Mice. J. Biol. Chem. 2006, 281, 32072–32080. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, J.M.; Gainetdinov, R.R.; Caron, M.G. Akt/GSK3 Signaling in the Action of Psychotropic Drugs. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 327–347. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, J.M.; Tirotta, E.; Sotnikova, T.D.; Masri, B.; Salahpour, A.; Gainetdinov, R.R.; Borrelli, E.; Caron, M.G. Regulation of Akt Signaling by D2 and D3 Dopamine Receptors In Vivo. J. Neurosci 2007, 27, 881–885. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.C.; Xi, D.; Roman, J.; Huang, Y.Q.; Gao, W.J. Activation of Glycogen Synthase Kinase-3β Is Required for Hyperdopamine and D2 Receptor-Mediated Inhibition of Synaptic NMDA Receptor Function in the Rat Prefrontal Cortex. J. Neurosci 2009, 29, 15551. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.R.; Sun, P.H.; Ren, Z.X.; Meltzer, H.Y.; Zhen, X.C. GSK-3β Interacts with Dopamine D1 Receptor to Regulate Receptor Function: Implication for Prefrontal Cortical D1 Receptor Dysfunction in Schizophrenia. CNS Neurosci. Ther. 2017, 23, 174–187. [Google Scholar] [CrossRef]
- Li, X.; Zhu, W.; Roh, M.S.; Friedman, A.B.; Rosborough, K.; Jope, R.S. In Vivo Regulation of Glycogen Synthase Kinase-3beta (GSK3beta) by Serotonergic Activity in Mouse Brain. Neuropsychopharmacology 2004, 29, 1426–1431. [Google Scholar] [CrossRef] [Green Version]
- Polter, A.M.; Li, X. Glycogen Synthase Kinase-3 Is an Intermediate Modulator of Serotonin Neurotransmission. Front. Mol. Neurosci 2011, 4, 31. [Google Scholar] [CrossRef] [Green Version]
- Lu, F.F.; Su, P.; Liu, F.; Daskalakis, Z.J. Activation of GABAB Receptors Inhibits Protein Kinase B/Glycogen Synthase Kinase 3 Signaling. Mol. Brain 2012, 5, 41. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.A.; Marshall, J.F. Molecular Substrates for Retrieval and Reconsolidation of Cocaine-Associated Contextual Memory. Neuron 2005, 47, 873–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiflett, M.W.; Mauna, J.C.; Chipman, A.M.; Peet, E.; Thiels, E. Appetitive Pavlovian Conditioned Stimuli Increase CREB Phosphorylation in the Nucleus Accumbens. Neurobiol. Learn. Mem. 2009, 92, 451. [Google Scholar] [CrossRef] [Green Version]
- Hur, E.M.; Zhou, F.Q. GSK3 Signalling in Neural Development. Nat. Rev. Neurosci. 2010, 11, 539–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandesquille, M.; Baudonnat, M.; Decorte, L.; Louis, C.; Lestage, P.; Béracochéa, D. Working Memory Deficits and Related Disinhibition of the CAMP/PKA/CREB Are Alleviated by Prefrontal A4β2*-NAChRs Stimulation in Aged Mice. Neurobiol. Aging 2013, 34, 1599–1609. [Google Scholar] [CrossRef]
- Parsons, R.G.; Gafford, G.M.; Helmstetter, F.J. Translational Control via the Mammalian Target of Rapamycin Pathway Is Critical for the Formation and Stability of Long-Term Fear Memory in Amygdala Neurons. J. Neurosci. 2006, 26, 12977–12983. [Google Scholar] [CrossRef] [PubMed]
- Stoica, L.; Zhu, P.J.; Huang, W.; Zhou, H.; Kozma, S.C.; Costa-Mattioli, M. Selective Pharmacogenetic Inhibition of Mammalian Target of Rapamycin Complex I (MTORC1) Blocks Long-Term Synaptic Plasticity and Memory Storage. Proc. Natl. Acad. Sci. USA 2011, 108, 3791–3796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, N.; Sonenberg, N. Upstream and Downstream of MTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.M.; Blenis, J. Molecular Mechanisms of MTOR-Mediated Translational Control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318. [Google Scholar] [CrossRef]
- Wu, D.; Pan, W. GSK3: A Multifaceted Kinase in Wnt Signaling. Trends Biochem. Sci. 2010, 35, 161. [Google Scholar] [CrossRef] [Green Version]
- Doble, B.W.; Patel, S.; Wood, G.A.; Kockeritz, L.K.; Woodgett, J.R. Functional Redundancy of GSK-3alpha and GSK-3beta in Wnt/Beta-Catenin Signaling Shown by Using an Allelic Series of Embryonic Stem Cell Lines. Dev. Cell 2007, 12, 957–971. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Rubin, J.S.; Kimmel, A.R. Rapid, Wnt-Induced Changes in GSK3beta Associations That Regulate Beta-Catenin Stabilization Are Mediated by Galpha Proteins. Curr. Biol. 2005, 15, 1989–1997. [Google Scholar] [CrossRef] [Green Version]
- Mao, Y.; Ge, X.; Frank, C.L.; Madison, J.M.; Koehler, A.N.; Doud, M.K.; Tassa, C.; Berry, E.M.; Soda, T.; Singh, K.K.; et al. Disrupted in Schizophrenia 1 Regulates Neuronal Progenitor Proliferation via Modulation of GSK3β/β-Catenin Signaling. Cell 2009, 136, 1017–1031. [Google Scholar] [CrossRef] [Green Version]
- Young, C.S.; Kitamura, M.; Hardy, S.; Kitajewski, J. Wnt-1 Induces Growth, Cytosolic Beta-Catenin, and Tcf/Lef Transcriptional Activation in Rat-1 Fibroblasts. Mol. Cell. Biol. 1998, 18, 2474–2485. [Google Scholar] [CrossRef] [Green Version]
- Orford, K.; Crockett, C.; Jensen, J.P.; Weissman, A.M.; Byers, S.W. Serine Phosphorylation-Regulated Ubiquitination and Degradation of Beta-Catenin. J. Biol. Chem. 1997, 272, 24735–24738. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.R.; Moon, R.T. Signal Transduction through Beta-Catenin and Specification of Cell Fate during Embryogenesis. Genes Dev. 1996, 10, 2527–2539. [Google Scholar] [CrossRef] [Green Version]
- Im, H.I.; Kenny, P.J. MicroRNAs in Neuronal Function and Dysfunction. Trends Neurosci. 2012, 35, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Ballou, L.M.; Tian, P.Y.; Lin, H.Y.; Jiang, Y.P.; Lin, R.Z. Dual Regulation of Glycogen Synthase Kinase-3beta by the Alpha1A-Adrenergic Receptor. J. Biol. Chem. 2001, 276, 40910–40916. [Google Scholar] [CrossRef] [Green Version]
- Xing, B.; Li, Y.C.; Gao, W.J. Norepinephrine versus Dopamine and Their Interaction in Modulating Synaptic Function in the Prefrontal Cortex. Brain Res. 2016, 1641, 217. [Google Scholar] [CrossRef] [Green Version]
- Morioka, N.; Kodama, K.; Tsuruta, M.; Hashizume, H.; Kochi, T.; Nakamura, Y.; Zhang, F.F.; Hisaoka-Nakashima, K. Stimulation of Nuclear Receptor REV-ERBs Suppresses Inflammatory Responses in Spinal Microglia. Neurochem. Int. 2021, 151, 105216. [Google Scholar] [CrossRef]
- Luo, H.R.; Hattori, H.; Hossain, M.A.; Hester, L.; Huang, Y.; Lee-Kwon, W.; Donowitz, M.; Nagata, E.; Snyder, S.H. Akt as a Mediator of Cell Death. Proc. Natl. Acad. Sci. USA 2003, 100, 11712–11717. [Google Scholar] [CrossRef] [Green Version]
- Szatmari, E.; Habas, A.; Yang, P.; Zheng, J.J.; Hagg, T.; Hetman, M. A Positive Feedback Loop between Glycogen Synthase Kinase 3beta and Protein Phosphatase 1 after Stimulation of NR2B NMDA Receptors in Forebrain Neurons. J. Biol. Chem. 2005, 280, 37526–37535. [Google Scholar] [CrossRef] [Green Version]
- Chen, P.; Gu, Z.; Liu, W.; Yan, Z. Glycogen Synthase Kinase 3 Regulates N-Methyl-D-Aspartate Receptor Channel Trafficking and Function in Cortical Neurons. Mol. Pharmacol. 2007, 72, 40–51. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Xiong, Z.; Chen, P.; Wei, J.; Chen, S.; Yan, Z. β-Amyloid Impairs the Regulation of N-Methyl-D-Aspartate Receptors by Glycogen Synthase Kinase 3. Neurobiol. Aging 2014, 35, 449–459. [Google Scholar] [CrossRef]
- Li, L.; Stefan, M.I.; le Novère, N. Calcium Input Frequency, Duration and Amplitude Differentially Modulate the Relative Activation of Calcineurin and CaMKII. PLoS ONE 2012, 7, e43810. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Lai, B.; Zheng, Z.; Zhang, Y.; Luo, J.; Wang, C.; Chen, Y.; Woodgett, J.R.; Li, M. Inhibitory Phosphorylation of GSK-3 by CaMKII Couples Depolarization to Neuronal Survival. J. Biol. Chem. 2010, 285, 41122–41134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Lee, Y.; Seo, M.; Kim, S.Y.; Lee, J.E.; Youn, H.D.; Kim, Y.S.; Juhnn, Y.S. Calcineurin Dephosphorylates Glycogen Synthase Kinase-3 Beta at Serine-9 in Neuroblast-Derived Cells. J. Neurochem. 2009, 111, 344–354. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.J.; Zukin, R.S. Ca2+-Permeable AMPA Receptors in Synaptic Plasticity and Neuronal Death. Trends Neurosci. 2007, 30, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Bats, C.; Groc, L.; Choquet, D. The Interaction between Stargazin and PSD-95 Regulates AMPA Receptor Surface Trafficking. Neuron 2007, 53, 719–734. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Liu, W.; Yan, Z. Regulation of AMPA Receptor Trafficking and Function by Glycogen Synthase Kinase 3. J. Biol. Chem. 2010, 285, 26369–26376. [Google Scholar] [CrossRef] [Green Version]
- Doble, B.W.; Woodgett, J.R. GSK-3: Tricks of the Trade for a Multi-Tasking Kinase. J. Cell Sci. 2003, 116, 1175–1186. [Google Scholar] [CrossRef] [Green Version]
- Duda, P.; Hajka, D.; Wójcicka, O.; Rakus, D.; Gizak, A. GSK3β: A Master Player in Depressive Disorder Pathogenesis and Treatment Responsiveness. Cells 2020, 9, 727. [Google Scholar] [CrossRef] [Green Version]
- Balsi, G.; Napolitano, F.; Ursini, G.; Di Giorgio, A.; Caforio, G.; Taurisano, P.; Fazio, L.; Gelao, B.; Attorotto, M.T.; Colagiorgio, L.; et al. Association of GSK-3β Genetic Variation with GSK-3β Expression, Prefrontal Cortical Thickness, Prefrontal Physiology, and Schizophrenia. Am. J. Psychiatry 2013, 170, 868–876. [Google Scholar] [CrossRef]
- Khan, I.; Tantray, M.A.; Alam, M.A.S.; Hamid, H. Natural and Synthetic Bioactive Inhibitors of Glycogen Synthase Kinase. Eur. J. Med. Chem. 2017, 125, 464–477. [Google Scholar] [CrossRef]
- Olds, J.; Milner, P. Positive Reinforcement Produced by Electrical Stimulation of Septal Area and Other Regions of Rat Brain. J. Comp. Physiol. Psychol. 1954, 47, 419–427. [Google Scholar] [CrossRef]
- Ferenczi, E.A.; Zalocusky, K.A.; Liston, C.; Grosenick, L.; Warden, M.R.; Amatya, D.; Katovich, K.; Mehta, H.; Patenaude, B.; Ramakrishnan, C.; et al. Prefrontal Cortical Regulation of Brainwide Circuit Dynamics and Reward-Related Behavior. Science 2016, 351, 6268. [Google Scholar] [CrossRef] [Green Version]
- Drugs, Brains, and Behavior: The Science of Addiction: Preface|NIDA. Available online: https://www.drugabuse.gov/publications/drugs-brains-behavior-science-addiction/preface (accessed on 13 November 2021).
- Robbins, T.W.; Everitt, B.J. Limbic-Striatal Memory Systems and Drug Addiction. Neurobiol. Learn. Mem. 2002, 78, 625–636. [Google Scholar] [CrossRef]
- Mitchell, J.M.; O’Neil, J.P.; Janabi, M.; Marks, S.M.; Jagust, W.J.; Fields, H.L. Alcohol Consumption Induces Endogenous Opioid Release in the Human Orbitofrontal Cortex and Nucleus Accumbens. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef] [Green Version]
- Wise, R.A. Roles for Nigrostriatal--Not Just Mesocorticolimbic—Dopamine in Reward and Addiction. Trends Neurosci. 2009, 32, 517–524. [Google Scholar] [CrossRef] [Green Version]
- Volkow, N.D.; Wang, G.J.; Telang, F.; Fowler, J.S.; Logan, J.; Jayne, M.; Ma, Y.; Pradhan, K.; Wong, C. Profound Decreases in Dopamine Release in Striatum in Detoxified Alcoholics: Possible Orbitofrontal Involvement. J. Neurosci. 2007, 27, 12700–12706. [Google Scholar] [CrossRef] [Green Version]
- Wise, R.A.; Bozarth, M.A. Brain Reward Circuitry: Four Circuit Elements “Wired” in Apparent Series. Brain Res. Bulletin 1984, 12, 203–208. [Google Scholar] [CrossRef]
- Gallistel, C.R.; Shizgal, P.; Yeomans, J.S. A Portrait of the Substrate for Self-Stimulation. Psychol. Rev. 1981, 88, 228–273. [Google Scholar] [CrossRef]
- Gardner, E.L. Addiction and Brain Reward and Antireward Pathways. Adv. Psychosom. Med. 2011, 30, 22–60. [Google Scholar] [CrossRef] [Green Version]
- Alheid, G.F.; Heimer, L. New Perspectives in Basal Forebrain Organization of Special Relevance for Neuropsychiatric Disorders: The Striatopallidal, Amygdaloid, and Corticopetal Components of Substantia Innominata. Neuroscience 1988, 27, 1–39. [Google Scholar] [CrossRef]
- Wise, R.A. Addictive Drugs and Brain Stimulation Reward. Annu. Rev. Neurosci. 1996, 19, 319–340. [Google Scholar] [CrossRef]
- McBride, W.J.; Murphy, J.M.; Ikemoto, S. Localization of Brain Reinforcement Mechanisms: Intracranial Self-Administration and Intracranial Place-Conditioning Studies. Behav. Brain Res. 1999, 101, 129–152. [Google Scholar] [CrossRef]
- Nauta, W.J.H.; Mehler, W.R. Projections of the Lentiform Nucleus in the Monkey. Brain Res. 1966, 1, 3–42. [Google Scholar] [CrossRef]
- Lammel, S.; Lim, B.K.; Malenka, R.C. Reward and Aversion in a Heterogeneous Midbrain Dopamine System. Neuropharmacology 2014, 76 Pt B, 351–359. [Google Scholar] [CrossRef] [Green Version]
- Lammel, S.; Hetzel, A.; Häckel, O.; Jones, I.; Liss, B.; Roeper, J. Unique Properties of Mesoprefrontal Neurons within a Dual Mesocorticolimbic Dopamine System. Neuron 2008, 57, 760–773. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, E.E.; Boivin, J.R.; Saunders, B.T.; Witten, I.B.; Deisseroth, K.; Janak, P.H. Positive Reinforcement Mediated by Midbrain Dopamine Neurons Requires D1 and D2 Receptor Activation in the Nucleus Accumbens. PLoS ONE 2014, 9, e94771. [Google Scholar] [CrossRef] [Green Version]
- Phelps, E.A. Human Emotion and Memory: Interactions of the Amygdala and Hippocampal Complex. Curr. Opin. Neurobiol. 2004, 14, 198–202. [Google Scholar] [CrossRef]
- Stephan, K.E.; Bach, D.R.; Fletcher, P.C.; Flint, J.; Frank, M.J.; Friston, K.J.; Heinz, A.; Huys, Q.J.M.; Owen, M.J.; Binder, E.B.; et al. Charting the Landscape of Priority Problems in Psychiatry, Part 1: Classification and Diagnosis. Lancet Psychiatry 2016, 3, 77–83. [Google Scholar] [CrossRef] [Green Version]
- Gardner, E.L. What We Have Learned about Addiction from Animal Models of Drug Self-Administration. Am. J. Addict. 2000, 9, 285–313. [Google Scholar] [CrossRef] [PubMed]
- Anagnostaras, S.G.; Robinson, T.E. Sensitization to the Psychomotor Stimulant Effects of Amphetamine: Modulation by Associative Learning. Behav. Neurosci. 1996, 110, 1397–1414. [Google Scholar] [CrossRef] [PubMed]
- Badiani, A.; Anagnostaras, S.G.; Robinson, T.E. The Development of Sensitization to the Psychomotor Stimulant Effects of Amphetamine Is Enhanced in a Novel Environment. Psychopharmacology 1995, 117, 443–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nestler, E.J. Under Siege: The Brain on Opiates. Neuron 1996, 16, 897–900. [Google Scholar] [CrossRef] [Green Version]
- Hope, B.T.; Nye, H.E.; Kelz, M.B.; Self, D.W.; Iadarola, M.J.; Nakabeppu, Y.; Duman, R.S.; Nestler, E.J. Induction of a Long-Lasting AP-1 Complex Composed of Altered Fos-like Proteins in Brain by Chronic Cocaine and Other Chronic Treatments. Neuron 1994, 13, 1235–1244. [Google Scholar] [CrossRef]
- Koob, G.F.; le Moal, M. Plasticity of Reward Neurocircuitry and the “dark Side” of Drug Addiction. Nat. Neurosci. 2005, 8, 1442–1444. [Google Scholar] [CrossRef]
- Gerfen, C.R.; Engber, T.M.; Mahan, L.C.; Susel, Z.; Chase, T.N.; Monsma, F.J.; Sibley, D.R. D1 and D2 Dopamine Receptor-Regulated Gene Expression of Striatonigral and Striatopallidal Neurons. Science 1990, 250, 1429–1432. [Google Scholar] [CrossRef]
- Lobo, M.K.; Covington, H.E.; Chaudhury, D.; Friedman, A.K.; Sun, H.S.; Damez-Werno, D.; Dietz, D.M.; Zaman, S.; Koo, J.W.; Kennedy, P.J.; et al. Cell Type-Specific Loss of BDNF Signaling Mimics Optogenetic Control of Cocaine Reward. Science 2010, 330, 385–390. [Google Scholar] [CrossRef] [Green Version]
- Volkow, N.D.; Fowler, J.S.; Wang, G.-J.; Hitzemann, R.; Logan, J.; Schlyer, D.J.; Dewey, S.L.; Wolf, A.P. Decreased Dopamine D2 Receptor Availability Is Associated with Reduced Frontal Metabolism in Cocaine Abusers. Synapse 1993, 14, 169–177. [Google Scholar] [CrossRef]
- Davey, C.G.; Yücel, M.; Allen, N.B. The Emergence of Depression in Adolescence: Development of the Prefrontal Cortex and the Representation of Reward. Neurosci. Biobehav. Rev. 2008, 32, 1–19. [Google Scholar] [CrossRef]
- Schultz, W.; Dayan, P.; Montague, P.R. A Neural Substrate of Prediction and Reward. Science 1997, 275, 1593–1599. [Google Scholar] [CrossRef] [Green Version]
- Waelti, P.; Dickinson, A.; Schultz, W. Dopamine Responses Comply with Basic Assumptions of Formal Learning Theory. Nature 2001, 412, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Garbusow, M.; Schad, D.J.; Sebold, M.; Friedel, E.; Bernhardt, N.; Koch, S.P.; Steinacher, B.; Kathmann, N.; Geurts, D.E.M.; Sommer, C.; et al. Pavlovian-to-Instrumental Transfer Effects in the Nucleus Accumbens Relate to Relapse in Alcohol Dependence. Addict. Biol. 2016, 21, 719–731. [Google Scholar] [CrossRef]
- LeBlanc, K.H.; Ostlund, S.B.; Maidment, N.T. Pavlovian-to-Instrumental Transfer in Cocaine Seeking Rats. Behav. Neurosci. 2012, 126, 681–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nestler, E.J.; Barrot, M.; Self, D.W. DeltaFosB: A Sustained Molecular Switch for Addiction. Proc. Natl. Acad. Sci. USA 2001, 98, 11042–11046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyman, S.E.; Malenka, R.C. Addiction and the Brain: The Neurobiology of Compulsion and Its Persistence. Nat. Rev. Neurosci. 2001, 2, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Werme, M.; Messer, C.; Olson, L.; Gilden, L.; Thorén, P.; Nestler, E.J.; Brené, S. Delta FosB Regulates Wheel Running. J. Neurosci. 2002, 22, 8133–8138. [Google Scholar] [CrossRef] [Green Version]
- Kelz, M.B.; Chen, J.; Carlezon, W.A.; Whisler, K.; Gilden, L.; Beckmann, A.M.; Steffen, C.; Zhang, Y.J.; Marotti, L.; Self, D.W.; et al. Expression of the Transcription Factor DeltaFosB in the Brain Controls Sensitivity to Cocaine. Nature 1999, 401, 272–276. [Google Scholar] [CrossRef]
- Teegarden, S.L.; Nestler, E.J.; Bale, T.L. Delta FosB-Mediated Alterations in Dopamine Signaling Are Normalized by a Palatable High-Fat Diet. Biol. Psychiatry 2008, 64, 941–950. [Google Scholar] [CrossRef] [Green Version]
- Teegarden, S.L.; Bale, T.L. Decreases in Dietary Preference Produce Increased Emotionality and Risk for Dietary Relapse. Biol. Psychiatry 2007, 61, 1021–1029. [Google Scholar] [CrossRef]
- Dobrazanski, P.; Noguchi, T.; Kovary, K.; Rizzo, C.A.; Lazo, P.S.; Bravo, R. Both Products of the FosB Gene, FosB and Its Short Form, FosB/SF, Are Transcriptional Activators in Fibroblasts. Mol. Cell. Biol. 1991, 11, 5470–5478. [Google Scholar] [CrossRef]
- White, F.J.; Hu, X.T.; Zhang, X.F.; Wolf, M.E. Repeated Administration of Cocaine or Amphetamine Alters Neuronal Responses to Glutamate in the Mesoaccumbens Dopamine System. J. Pharmacol. Exp. Ther. 1995, 273, 445–454. [Google Scholar]
- Carlezon, W.A.; Thome, J.; Olson, V.G.; Lane-Ladd, S.B.; Brodkin, E.S.; Hiroi, N.; Duman, R.S.; Neve, R.L.; Nestler, E.J. Regulation of Cocaine Reward by CREB. Science 1998, 282, 2272–2275. [Google Scholar] [CrossRef] [Green Version]
- Todtenkopf, M.S.; Marcus, J.F.; Portoghese, P.S.; Carlezon, W.A. Effects of Kappa-Opioid Receptor Ligands on Intracranial Self-Stimulation in Rats. Psychopharmacology 2004, 172, 463–470. [Google Scholar] [CrossRef]
- Svensson, P.; Hurd, Y.L. Specific Reductions of Striatal Prodynorphin and D1 Dopamine Receptor Messenger RNAs during Cocaine Abstinence. Brain Res. Mol. Brain Res. 1998, 56, 162–168. [Google Scholar] [CrossRef]
- Uhl, G.R. Molecular Genetics of Substance Abuse Vulnerability: Remarkable Recent Convergence of Genome Scan Results. Ann. N. Y. Acad. Sci. 2004, 1025, 1–13. [Google Scholar] [CrossRef]
- Uhl, G.R.; Drgon, T.; Johnson, C.; Fatusin, O.O.; Liu, Q.R.; Contoreggi, C.; Li, C.Y.; Buck, K.; Crabbe, J. “Higher Order” Addiction Molecular Genetics: Convergent Data from Genome-Wide Association in Humans and Mice. Biochem. Pharmacol. 2008, 75, 98–111. [Google Scholar] [CrossRef] [Green Version]
- Rennels, M.L.; Gregory, T.F.; Blaumanis, O.R.; Fujimoto, K.; Grady, P.A. Evidence for a ‘Paravascular’ Fluid Circulation in the Mammalian Central Nervous System, Provided by the Rapid Distribution of Tracer Protein throughout the Brain from the Subarachnoid Space. Brain Res. 1985, 326, 47–63. [Google Scholar] [CrossRef]
- Blum, K.; Braverman, E.R.; Holder, J.M.; Lubar, J.F.; Monastra, V.I.; Miller, D.; Lubar, J.O.; Chen, T.J.H.; Comings, D.E. Reward Deficiency Syndrome: A Biogenetic Model for the Diagnosis and Treatment of Impulsive, Addictive, and Compulsive Behaviors. J. Psychoact. Drugs 2000, 32 (Suppl. 1), 1–112. [Google Scholar] [CrossRef]
- Comings, D.E.; Blum, K. Reward Deficiency Syndrome: Genetic Aspects of Behavioral Disorders. Prog. Brain Res. 2000, 126, 325–341. [Google Scholar] [CrossRef]
- Blum, K.; Sheridan, P.J.; Wood, R.C.; Braverman, E.R.; Chen, T.J.H.; Cull, J.G.; Comings, D.E. The D2 Dopamine Receptor Gene as a Determinant of Reward Deficiency Syndrome. J. R. Soc. Med. 1996, 89, 396. [Google Scholar] [CrossRef] [Green Version]
- Leroy, K.; Brion, J.P. Developmental Expression and Localization of Glycogen Synthase Kinase-3beta in Rat Brain. J. Chem. Neuroanat. 1999, 16, 279–293. [Google Scholar] [CrossRef]
- Latapy, C.; Rioux, V.; Guitton, M.J.; Beaulieu, J.M. Selective Deletion of Forebrain Glycogen Synthase Kinase 3β Reveals a Central Role in Serotonin-Sensitive Anxiety and Social Behaviour. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 2460–2474. [Google Scholar] [CrossRef] [Green Version]
- Pandey, G.N.; Dwivedi, Y.; Rizavi, H.S.; Teppen, T.; Gaszner, G.L.; Roberts, R.C.; Conley, R.R. GSK-3beta Gene Expression in Human Postmortem Brain: Regional Distribution, Effects of Age and Suicide. Neurochem. Res. 2009, 34, 274–285. [Google Scholar] [CrossRef]
- Wilkinson, M.B.; Dias, C.; Magida, J.; Mazei-Robison, M.; Lobo, M.; Kennedy, P.; Dietz, D.; Covington, H.; Russo, S.; Neve, R.; et al. A Novel Role of the WNT-Dishevelled-GSK3β Signaling Cascade in the Mouse Nucleus Accumbens in a Social Defeat Model of Depression. J. Neurosci. 2011, 31, 9084–9092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucas, J.J.; Hernández, F.; Gómez-Ramos, P.; Morán, M.A.; Hen, R.; Avila, J. Decreased Nuclear Beta-Catenin, Tau Hyperphosphorylation and Neurodegeneration in GSK-3beta Conditional Transgenic Mice. EMBO J. 2001, 20, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Spittaels, K.; van den Haute, C.; van Dorpe, J.; Terwel, D.; Vandezande, K.; Lasrado, R.; Bruynseels, K.; Irizarry, M.; Verhoye, M.; van Lint, J.; et al. Neonatal Neuronal Overexpression of Glycogen Synthase Kinase-3 Beta Reduces Brain Size in Transgenic Mice. Neuroscience 2002, 113, 797–808. [Google Scholar] [CrossRef]
- Tan, M.; Ma, S.; Huang, Q.; Hu, K.; Song, B.; Li, M. GSK-3α/β-Mediated Phosphorylation of CRMP-2 Regulates Activity-Dependent Dendritic Growth. J. Neurochem. 2013, 125, 685–697. [Google Scholar] [CrossRef]
- Rippin, I.; Eldar-Finkelman, H. Mechanisms and Therapeutic Implications of GSK-3 in Treating Neurodegeneration. Cells 2021, 10, 262. [Google Scholar] [CrossRef]
- Yoshimura, T.; Kawano, Y.; Arimura, N.; Kawabata, S.; Kikuchi, A.; Kaibuchi, K. GSK-3beta Regulates Phosphorylation of CRMP-2 and Neuronal Polarity. Cell 2005, 120, 137–149. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; Barr, J.L.; von Weltin, E.; Wolsh, C.; Unterwald, E.M. Differential Roles of Accumbal GSK3 β in Cocaine versus Morphine-Induced Place Preference, U50,488H-Induced Place Aversion, and Object Memory. J. Pharmacol. Exp. Ther. 2019, 371, 339–347. [Google Scholar] [CrossRef]
- Xu, C.M.; Wang, J.; Wu, P.; Zhu, W.L.; Li, Q.Q.; Xue, Y.X.; Zhai, H.F.; Shi, J.; Lu, L. Glycogen Synthase Kinase 3beta in the Nucleus Accumbens Core Mediates Cocaine-Induced Behavioral Sensitization. J. Neurochem. 2009, 111, 1357–1368. [Google Scholar] [CrossRef]
- Crofton, E.J.; Nenov, M.N.; Zhang, Y.; Scala, F.; Page, S.A.; McCue, D.L.; Li, D.; Hommel, J.D.; Laezza, F.; Green, T.A. Glycogen Synthase Kinase 3 Beta Alters Anxiety-, Depression-, and Addiction-Related Behaviors and Neuronal Activity in the Nucleus Accumbens Shell. Neuropharmacology 2017, 117, 49–60. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Wei, Y.; Liu, L.; Wang, Y.; Khairova, R.; Blumenthal, R.; Tragon, T.; Hunsberger, J.G.; Machado-Vieira, R.; Drevets, W.; et al. A Kinesin Signaling Complex Mediates the Ability of GSK-3beta to Affect Mood-Associated Behaviors. Proc. Natl. Acad. Sci. USA 2010, 107, 11573–11578. [Google Scholar] [CrossRef] [Green Version]
- Hooper, C.; Markevich, V.; Plattner, F.; Killick, R.; Schofield, E.; Engel, T.; Hernandez, F.; Anderton, B.; Rosenblum, K.; Bliss, T.; et al. Glycogen Synthase Kinase-3 Inhibition Is Integral to Long-Term Potentiation. Eur. J. Neurosci 2007, 25, 81–86. [Google Scholar] [CrossRef]
- Peineau, S.; Taghibiglou, C.; Bradley, C.; Wong, T.P.; Liu, L.; Lu, J.; Lo, E.; Wu, D.; Saule, E.; Bouschet, T.; et al. LTP Inhibits LTD in the Hippocampus via Regulation of GSK3beta. Neuron 2007, 53, 703–717. [Google Scholar] [CrossRef] [Green Version]
- Mulkey, R.M.; Endo, S.; Shenolikar, S.; Malenka, R.C. Involvement of a Calcineurin/Inhibitor-1 Phosphatase Cascade in Hippocampal Long-Term Depression. Nature 1994, 369, 486–488. [Google Scholar] [CrossRef]
- Zhu, L.Q.; Wang, S.H.; Liu, D.; Yin, Y.Y.; Tian, Q.; Wang, X.C.; Wang, Q.; Chen, J.G.; Wang, J.Z. Activation of Glycogen Synthase Kinase-3 Inhibits Long-Term Potentiation with Synapse-Associated Impairments. J. Neurosci. 2007, 27, 12211–12220. [Google Scholar] [CrossRef]
- Hernández, F.; Borrell, J.; Guaza, C.; Avila, J.; Lucas, J.J. Spatial Learning Deficit in Transgenic Mice That Conditionally Over-Express GSK-3beta in the Brain but Do Not Form Tau Filaments. J. Neurochem. 2002, 83, 1529–1533. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Yamashita, S.; Nakao, S.; Park, J.M.; Murayama, M.; Mizoroki, T.; Yoshiike, Y.; Sahara, N.; Takashima, A. GSK-3beta Is Required for Memory Reconsolidation in Adult Brain. PLoS ONE 2008, 3, e3540. [Google Scholar] [CrossRef] [Green Version]
- Jope, R.S.; Cheng, Y.; Lowell, J.A.; Worthen, R.J.; Sitbon, Y.H.; Beurel, E. Stressed and Inflamed, Can GSK3 Be Blamed? Trends Biochem. Sci. 2017, 42, 180–192. [Google Scholar] [CrossRef] [Green Version]
- Brami-Cherrier, K.; Valjent, E.; Garcia, M.; Pagès, C.; Hipskind, R.A.; Caboche, J. Dopamine Induces a PI3-Kinase-Independent Activation of Akt in Striatal Neurons: A New Route to CAMP Response Element-Binding Protein Phosphorylation. J. Neurosci. 2002, 22, 8911–8921. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, J.M.; Sotnikova, T.D.; Yao, W.D.; Kockeritz, L.; Woodgett, J.R.; Gainetdinov, R.R.; Caron, M.G. Lithium Antagonizes Dopamine-Dependent Behaviors Mediated by an AKT/Glycogen Synthase Kinase 3 Signaling Cascade. Proc. Natl. Acad. Sci. USA 2004, 101, 5099–5104. [Google Scholar] [CrossRef] [Green Version]
- Beaulieu, J.M.; Sotnikova, T.D.; Marion, S.; Lefkowitz, R.J.; Gainetdinov, R.R.; Caron, M.G. An Akt/Beta-Arrestin 2/PP2A Signaling Complex Mediates Dopaminergic Neurotransmission and Behavior. Cell 2005, 122, 261–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urs, N.M.; Daigle, T.L.; Caron, M.G. A Dopamine D1 Receptor-Dependent β-Arrestin Signaling Complex Potentially Regulates Morphine-Induced Psychomotor Activation but Not Reward in Mice. Neuropsychopharmacology 2011, 36, 551–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salles, M.J.; Hervé, D.; Rivet, J.M.; Longueville, S.; Millan, M.J.; Girault, J.A.; Cour, C.M.L. Transient and Rapid Activation of Akt/GSK-3β and MTORC1 Signaling by D3 Dopamine Receptor Stimulation in Dorsal Striatum and Nucleus Accumbens. J. Neurochem. 2013, 125, 532–544. [Google Scholar] [CrossRef] [PubMed]
- La Cour, C.M.; Salles, M.J.; Pasteau, V.; Millan, M.J. Signaling Pathways Leading to Phosphorylation of Akt and GSK-3β by Activation of Cloned Human and Rat Cerebral D₂and D₃ Receptors. Mol. Pharmacol. 2011, 79, 91–105. [Google Scholar] [CrossRef]
- Li, S.X.; Wei, Y.M.; Shi, H.S.; Luo, Y.X.; Ding, Z.B.; Xue, Y.X.; Lu, L.; Yu, C.X. Glycogen Synthase Kinase-3β in the Ventral Tegmental Area Mediates Diurnal Variations in Cocaine-Induced Conditioned Place Preference in Rats. Addict. Biol. 2014, 19, 996–1005. [Google Scholar] [CrossRef]
- Khlghatyan, J.; Khlghatyan, J.; Beaulieu, J.M. CRISPR-Cas9-Mediated Intersectional Knockout of Glycogen Synthase Kinase 3β in D2 Receptor-Expressing Medial Prefrontal Cortex Neurons Reveals Contributions to Emotional Regulation. CRISPR J. 2020, 3, 198–210. [Google Scholar] [CrossRef]
- Fatahi, Z.; Zeinaddini-Meymand, A.; Karimi-Haghighi, S.; Moradi, M.; Khodagholi, F.; Haghparast, A. Naloxone-Precipitated Withdrawal Ameliorates Impairment of Cost-Benefit Decision Making in Morphine-Treated Rats: Involvement of BDNF, p-GSK3-β, and p-CREB in the Amygdala. Neurobiol. Learn. Mem. 2020, 167, 107138. [Google Scholar] [CrossRef]
- White, N.M. Addictive Drugs as Reinforcers: Multiple Partial Actions on Memory Systems. Addiction 1996, 91, 921–950. [Google Scholar] [CrossRef]
- Packard, M.G.; Teather, L.A. Amygdala Modulation of Multiple Memory Systems: Hippocampus and Caudate- Putamen. Neurobiol. Learn. Mem. 1998, 69, 163–203. [Google Scholar] [CrossRef] [Green Version]
- Udo, T.; Ugalde, F.; DiPietro, N.; Eichenbaum, H.B.; Kantak, K.M. Effect of Persistent Cocain Self-Administration on Amygdala-Dependent and Dorsal Striatum-Dependent Learning in Rats. Psychopharmacology 2004, 174, 237–245. [Google Scholar] [CrossRef]
- Dickinson, A.; Wood, N.; Smith, J.W. Alcohol Seeking by Rats: Action or Habit? Q. J. Exp. Psychol 2002, 55, 331–348. [Google Scholar] [CrossRef]
- Wood, S.C.; Fay, J.; Sage, J.R.; Anagnostaras, S.G. Cocaine and Pavlovian Fear Conditioning: Dose-Effect Analysis. Behav. Brain Res. 2007, 176, 244–250. [Google Scholar] [CrossRef] [Green Version]
- Wood, S.C.; Anagnostaras, S.G. Memory and Psychostimulants: Modulation of Pavlovian Fear Conditioning by Amphetamine in C57BL/6 Mice. Psychopharmacology 2009, 202, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Iñiguez, S.D.; Charntikov, S.; Baella, S.A.; Herbert, M.S.; Bolaños-Guzmán, C.A.; Crawford, C.A. Post-Training Cocaine Exposure Facilitates Spatial Memory Consolidation in C57BL/6 Mice. Hippocampus 2012, 22, 802–813. [Google Scholar] [CrossRef] [Green Version]
- Leri, F.; Nahas, E.; Henderson, K.; Limebeer, C.L.; Parker, L.A.; White, N.M. Effects of Post-Training Heroin and d-Amphetamine on Consolidation of Win-Stay Learning and Fear Conditioning. J. Psychopharmacol. 2013, 27, 292–301. [Google Scholar] [CrossRef]
- DePoy, L.; Daut, R.; Brigman, J.L.; MacPherson, K.; Crowley, N.; Gunduz-Cinar, O.; Pickens, C.L.; Cinar, R.; Saksida, L.M.; Kunos, G.; et al. Chronic Alcohol Produces Neuroadaptations to Prime Dorsal Striatal Learning. Proc. Natl. Acad. Sci. USA 2013, 110, 14783–14788. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.E.; Tseng, K.Y. Calcium-Permeable AMPA Receptors in the VTA and Nucleus Accumbens after Cocaine Exposure: When, How, and Why? Front. Mol. Neurosci. 2012, 5, 72. [Google Scholar] [CrossRef] [Green Version]
- Lüscher, C.; Malenka, R.C. Drug-Evoked Synaptic Plasticity in Addiction: From Molecular Changes to Circuit Remodeling. Neuron 2011, 69, 650–663. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.E. The Bermuda Triangle of Cocaine-Induced Neuroadaptations. Trends Neurosci. 2010, 33, 391–398. [Google Scholar] [CrossRef] [Green Version]
- Gil, M.; Zhen, X.; Friedman, E. Prenatal Cocaine Exposure Alters Glycogen Synthase Kinase-3beta (GSK3beta) Pathway in Select Rabbit Brain Areas. Neurosci. Lett. 2003, 349, 143–146. [Google Scholar] [CrossRef]
- Svenningsson, P.; Tzavara, E.T.; Carruthers, R.; Rachleff, I.; Wattler, S.; Nehls, M.; McKinzie, D.L.; Fienberg, A.A.; Nomikos, G.G.; Greengard, P. Diverse Psychotomimetics Act through a Common Signaling Pathway. Science 2003, 302, 1412–1415. [Google Scholar] [CrossRef] [PubMed]
- Nwaneshiudu, C.A.; Unterwald, E.M. NK-3 Receptor Antagonism Prevents Behavioral Sensitization to Cocaine: A Role of Glycogen Synthase Kinase-3 in the Nucleus Accumbens. J. Neurochem. 2010, 115, 635–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, X.; McGinty, J.F. Repeated Amphetamine Treatment Increases Phosphorylation of Extracellular Signal-Regulated Kinase, Protein Kinase B, and Cyclase Response Element-Binding Protein in the Rat Striatum. J. Neurochem. 2007, 103, 706–713. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.S.; Tallarida, R.J.; Unterwald, E.M. Cocaine-Induced Hyperactivity and Sensitization Are Dependent on GSK3. Neuropharmacology 2009, 56, 1116–1123. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.S.; Barr, J.L.; Harper, L.J.; Poole, R.L.; Gould, T.J.; Unterwald, E.M. The GSK3 Signaling Pathway Is Activated by Cocaine and Is Critical for Cocaine Conditioned Reward in Mice. PLoS ONE 2014, 9, e88026. [Google Scholar] [CrossRef] [Green Version]
- Shi, X.; McGinty, J.F. D1 and D2 Dopamine Receptors Differentially Mediate the Activation of Phosphoproteins in the Striatum of Amphetamine-Sensitized Rats. Psychopharmacology 2011, 214, 653–663. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.M.; Wang, J.; Wu, P.; Xue, Y.X.; Zhu, W.L.; Li, Q.Q.; Zhai, H.F.; Shi, J.; Lu, L. Glycogen Synthase Kinase 3β in the Nucleus Accumbens Core Is Critical for Methamphetamine-Induced Behavioral Sensitization. J. Neurochem. 2011, 118, 126–139. [Google Scholar] [CrossRef]
- Xing, B.; Liang, X.P.; Liu, P.; Zhao, Y.; Chu, Z.; Dang, Y.H. Valproate Inhibits Methamphetamine Induced Hyperactivity via Glycogen Synthase Kinase 3β Signaling in the Nucleus Accumbens Core. PLoS ONE 2015, 10, e0128068. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Sun, L.L.; Zhu, W.L.; Sun, Y.; Liu, J.F.; Lu, L.; Shi, J. Role of Calcineurin in the VTA in Rats Behaviorally Sensitized to Methamphetamine. Psychopharmacology 2012, 220, 117–128. [Google Scholar] [CrossRef]
- Park, H.J.; Cui, F.J.; Hwang, J.Y.; Kang, U.G. Effects of Clozapine on Behavioral Sensitization Induced by Cocaine. Psychiatry Res. 2010, 175, 165–170. [Google Scholar] [CrossRef]
- Perrine, S.A.; Miller, J.S.; Unterwald, E.M. Cocaine Regulates Protein Kinase B and Glycogen Synthase Kinase-3 Activity in Selective Regions of Rat Brain. J. Neurochem. 2008, 107, 570–577. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Liu, L.; Zhang, R.; Li, J.; Leung, C.K.; Huang, J.; Li, Y.; Shen, B.; Zeng, X.; Zhang, D. Cannabidiol Attenuates Methamphetamine-Induced Conditioned Place Preference via the Sigma1R/AKT/GSK-3β/CREB Signaling Pathway in Rats. Toxicol. Res. 2020, 9, 202–211. [Google Scholar] [CrossRef]
- Kalivas, P.W.; Stewart, J. Dopamine Transmission in the Initiation and Expression of Drug- and Stress-Induced Sensitization of Motor Activity. Brain Res. Rev. 1991, 16, 223–244. [Google Scholar] [CrossRef]
- Hyman, S.E. Addiction to Cocaine and Amphetamine. Neuron 1996, 16, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Enman, N.M.; Unterwald, E.M. Inhibition of GSK3 Attenuates Amphetamine-Induced Hyperactivity and Sensitization in the Mouse. Behav. Brain Res. 2012, 231, 217–225. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.Y.; Jang, J.K.; Lee, J.W.; Jang, H.; Kim, J.H. Decrease of GSK3β Phosphorylation in the Rat Nucleus Accumbens Core Enhances Cocaine-Induced Hyper-Locomotor Activity. J. Neurochem. 2013, 125, 642–648. [Google Scholar] [CrossRef]
- Shi, X.; Miller, J.S.; Harper, L.J.; Poole, R.L.; Gould, T.J.; Unterwald, E.M. Reactivation of Cocaine Reward Memory Engages the Akt/GSK3/MTOR Signaling Pathway and Can Be Disrupted by GSK3 Inhibition. Psychopharmacology 2014, 231, 3109–3118. [Google Scholar] [CrossRef] [Green Version]
- Bowers, M.S.; Chen, B.T.; Chou, J.K.; Osborne, M.P.H.; Gass, J.T.; See, R.E.; Bonci, A.; Janak, P.H.; Olive, M.F. Acamprosate Attenuates Cocaine- and Cue-Induced Reinstatement of Cocaine-Seeking Behavior in Rats. Psychopharmacology 2007, 195, 397–406. [Google Scholar] [CrossRef]
- Liddie, S.; Itzhak, Y. Variations in the Stimulus Salience of Cocaine Reward Influences Drug-Associated Contextual Memory. Addict. Biol. 2016, 21, 242–254. [Google Scholar] [CrossRef] [Green Version]
- Alaghband, Y.; Marshall, J.F. Common Influences of Non-Competitive NMDA Receptor Antagonists on the Consolidation and Reconsolidation of Cocaine-Cue Memory. Psychopharmacology 2013, 226, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Itzhak, Y. Role of the NMDA Receptor and Nitric Oxide in Memory Reconsolidation of Cocaine-Induced Conditioned Place Preference in Mice. Ann. N. Y. Acad. Sci. 2008, 1139, 350–357. [Google Scholar] [CrossRef]
- Shi, X.; von Weltin, E.; Barr, J.L.; Unterwald, E.M. Activation of GSK3β Induced by Recall of Cocaine Reward Memories Is Dependent on GluN2A/B NMDA Receptor Signaling. J. Neurochem. 2019, 151, 91–102. [Google Scholar] [CrossRef]
- Prickaerts, J.; Moechars, D.; Cryns, K.; Lenaerts, I.; van Craenendonck, H.; Goris, I.; Daneels, G.; Bouwknecht, J.A.; Steckler, T. Transgenic Mice Overexpressing Glycogen Synthase Kinase 3beta: A Putative Model of Hyperactivity and Mania. J. Neurosci. 2006, 26, 9022–9029. [Google Scholar] [CrossRef]
- German, P.W.; Fields, H.L. Rat Nucleus Accumbens Neurons Persistently Encode Locations Associated with Morphine Reward. J. Neuro-Physiol. 2007, 97, 2094–2106. [Google Scholar] [CrossRef]
- Mazei-Robison, M.S.; Appasani, R.; Edwards, S.; Wee, S.; Taylor, S.R.; Picciotto, M.R.; Koob, G.F.; Nestler, E.J. Self-Administration of Ethanol, Cocaine, or Nicotine Does Not Decrease the Soma Size of Ventral Tegmental Area Dopamine Neurons. PLoS ONE 2014, 9, e95962. [Google Scholar] [CrossRef] [Green Version]
- Neasta, J.; ben Hamida, S.; Yowell, Q.V.; Carnicella, S.; Ron, D. AKT Signaling Pathway in the Nucleus Accumbens Mediates Excessive Alcohol Drinking Behaviors. Biol. Psychiatry 2011, 70, 575–582. [Google Scholar] [CrossRef] [Green Version]
- Qiao, X.; Gai, H.; Su, R.; Deji, C.; Cui, J.; Lai, J.; Zhu, Y. PI3K-AKT-GSK3β-CREB Signaling Pathway Regulates Anxiety-like Behavior in Rats Following Alcohol Withdrawal. J. Affect. Disord. 2018, 235, 96–104. [Google Scholar] [CrossRef]
- Acquaah-Mensah, G.K.; Kehrer, J.P.; Leslie, S.W. In Utero Ethanol Suppresses Cerebellar Activator Protein-1 and Nuclear Factor-Kappa B Transcriptional Activation in a Rat Fetal Alcohol Syndrome Model. J. Pharmacol. Exp. Ther. 2002, 301, 277–283. [Google Scholar] [CrossRef] [Green Version]
- De la Monte, S.M.; Wands, J.R. Chronic Gestational Exposure to Ethanol Impairs Insulin-Stimulated Survival and Mitochondrial Function in Cerebellar Neurons. Cell. Mol. Life Sci. 2002, 59, 882–893. [Google Scholar] [CrossRef]
- Duverne, S.; Koechlin, E. Rewards and Cognitive Control in the Human Prefrontal Cortex. Cereb. Cortex 2017, 27, 5024–5039. [Google Scholar] [CrossRef] [Green Version]
- Stefani, M.R.; Moghaddam, B. Rule Learning and Reward Contingency Are Associated with Dissociable Patterns of Dopamine Activation in the Rat Prefrontal Cortex, Nucleus Accumbens, and Dorsal Striatum. J. Neurosci. 2006, 26, 8810–8818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, A.A.; Treit, D. Excitotoxic Lesions of the Medial Prefrontal Cortex Attenuate Fear Responses in the Elevated-plus Maze, Social Interaction and Shock Probe Burying Tests. Brain Res. 2003, 969, 183–194. [Google Scholar] [CrossRef]
- Klenowski, P.M. Emerging Role for the Medial Prefrontal Cortex in Alcohol-Seeking Behaviors. Addict. Behav. 2018, 77, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.A.; LeDoux, J.E. Differential Contribution of Dorsal and Ventral Medial Prefrontal Cortex to the Acquisition and Extinction of Conditioned Fear in Rats. Behav. Neurosci. 1995, 109, 681–688. [Google Scholar] [CrossRef]
- Quirk, G.J.; Beer, J.S. Prefrontal Involvement in the Regulation of Emotion: Convergence of Rat and Human Studies. Curr. Opin. Neurobiol. 2006, 16, 723–727. [Google Scholar] [CrossRef]
- Malizia, A.L. What Do Brain Imaging Studies Tell Us about Anxiety Disorders? J. Psychopharmacol. 1999, 13, 372–378. [Google Scholar] [CrossRef]
- Luo, J. GSK3beta in Ethanol Neurotoxicity. Mol. Neurobiol. 2009, 40, 108–121. [Google Scholar] [CrossRef] [Green Version]
- Logrip, M.L.; Barak, S.; Warnault, V.; Ron, D. Corticostriatal BDNF and Alcohol Addiction. Brain Res. 2015, 1628, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Van der Vaart, A.; Meng, X.; Bowers, M.S.; Batman, A.M.; Aliev, F.; Farris, S.P.; Hill, J.S.; Green, T.A.; Dick, D.; Wolstenholme, J.T.; et al. Glycogen Synthase Kinase 3 Beta Regulates Ethanol Consumption and Is a Risk Factor for Alcohol Dependence. Neuropsychopharmacology 2018, 43, 2521–2531. [Google Scholar] [CrossRef]
- Cheng, Y.; Huang, C.C.Y.; Ma, T.; Wei, X.; Wang, X.; Lu, J.; Wang, J. Distinct Synaptic Strengthening of the Striatal Direct and Indirect Pathways Drives Alcohol Consumption. Biol. Psychiatry 2017, 81, 918–929. [Google Scholar] [CrossRef]
- Tsurutani, J.; Castillo, S.S.; Brognard, J.; Granville, C.A.; Zhang, C.; Gills, J.J.; Sayyah, J.; Dennis, P.A. Tobacco Components Stimulate Akt-Dependent Proliferation and NFkappaB-Dependent Survival in Lung Cancer Cells. Carcinogenesis 2005, 26, 1182–1195. [Google Scholar] [CrossRef] [Green Version]
- Hudson, R.; Green, M.; Wright, D.J.; Renard, J.; Jobson, C.E.L.; Jung, T.; Rushlow, W.; Laviolette, S.R. Adolescent Nicotine Induces Depressive and Anxiogenic Effects through ERK 1-2 and Akt-GSK-3 Pathways and Neuronal Dysregulation in the Nucleus Accumbens. Addict. Biol. 2021, 26, e12891. [Google Scholar] [CrossRef]
- Hermann, D.; Sartorius, A.; Welzel, H.; Walter, S.; Skopp, G.; Ende, G.; Mann, K. Dorsolateral Prefrontal Cortex N-Acetylaspartate/Total Creatine (NAA/TCr) Loss in Male Recreational Cannabis Users. Biol. Psychiatry 2007, 61, 1281–1289. [Google Scholar] [CrossRef]
- Sarne, Y.; Mechoulam, R. Cannabinoids: Between Neuroprotection and Neurotoxicity. CNS Neurol. Disord. Drug Targets 2005, 4, 677–684. [Google Scholar] [CrossRef]
- García-Arencibia, M.; González, S.; de Lago, E.; Ramos, J.A.; Mechoulam, R.; Fernández-Ruiz, J. Evaluation of the Neuroprotective Effect of Cannabinoids in a Rat Model of Parkinson’s Disease: Importance of Antioxidant and Cannabinoid Receptor-Independent Properties. Brain Res. 2007, 1134, 162–170. [Google Scholar] [CrossRef]
- Möller, T. Neuroinflammation in Huntington’s Disease. J. Neural Transm. Suppl. 2010, 117, 1001–1008. [Google Scholar] [CrossRef]
- Esposito, G.; de Filippis, D.; Maiuri, M.C.; de Stefano, D.; Carnuccio, R.; Iuvone, T. Cannabidiol Inhibits Inducible Nitric Oxide Synthase Protein Expression and Nitric Oxide Production in β-Amyloid Stimulated PC12 Neurons through P38 MAP Kinase and NF-ΚB Involvement. Neurosci. Lett. 2006, 399, 91–95. [Google Scholar] [CrossRef]
- Esposito, G.; de Filippis, D.; Carnuccio, R.; Izzo, A.A.; Iuvone, T. The Marijuana Component Cannabidiol Inhibits β-Amyloid-Induced Tau Protein Hyperphosphorylation through Wnt/β-Catenin Pathway Rescue in PC12 Cells. J. Mol. Med. 2006, 84, 253–258. [Google Scholar] [CrossRef]
- Molina-Holgado, F.; Pinteaux, E.; Heenan, L.; Moore, J.D.; Rothwell, N.J.; Gibson, R.M. Neuroprotective Effects of the Synthetic Cannabinoid HU-210 in Primary Cortical Neurons Are Mediated by Phosphatidylinositol 3-Kinase/AKT Signaling. Mol. Cell. Neurosci. 2005, 28, 189–194. [Google Scholar] [CrossRef]
- Molina-Holgado, E.; Vela, J.M.; Arévalo-Martín, A.; Almazán, G.; Molina-Holgado, F.; Borrell, J.; Guaza, C. Cannabinoids Promote Oligodendrocyte Progenitor Survival: Involvement of Cannabinoid Receptors and Phosphatidylinositol-3 Kinase/Akt Signaling. J. Neurosci. 2002, 22, 9742–9753. [Google Scholar] [CrossRef]
- Del Pulgar, T.G.; de Ceballos, M.L.; Guzmán, M.; Velasco, G. Cannabinoids Protect Astrocytes from Ceramide-Induced Apoptosis through the Phosphatidylinositol 3-Kinase/Protein Kinase B Pathway. J. Biol. Chem. 2002, 277, 36527–36533. [Google Scholar] [CrossRef] [Green Version]
- Sánchez, M.G.; Ruiz-Llorente, L.; Sánchez, A.M.; Díaz-Laviada, I. Activation of Phosphoinositide 3-Kinase/PKB Pathway by CB(1) and CB(2) Cannabinoid Receptors Expressed in Prostate PC-3 Cells. Involvement in Raf-1 Stimulation and NGF Induction. Cell. Signal. 2003, 15, 851–859. [Google Scholar] [CrossRef]
- Ozaita, A.; Puighermanal, E.; Maldonado, R. Regulation of PI3K/Akt/GSK-3 Pathway by Cannabinoids in the Brain. J. Neurochem. 2007, 102, 1105–1114. [Google Scholar] [CrossRef]
- Valjent, E.; Pagès, C.; Hervé, D.; Girault, J.A.; Caboche, J. Addictive and Non-Addictive Drugs Induce Distinct and Specific Patterns of ERK Activation in Mouse Brain. Eur. J. Neurosci. 2004, 19, 1826–1836. [Google Scholar] [CrossRef]
- Valjent, E.; Pagès, C.; Rogard, M.; Besson, M.J.; Maldonado, R.; Caboche, J. Delta 9-Tetrahydrocannabinol-Induced MAPK/ERK and Elk-1 Activation in Vivo Depends on Dopaminergic Transmission. Eur. J. Neurosci. 2001, 14, 342–352. [Google Scholar] [CrossRef]
- Ackermann, T.F.; Kempe, D.S.; Lang, F.; Lang, U.E. Hyperactivity and Enhanced Curiosity of Mice Expressing PKB/SGK-Resistant Glycogen Synthase Kinase-3 (GSK-3). Cell. Physiol. Biochem. 2010, 25, 775–786. [Google Scholar] [CrossRef]
- Rösner, S.; Leucht, S.; Lehert, P.; Soyka, M. Acamprosate Supports Abstinence, Naltrexone Prevents Excessive Drinking: Evidence from a Meta-Analysis with Unreported Outcomes. J. Psychopharmacol. 2008, 22, 11–23. [Google Scholar] [CrossRef]
- Mason, B.J.; Goodman, A.M.; Dixon, R.M.; Hameed, M.H.A.; Hulot, T.; Wesnes, K.; Hunter, J.A.; Boyeson, M.G. A Pharmacokinetic and Pharmacodynamic Drug Interaction Study of Acamprosate and Naltrexone. Neuropsychopharmacology 2002, 27, 596–606. [Google Scholar] [CrossRef] [Green Version]
- Allgaier, C.; Franke, H.; Sobottka, H.; Scheibler, P. Acamprosate Inhibits Ca2+ Influx Mediated by NMDA Receptors and Voltage-Sensitive Ca2+ Channels in Cultured Rat Mesencephalic Neurones. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2000, 362, 440–443. [Google Scholar] [CrossRef]
- Littleton, J.M. Acamprosate in Alcohol Dependence: Implications of a Unique Mechanism of Action. J. Addict. Med. 2007, 1, 115–125. [Google Scholar] [CrossRef]
- Skinner, M.D.; Lahmek, P.; Pham, H.; Aubin, H.J. Disulfiram Efficacy in the Treatment of Alcohol Dependence: A Meta-Analysis. PLoS ONE 2014, 9, e87366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gossop, M.; Caroll, K.M. Disulfiram, Cocaine, and Alcohol: Two Outcomes for the Price of One? Alcohol. Alcohol. 2006, 41, 119–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carroll, K.M.; Fenton, L.R.; Ball, S.A.; Nich, C.; Frankforter, T.L.; Shi, J.; Rounsaville, B.J. Efficacy of Disulfiram and Cognitive Behavior Therapy in Cocaine-DependentOutpatients: A Randomized Placebo-Controlled Trial. Arch. Gen. Psychiatry 2004, 61, 264–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoller, K.B.; Bigelow, G.E.; Walsh, S.L.; Strain, E.C. Effects of Buprenorphine/Naloxone in Opioid-Dependent Humans. Psychopharmacology 2001, 154, 230–242. [Google Scholar] [CrossRef]
- Fudala, P.J.; Bridge, T.P.; Herbert, S.; Williford, W.O.; Chiang, C.N.; Jones, K.; Collins, J.; Raisch, D.; Casadonte, P.; Goldsmith, R.J.; et al. Office-Based Treatment of Opiate Addiction with a Sublingual-Tablet Formulation of Buprenorphine and Naloxone. N. Engl. J. Med. 2003, 349, 949–958. [Google Scholar] [CrossRef]
- Feng, B.; Obach, R.S.; Burstein, A.H.; Clark, D.J.; de Morais, S.M.; Faessel, H.M. Effect of Human Renal Cationic Transporter Inhibition on the Pharmacokinetics of Varenicline, a New Therapy for Smoking Cessation: An In Vitro–In Vivo Study. Clin. Pharmacol. Ther. 2008, 83, 567–576. [Google Scholar] [CrossRef]
- Baker, T.B.; Piper, M.E.; Stein, J.H.; Smith, S.S.; Bolt, D.M.; Fraser, D.L.; Fiore, M.C. Effects of Nicotine Patch vs. Varenicline vs. Combination Nicotine Replacement Therapy on Smoking Cessation at 26 Weeks: A Randomized Clinical Trial. JAMA 2016, 315, 371–379. [Google Scholar] [CrossRef] [Green Version]
- Anthenelli, R.M.; Benowitz, N.L.; West, R.; St Aubin, L.; McRae, T.; Lawrence, D.; Ascher, J.; Russ, C.; Krishen, A.; Evins, A.E. Neuropsychiatric Safety and Efficacy of Varenicline, Bupropion, and Nicotine Patch in Smokers with and without Psychiatric Disorders (EAGLES): A Double-Blind, Randomised, Placebo-Controlled Clinical Trial. Lancet 2016, 387, 2507–2520. [Google Scholar] [CrossRef] [Green Version]
- Benowitz, N.L.; Pipe, A.; West, R.; Hays, J.T.; Tonstad, S.; McRae, T.; Lawrence, D.; St Aubin, L.; Anthenelli, R.M. Cardiovascular Safety of Varenicline, Bupropion, and Nicotine Patch in Smokers: A Randomized Clinical Trial. JAMA Intern. Med. 2018, 178, 622–631. [Google Scholar] [CrossRef] [Green Version]
- Burnsed, J.C.; Heinan, K.; Letzkus, L.; Zanelli, S. Gabapentin for Pain, Movement Disorders, and Irritability in Neonates and Infants. Dev. Med. Child 2020, 62, 386–389. [Google Scholar] [CrossRef]
- Anton, R.F.; Latham, P.; Voronin, K.; Book, S.; Hoffman, M.; Prisciandaro, J.; Bristol, E. Efficacy of Gabapentin for the Treatment of Alcohol Use Disorder in Patients With Alcohol Withdrawal Symptoms: A Randomized Clinical Trial. JAMA Intern. Med. 2020, 180, 728–736. [Google Scholar] [CrossRef]
- Hendrich, J.; van Minh, A.T.; Heblich, F.; Nieto-Rostro, M.; Watschinger, K.; Striessnig, J.; Wratten, J.; Davies, A.; Dolphin, A.C. Pharmacological Disruption of Calcium Channel Trafficking by the A2δ Ligand Gabapentin. Proc. Natl. Acad. Sci. USA 2008, 105, 3628–3633. [Google Scholar] [CrossRef] [Green Version]
- Sarhill, N.; Davis, M.P.; Walsh, D.; Nouneh, C. Methadone-Induced Myoclonus in Advanced Cancer. Am. J. Hosp. Palliat. Care 2001, 18, 51–53. [Google Scholar] [CrossRef]
- Inturrisi, C.E.; Colburn, W.A.; Kaiko, R.F.; Houde, R.W.; Foley, K.M. Pharmacokinetics and Pharmacodynamics of Methadone in Patients with Chronic Pain. Clin. Pharmacol. Ther. 1987, 41, 392–401. [Google Scholar] [CrossRef]
- Ferrari, A.; Coccia, C.P.R.; Bertolini, A.; Sternieri, E. Methadone--Metabolism, Pharmacokinetics and Interactions. Pharmacol. Res. 2004, 50, 551–559. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, R.; Wu, T.; Shi, Y.; Zhou, X.; Tang, D.; Yu, W.; So, E.C.; Wu, X.; Pan, Z.; et al. Successive Treatment with Naltrexone Induces Epithelial–Mesenchymal Transition and Facilitates the Malignant Biological Behaviors of Bladder Cancer Cells. Acta Biochim. Biophys. Sin. 2021, 53, 238–248. [Google Scholar] [CrossRef]
- Romeo-Guitart, D.; Marcos-DeJuana, C.; Marmolejo-Martínez-Artesero, S.; Navarro, X.; Casas, C. Novel Neuroprotective Therapy with NeuroHeal by Autophagy Induction for Damaged Neonatal Motoneurons. Theranostics 2020, 10, 5154–5168. [Google Scholar] [CrossRef]
- Zhao, Y.; Lin, Z.; Lin, Z.; Zhou, C.; Liu, G.; Lin, J.; Zhang, D.; Lin, D. Overexpression of Mucin 1 Suppresses the Therapeutical Efficacy of Disulfiram against Canine Mammary Tumor. Animals 2020, 11, 37. [Google Scholar] [CrossRef]
- Zamek-Gliszczynski, M.J.; Mohutsky, M.A.; Rehmel, J.L.F.; Ke, A.B. Investigational Small-Molecule Drug Selectively Suppresses Constitutive CYP2B6 Activity at the Gene Transcription Level: Physiologically Based Pharmacokinetic Model Assessment of Clinical Drug Interaction Risk. Drug Metab. Dispos. 2014, 42, 1008–1015. [Google Scholar] [CrossRef] [Green Version]
- Benowitz, N.L. Pharmacology of Nicotine: Addiction, Smoking-Induced Disease, and Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 57–71. [Google Scholar] [CrossRef] [Green Version]
- Lutfy, K.; Eitan, S.; Bryant, C.D.; Yang, Y.C.; Saliminejad, N.; Walwyn, W.; Kieffer, B.L.; Takeshima, H.; Carroll, F.I.; Maidment, N.T.; et al. Buprenorphine-Induced Antinociception Is Mediated by μ-Opioid Receptors and Compromised by Concomitant Activation of Opioid Receptor-Like Receptors. J. Neurosci. 2003, 23, 10331–10337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, B.C.; Wang, J.; Rui, Y.; Cao, J.; Xu, P.; Jiang, D.; Zhu, X.; Won, M.H.; Bo, P.; Su, P. Neuroprotective Effects of Gabapentin Against Cerebral Ischemia Reperfusion-Induced Neuronal Autophagic Injury via Regulation of the PI3K/Akt/MTOR Signaling Pathways. J. Neuropathol. Exp. Neurol. 2019, 78, 157–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nezamoleslami, S.; Sheibani, M.; Mumtaz, F.; Esmaeili, J.; Shafaroodi, H.; Dehpour, A.R. Lithium Reverses the Effect of Opioids on ENOS/Nitric Oxide Pathway in Human Umbilical Vein Endothelial Cells. Mol. Biol. Rep. 2020, 47, 6829–6840. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Molecular Activity | Type of Addiction | Impact on GSK3β |
|---|---|---|---|
| Naltrexone | Opioid-agonist | Alcohol | Decreases pSer9 GSK3β amount [227] |
| Acamprosate | Glutamate receptors antagonist, inhibits upregulation of Ca2+ channels | Alcohol | Sustains Akt activation [231] |
| Disulfiram | Blocks the enzyme aldehyde dehydrogenase | Cocaine Alcohol | Inhibits PI3K/Akt/mTOR Pathway [229] |
| Bupropion | Inhibits the reuptake of monoamine through DAT | Methamphetamine | Inhibits GSK3β [218,219,220] |
| Buprenorphine | Opioid agonist-antagonist | Opiate | Activates Akt via ORL-1 receptor [232] |
| Varenicline | Partial agonist of nAChR | Nicotine | Decreases pSer9 GSK3β 1 [231] |
| Gabapentin | Influences GABA and glutamate activity | Alcohol | Activates PI3K/Akt/mTOR pathway [233] |
| Methadone | 𝜇OR agonist, NMDAR antagonist | Heroine | Decreases pSer9 GSK3β amount 2 [234] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turlik, J.; Wąsikiewicz, E.; Domaradzka, A.; Chrostek, G.; Gniadzik, W.; Domagalski, M.; Duda, P. GSK3β Activity in Reward Circuit Functioning and Addiction. NeuroSci 2021, 2, 443-466. https://doi.org/10.3390/neurosci2040033
Turlik J, Wąsikiewicz E, Domaradzka A, Chrostek G, Gniadzik W, Domagalski M, Duda P. GSK3β Activity in Reward Circuit Functioning and Addiction. NeuroSci. 2021; 2(4):443-466. https://doi.org/10.3390/neurosci2040033
Chicago/Turabian StyleTurlik, Jakub, Ewa Wąsikiewicz, Aleksandra Domaradzka, Gabriela Chrostek, Weronika Gniadzik, Mikołaj Domagalski, and Przemysław Duda. 2021. "GSK3β Activity in Reward Circuit Functioning and Addiction" NeuroSci 2, no. 4: 443-466. https://doi.org/10.3390/neurosci2040033
APA StyleTurlik, J., Wąsikiewicz, E., Domaradzka, A., Chrostek, G., Gniadzik, W., Domagalski, M., & Duda, P. (2021). GSK3β Activity in Reward Circuit Functioning and Addiction. NeuroSci, 2(4), 443-466. https://doi.org/10.3390/neurosci2040033