
Synthesis and Characterization of the Conjugated Peptide Lunatin-Folate



Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
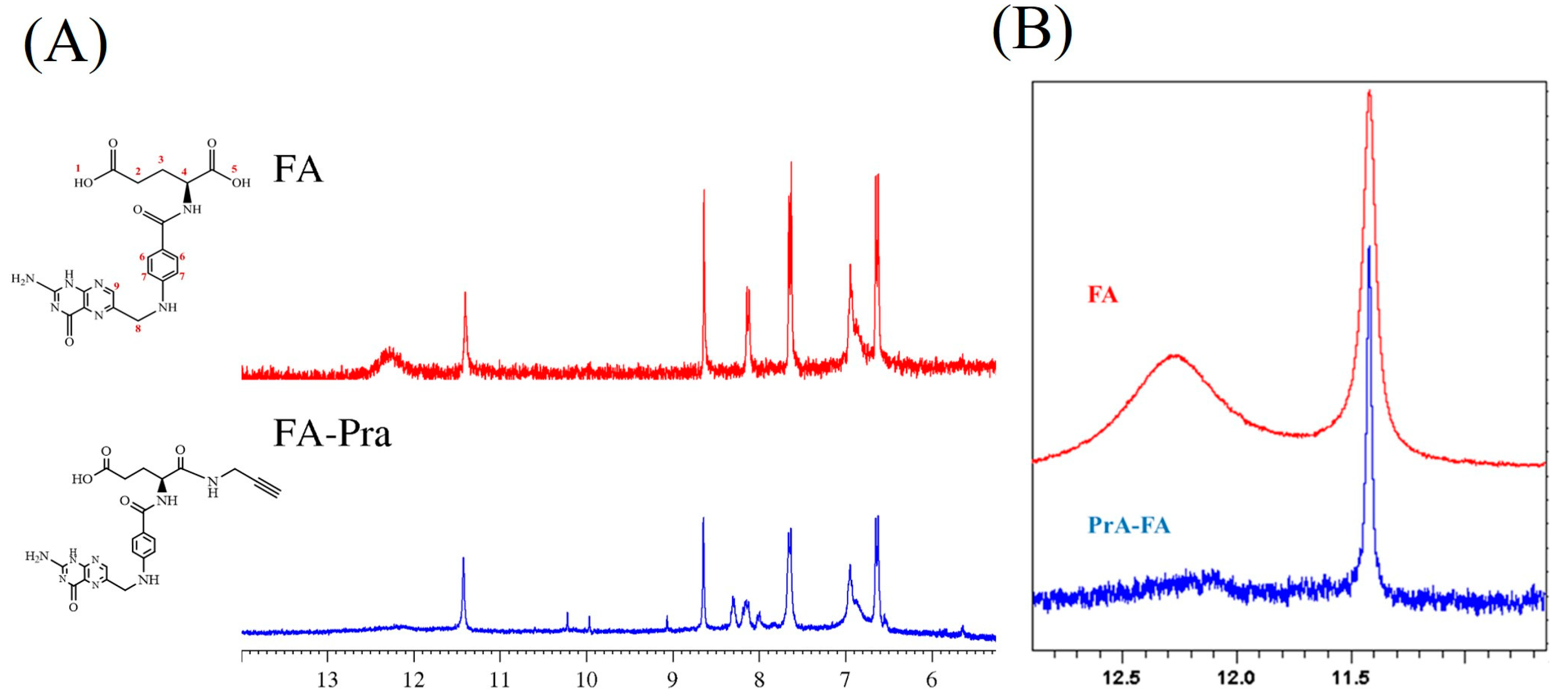
2.2. Synthesis of the Propargylated Derivative of Folic Acid
2.3. Solid-Phase Peptide Synthesis of Lunatin-1 and Tripeptide FIG-NH2
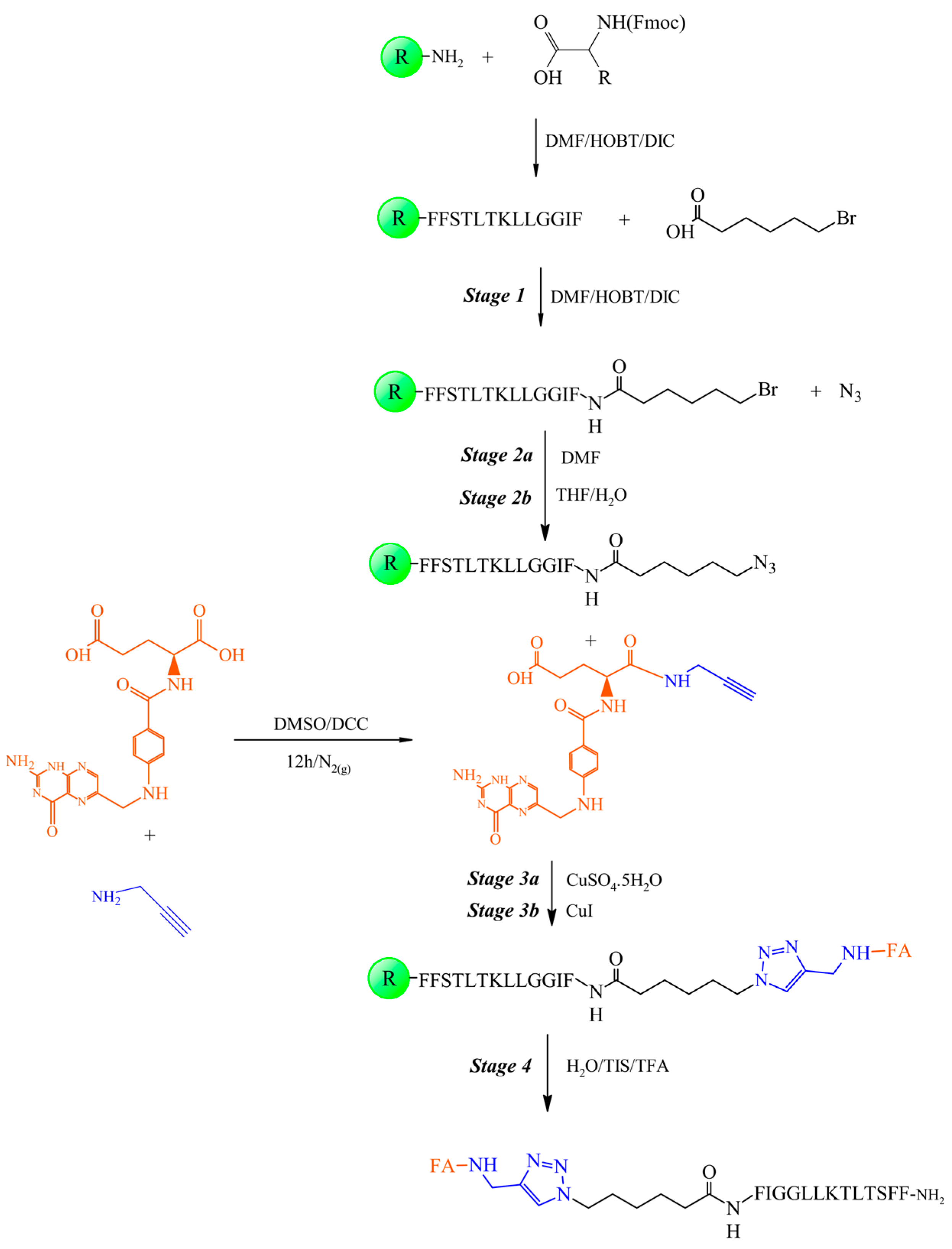
2.4. Synthesis of the Peptide-Folate Conjugate
2.4.1. Coupling of 6-Bromohexanoic Acid to the Peptidyl-Resin (Step 1)
2.4.2. Substitution of Bromine with Azide in Bromohexamide-Peptidyl-Resin (Step 2)
2.4.3. Obtaining Folate-Peptidyl-Resin (Step 3)
2.4.4. Cleavage Step of Folate-Peptidyl Resin
2.5. Characterization of Intermediates and Products of the Synthesis
2.5.1. Mass Spectrometry
2.5.2. Fourier Transform Infrared Region Spectroscopy
2.5.3. Nuclear Magnetic Resonance (NMR)
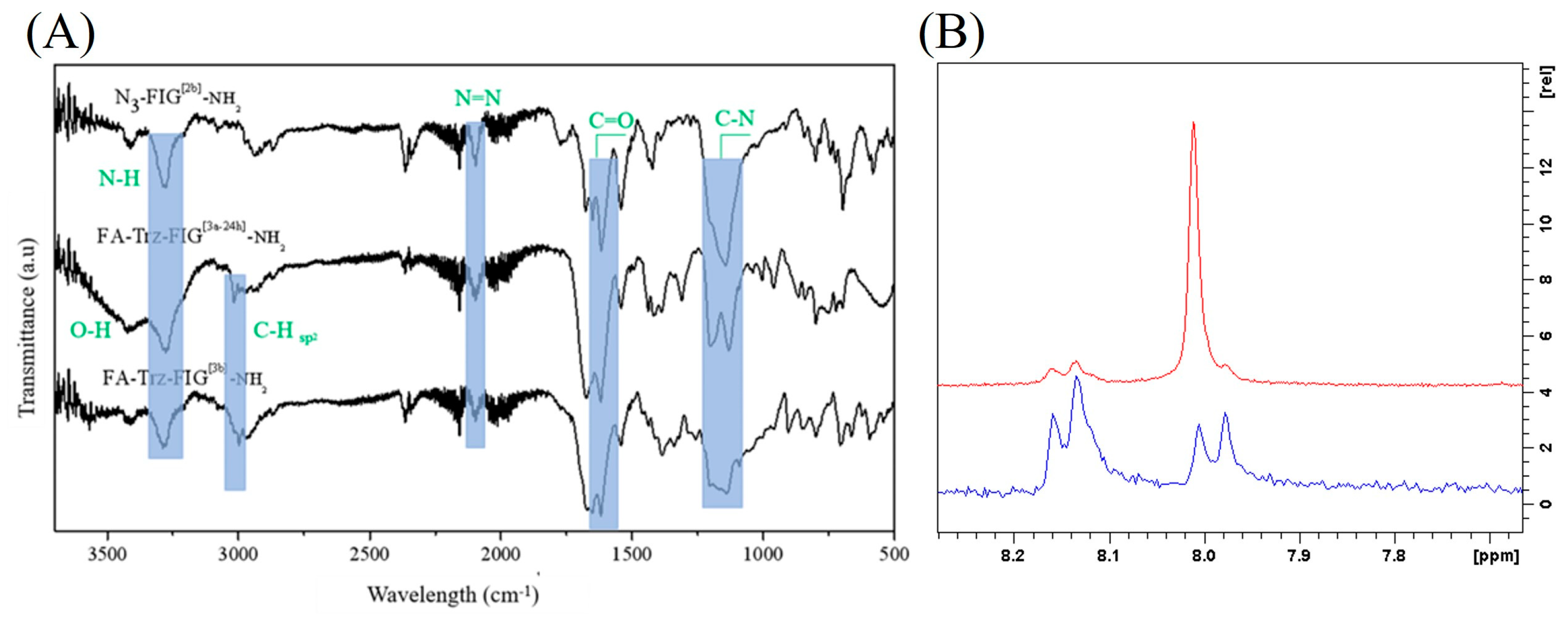
3. Results and Discussion
3.1. Synthesis of the Propargylated Folic Acid Derivative
3.2. Synthesis and Characterization of the Peptide-Conjugates FA-Trz-FIG and FA-Trz-Luna
3.2.1. Synthesis of the Tripeptide FIG-NH2
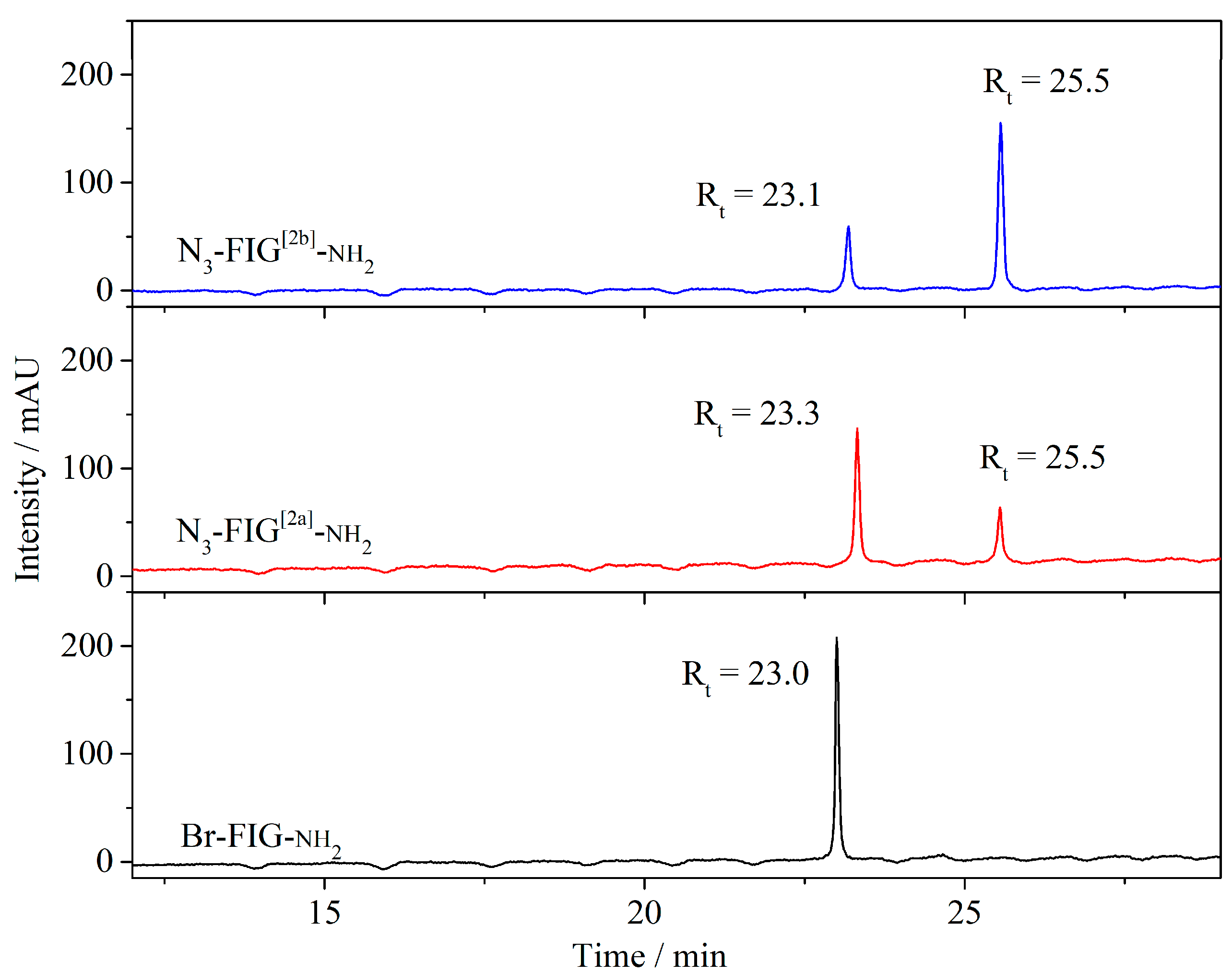
3.2.2. Synthesis of the Bromo-Hexamide-Peptide-Resin (Step 1)
3.2.3. Synthesis of Azide-Hexamide-Peptidyl-Resin (Step 2)
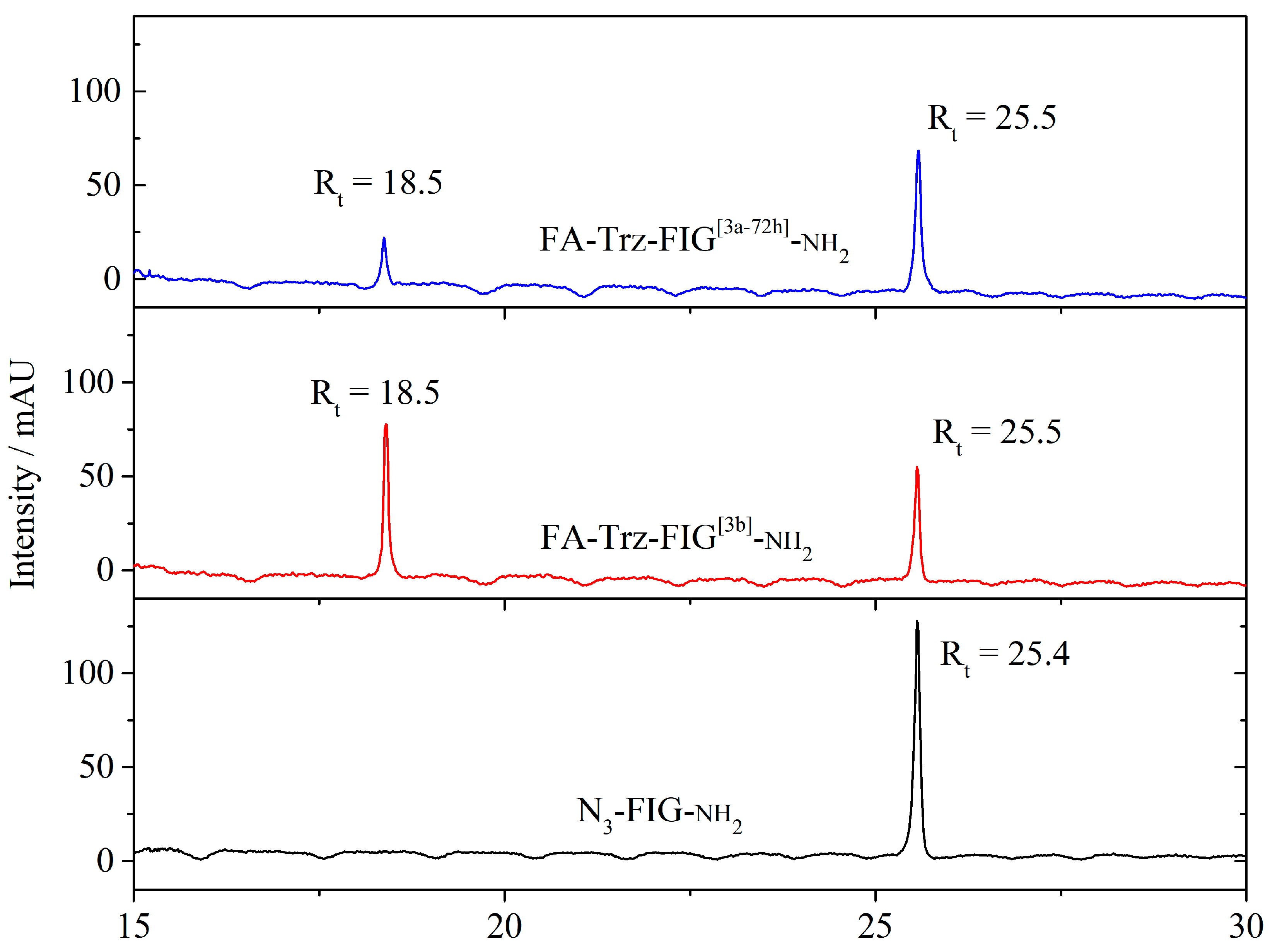
3.2.4. Synthesis of Folate-Peptidyl-Resin (Step 3)
3.3. Synthesis and Characterization of the FA-Trz-Luna Conjugate
Synthesis and Characterization of the Derivatives and the FA-Trz-Luna Conjugate
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chokshi, A.; Sifri, Z.; Cennimo, D.; Horng, H. Global contributors to antibiotic resistance. J. Glob. Infect. Dis. 2019, 11, 36. [Google Scholar] [CrossRef] [PubMed]
- Ragheb, M.N.; Thomason, M.K.; Hsu, C.; Nugent, P.; Gage, J.; Samadpour, A.N.; Kariisa, A.; Merrikh, C.N.; Miller, S.I.; Sherman, D.R.; et al. Inhibiting the Evolution of Antibiotic Resistance. Mol. Cell 2019, 73, 157–165.e5. [Google Scholar] [CrossRef]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.L.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2019, 48, D517–D525. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Chaudhary, K.; Gupta, S.; Singh, H.; Kumar, S.; Gautam, A.; Kapoor, P.; Raghava, G.P. CancerDR: Cancer Drug Resistance Database. Sci. Rep. 2013, 3, 1445. [Google Scholar] [CrossRef] [PubMed]
- Koehbach, J.; Craik, D.J. The Vast Structural Diversity of Antimicrobial Peptides. Trends Pharmacol. Sci. 2019, 40, 517–528. [Google Scholar] [CrossRef]
- Avci, F.G.; Sariyar Akbulut, B.; Ozkirimli, E. Membrane Active Peptides and Their Biophysical Characterization. Biomolecules 2018, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Marquette, A.; Bechinger, B. Biophysical Investigations Elucidating the Mechanisms of Action of Antimicrobial Peptides and Their Synergism. Biomolecules 2018, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Henriques, S.T.; Melo, M.N.; Castanho, M.A.R.B. How to address CPP and AMP translocation? Methods to detect and quantify peptide internalization in vitro and in vivo (Review). Mol. Membr. Biol. 2007, 24, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Wang, Q.; Ren, K.; Xu, T.; Zhang, Z.; Xu, M.; Rao, Z.; Zhang, X. A Review of Antimicrobial Peptides: Structure, Mechanism of Action, and Molecular Optimization Strategies. Fermentation 2024, 10, 540. [Google Scholar] [CrossRef]
- Gagandeep, K.R.; Narasingappa, R.B.; Vyas, G.V. Unveiling mechanisms of antimicrobial peptide: Actions beyond the membranes disruption. Heliyon 2024, 10, e38079. [Google Scholar] [CrossRef]
- Gupta, S.D.; Halder, B.; Gomes, A.; Gomes, A. Bengalin initiates autophagic cell death through ERK–MAPK pathway following suppression of apoptosis in human leukemic U937 cells. Life Sci. 2013, 93, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Felício, M.R.; Silva, O.N.; Gonçalves, S.; Santos, N.C.; Franco, O.L. Peptides with Dual Antimicrobial and Anticancer Activities. Front. Chem. 2017, 5, 5. [Google Scholar] [CrossRef]
- Junior, E.F.; Guimarães, C.F.; Franco, L.L.; Alves, R.J.; Kato, K.C.; Martins, H.R.; de Souza Filho, J.D.; Bemquerer, M.P.; Munhoz, V.H.; Resende, J.M.; et al. Glycotriazole-peptides derived from the peptide HSP1: Synergistic effect of triazole and saccharide rings on the antifungal activity. Amino Acids 2017, 49, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Cai, X.; Bi, X.; Liu, C.; Yue, N.; Zhu, Y.; Zhou, J.; Fu, M.; Huang, W.; Qian, H. Synthesis and anti-cancer evaluation of folic acid-peptide- paclitaxel conjugates for addressing drug resistance. Eur. J. Med. Chem. 2019, 171, 104–115. [Google Scholar] [CrossRef]
- Chen, J.; Li, S.; Shen, Q. Folic acid and cell-penetrating peptide conjugated PLGA–PEG bifunctional nanoparticles for vincristine sulfate delivery. Eur. J. Pharm. Sci. 2012, 47, 430–443. [Google Scholar] [CrossRef]
- Silva, A.R.; Guimarães, M.S.; Rabelo, J.; Belén, L.H.; Perecin, C.J.; Farías, J.G.; Santos, J.H.; Rangel-Yagui, C.O. Recent advances in the design of antimicrobial peptide conjugates. J. Mater. Chem. B 2022, 10, 3587–3600. [Google Scholar] [CrossRef]
- de Sousa Gomes, K. Relação Estrutura-Atividade do Peptídeo Sintético Lunatina-1 e Seus Derivados na Citotoxicidade de Diferentes Linhagens Celulares. Ph.D. Thesis, Universidade Federal de Minas Gerais, Belo Horizonte, Brazil, 2018. [Google Scholar]
- Puvvada, N.; Rajput, S.; Kumar, B.P.; Sarkar, S.; Konar, S.; Brunt, K.R.; Rao, R.R.; Mazumdar, A.; Das, S.K.; Basu, R.; et al. Novel ZnO hollow-nanocarriers containing paclitaxel targeting folate-receptors in a malignant pH-microenvironment for effective monitoring and promoting breast tumor regression. Sci. Rep. 2015, 5, 11760. [Google Scholar] [CrossRef]
- Chan, W.; White, P. Fmoc Solid Phase Peptide Synthesis: A Practical Approach; OUP Oxford: Oxford, UK, 1999. [Google Scholar]
- Kaiser, E.; Colescott, R.L.; Bossinger, C.D.; Cook, P.I. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal. Biochem. 1970, 34, 595–598. [Google Scholar] [CrossRef]
- Torres, L.M.F.C. Síntese de Nanoestruturas de Alumina Contendo o Peptídeo Antimicrobiano BP100 para Aplicações Biotecnológicas. Ph.D. Thesis, Universidade Federal dos Vales do Jequitinhonha e Mucuri, Diamantina, Brazil, 2018. [Google Scholar]
- Coessens, V.; Matyjaszewski, K. End group transformation of polymers prepared by atrp, substitution to azides. J. Macromol. Sci. Part A 1999, 36, 667–679. [Google Scholar] [CrossRef]
- Ghaffar, K.; Hussein, W.; Khalil, Z.; Capon, R.; Skwarczynski, M.; Toth, I. Levofloxacin and Indolicidin for Combination Antimicrobial Therapy. Curr. Drug Deliv. 2015, 12, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Bax, A.D.; Davis, D.G. MLEV-17-based two-dimensional homonuclear magnetization transfer spectroscopy. J. Magn. Reson. (1969) 1985, 65, 355–360. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Blevins, R.A. NMR View: A computer program for the visualization and analysis of NMR data. J. Biomol. NMR 1994, 4, 603–614. [Google Scholar] [CrossRef]
- Dutta, S.; Hartkopf-Fröder, C.; Witte, K.; Brocke, R.; Mann, U. Molecular characterization of fossil palynomorphs by transmission micro-FTIR spectroscopy: Implications for hydrocarbon source evaluation. Int. J. Coal Geol. 2013, 115, 13–23. [Google Scholar] [CrossRef]
- Müller, C.; Reuter, S.; Wille, J.; Xanthopoulou, K.; Stefanik, D.; Grundmann, H.; Higgins, P.G.; Seifert, H. A global view on carbapenem-resistant Acinetobacter baumannii. mBio 2023, 14, e02260-23. [Google Scholar] [CrossRef]
- Wu, Z.; Li, X.; Hou, C.; Qian, Y. Solubility of Folic Acid in Water at pH Values between 0 and 7 at Temperatures (298.15, 303.15, and 313.15) K. J. Chem. Eng. Data 2010, 55, 3958–3961. [Google Scholar] [CrossRef]
- Nascimento, C. Ressonância Magnética Nuclear; BlucherLTDA: São Paulo, Brazil, 2018. [Google Scholar]
- Mirkin, N.G.; Krimm, S. Structural Dependence of NH Stretch Mode Frequency Shifts in Amide and Peptide Systems. J. Phys. Chem. A 2004, 108, 5438–5448. [Google Scholar] [CrossRef]
- Surewicz, W.K.; Mantsch, H.H.; Chapman, D. Determination of protein secondary structure by Fourier transform infrared spectroscopy: A critical assessment. Biochemistry 1993, 32, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Bratu, I.; Grecu, R.; Constantinescu, R.; Iliescu, T. Molecular relaxation processes of 2-bromopropane in solutions from IR ν(C–Br) band shape analysis. Spectrochim. Acta A Mol. Biomol. Spectrosc. 1998, 54, 501–506. [Google Scholar] [CrossRef]
- Keeler, J. Understanding NMR Spectroscopy; Wiley: Chichester, UK, 2011. [Google Scholar]
- Li, K.; Xue, D. Estimation of Electronegativity Values of Elements in Different Valence States. J. Phys. Chem. A 2006, 110, 11332–11337. [Google Scholar] [CrossRef] [PubMed]
- Phelan, J.; Altharawi, A.; Chan, K.L.A. Tracking glycosylation in live cells using FTIR spectroscopy. Talanta 2020, 211, 120737. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, C.P.; Kumar, K.; Singh, S.K.; Singh, D.; Saini, S. Synthesis and antimicrobial evaluation of 1,4-disubstituted 1,2,3-triazoles with aromatic ester functionality. Arab. J. Chem. 2016, 9, 865–871. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reagents/Peptidyl | Acronyms |
|---|---|
| Folic Acid | FA |
| Propargylamine | Pra |
| 6-Bromohexanoic Acid | Br-Ac |
| Propargylated Derivative of Folic Acid | FA-Pra |
| Peptides | FIG-NH2/Luna |
| Stage 1 | Br-FIG-NH2/Br-Luna |
| Stage 2a | N3-FIG [2a]-NH2 |
| Stage 2b | N3-FIG[2b]-NH2/N3-Luna |
| Stage 3a | FA-Trz-FIG[3a_6h]-NH2 FA-Trz-FIG[3a_24h]-NH2 FA-Trz-FIG[3a_48h]-NH2 FA-Trz-FIG[3a_72h]-NH2 |
| Stage 3b | FA-Trz-FIG[3b]-NH2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Souza, A.N.; de Castro Pimenta, A.M.; Verly, R.M. Synthesis and Characterization of the Conjugated Peptide Lunatin-Folate. Organics 2025, 6, 8. https://doi.org/10.3390/org6010008
de Souza AN, de Castro Pimenta AM, Verly RM. Synthesis and Characterization of the Conjugated Peptide Lunatin-Folate. Organics. 2025; 6(1):8. https://doi.org/10.3390/org6010008
Chicago/Turabian Stylede Souza, Amanda Neves, Adriano Monteiro de Castro Pimenta, and Rodrigo Moreira Verly. 2025. "Synthesis and Characterization of the Conjugated Peptide Lunatin-Folate" Organics 6, no. 1: 8. https://doi.org/10.3390/org6010008
APA Stylede Souza, A. N., de Castro Pimenta, A. M., & Verly, R. M. (2025). Synthesis and Characterization of the Conjugated Peptide Lunatin-Folate. Organics, 6(1), 8. https://doi.org/10.3390/org6010008