
A New Rapid and Specific Iodination Reagent for Phenolic Compounds


Abstract
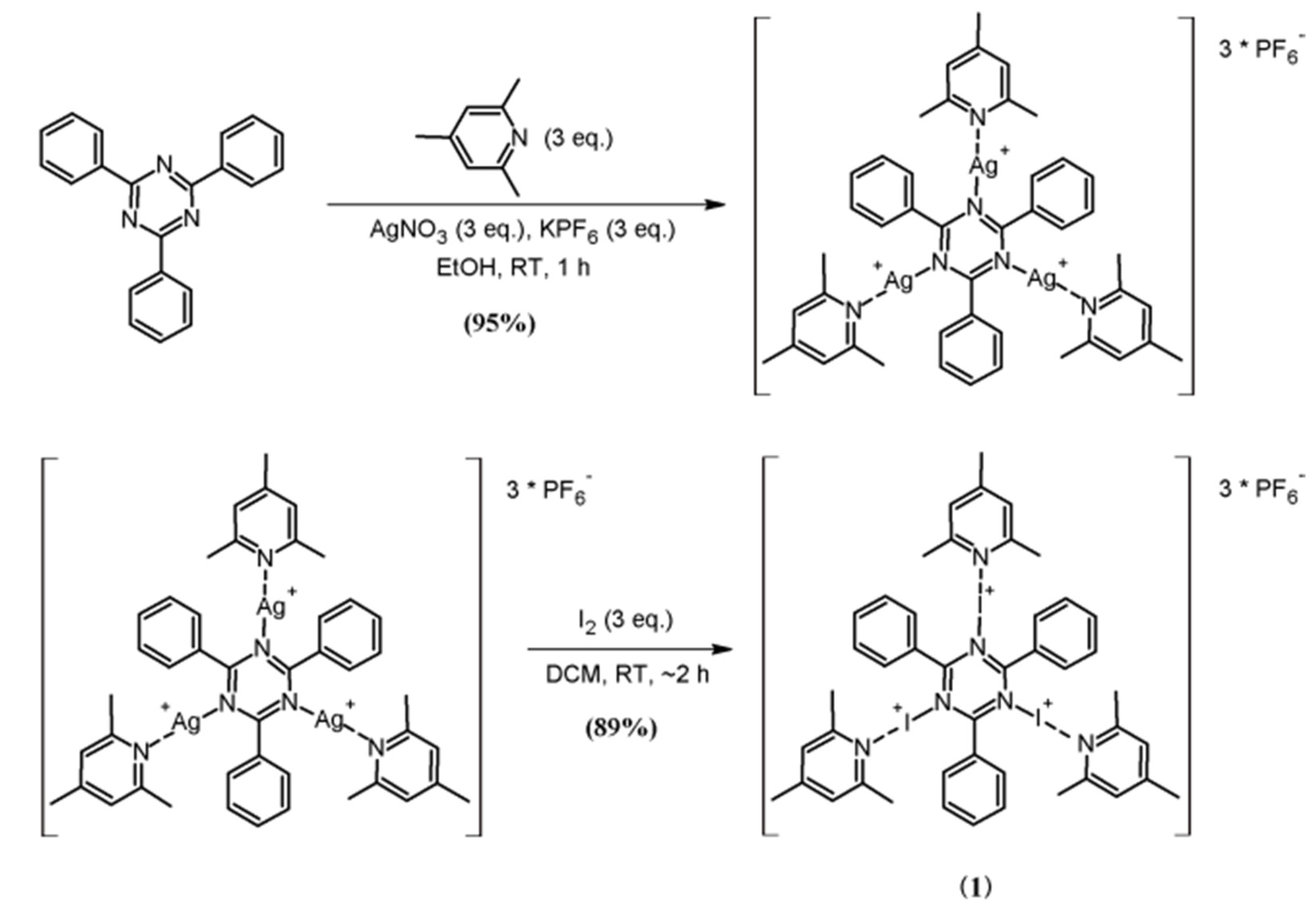
1. Introduction
2. Materials and Methods
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krasnokutskaya, E.A.; Semenischeva, N.I.; Filimonov, V.D.; Knochel, P. A New, One-Step, Effective Protocol for the Iodination of Aromatic and Heterocyclic Compounds via Aprotic Diazotization of Amines. Synthesis 2007, 1, 81–84. [Google Scholar] [CrossRef]
- Hubbard, A.; Okazaki, T.; Laali, K.K. Halo- and Azidodediazoniation of Arenediazonium Tetrafluoroborates with Trimethylsilyl Halides and Trimethylsilyl Azide and Sandmeyer-Type Bromodediazoniation with Cu(I)Br in [BMIM][PF6] Ionic Liquid. J. Org. Chem. 2008, 73, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Filimonov, V.D.; Trusova, M.; Postnikov, P.; Krasnokutskaya, E.A.; Lee, Y.M.; Hwang, H.Y.; Kim, H.; Chi, K.-W. Unusually Stable, Versatile, and Pure Arenediazonium Tosylates: Their Preparation, Structures, and Synthetic Applicability. Org. Lett. 2008, 10, 3961–3964. [Google Scholar] [CrossRef]
- Filimonov, V.D.; Semenischeva, N.I.; Krasnokutskaya, E.A.; Tretyakov, A.N.; Hwang, H.Y.; Chi, K.-W. Sulfonic Acid Based Cation-Exchange Resin: A Novel Proton Source for One-Pot Diazotization-Iodination of Aromatic Amines in Water. Synthesis 2008, 2, 185–187. [Google Scholar] [CrossRef]
- Leas, D.A.; Dong, Y.; Vennerstrom, J.L.; Stack, D.E. One-Pot, Metal-Free Conversion of Anilines to Aryl Bromides and Iodides. Org. Lett. 2017, 19, 2518–2521. [Google Scholar] [CrossRef]
- Hauenschild, T.; Hinderberger, D. A Platform of Phenol-Based Nitroxide Radicals as an “EPR Toolbox” in Supramolecular and Click Chemistry. ChemPlusChem 2019, 84, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Thiebes, C.; Surya Prakash, G.K.; Petasis, N.A.; Olah, G.A. Mild Preparation of Haloarenes by Ipso-Substitution of Arylboronic Acids with N-Halosuccinimides. Synlett 1998, 2, 141–142. [Google Scholar] [CrossRef]
- MacNeil, S.L.; Familoni, O.B.; Snieckus, V. Selective Ortho and Benzylic Functionalization of Secondary and Tertiary p-Tolylsulfonamides. Ipso-Bromo Desilylation and Suzuki Cross-Coupling Reactions. J. Org. Chem. 2001, 66, 3662–3670. [Google Scholar] [CrossRef]
- Klapars, A.; Buchwald, S.L. Copper-Catalyzed Halogen Exchange in Aryl Halides: An Aromatic Finkelstein Reaction. J. Am. Chem. Soc. 2002, 124, 14844–14845. [Google Scholar] [CrossRef]
- Stavber, S.; Kralj, P.; Zupan, M. Progressive Direct Iodination of Sterically Hindered Alkyl Substituted Benzenes. Synthesis 2002, 11, 1513–1518. [Google Scholar] [CrossRef]
- Castanet, A.-S.; Colobert, F.; Broutin, P.-E. Mild and regioselective iodination of electron-rich aromatics with N-iodosuccinimide and catalytic trifluoroacetic acid. Tetrahedron Lett. 2002, 43, 5047–5048. [Google Scholar] [CrossRef]
- Lulinski, P.; Kryska, A.; Sosnowski, M.; Skulski, L. Eco-friendly Oxidative Iodination of Various Arenes with a Urea-Hydrogen Peroxide Adduct (UHP) as the Oxidant [1]. Synthesis 2004, 3, 441–445. [Google Scholar] [CrossRef]
- Iskra, J.; Stavber, S.; Zupan, M. Nonmetal-Catalyzed Iodination of Arenes with Iodide and Hydrogen Peroxide. Synthesis 2004, 11, 1869–1873. [Google Scholar] [CrossRef]
- Prakash, G.K.S.; Mathew, T.; Hoole, D.; Esteves, P.M.; Wang, Q.; Rasul, G.; Olah, G.A. N-Halosuccinimide/BF3−H2O, Efficient Electrophilic Halogenating Systems for Aromatics. J. Am. Chem. Soc. 2004, 126, 15770–15776. [Google Scholar] [CrossRef]
- Sedelmeier, J.; Bolm, C. Efficient Copper-Catalyzed N-Arylation of Sulfoximines with Aryl Iodides and Aryl Bromides. J. Org. Chem. 2005, 70, 6904–6906. [Google Scholar] [CrossRef] [PubMed]
- Kalyani, D.; Dick, A.R.; Anani, W.Q.; Sanford, M.S. A Simple Catalytic Method for the Regioselective Halogenation of Arenes. Org. Lett. 2006, 8, 2523–2526. [Google Scholar] [CrossRef]
- Kraszkiewicz, L.; Sosnowski, M.; Skulski, L. Oxidative Iodination of Deactivated Arenes in Concentrated Sulfuric Acid with I2/NaIO4 and KI/NaIO4 Iodinating Systems. Synthesis 2006, 7, 1195–1199. [Google Scholar]
- Ganguly, N.C.; Barik, S.K.; Dutta, S. Ecofriendly Iodination of Activated Aromatics and Coumarins Using Potassium Iodide and Ammonium Peroxodisulfate. Synthesis 2010, 9, 1467–1472. [Google Scholar] [CrossRef]
- Qiu, D.; Mo, F.; Zheng, Z.; Zhang, Y.; Wang, J. Gold(III)-Catalyzed Halogenation of Aromatic Boronates with N-Halosuccinimides. Org. Lett. 2010, 12, 5474–5477. [Google Scholar] [CrossRef]
- Gallo, R.D.C.; Gebara, K.S.; Muzzi, R.M.; Raminelli, C. Efficient and selective iodination of phenols promoted by iodine and hydrogen peroxide in water. Braz. J. Chem. Soc. 2010, 21, 770–774. [Google Scholar] [CrossRef]
- Rodríguez-Lojo, D.; Cobas, A.; Peña, D.; Pérez, D.; Guitián, E. Aryne Insertion into I–I σ-Bonds. Org. Lett. 2012, 14, 1363–1365. [Google Scholar] [CrossRef]
- Jakab, G.; Hosseini, A.; Hausmann, H.; Schreiner, P.R. Mild and Selective Organocatalytic Iodination of Activated Aromatic Compounds. Synthesis 2013, 45, 1635–1640. [Google Scholar]
- Du, B.; Jiang, X.; Sun, P. Palladium-Catalyzed Highly Selective ortho-Halogenation (I, Br, Cl) of Arylnitriles via sp2 C–H Bond Activation Using Cyano as Directing Group. J. Org. Chem. 2013, 78, 2786–2791. [Google Scholar] [CrossRef]
- Partridge, B.M.; Hartwig, J.F. Sterically Controlled Iodination of Arenes via Iridium-Catalyzed C–H Borylation. Org. Lett. 2013, 15, 140–143. [Google Scholar] [CrossRef] [PubMed]
- Niu, L.; Zhang, H.; Yang, H.; Fu, H. Metal-Free Iodination of Arylboronic Acids and the Synthesis of Biaryl Derivatives. Synlett 2014, 25, 995–1000. [Google Scholar]
- Leboeuf, D.; Ciesielsk, J.; Frontier, A.J. Gold(I)-Catalyzed Iodination of Arenes. Synlett 2014, 25, 399–402. [Google Scholar] [CrossRef]
- Song, S.; Sun, X.; Li, X.; Yuan, Y.; Jiao, N. Efficient and Practical Oxidative Bromination and Iodination of Arenes and Heteroarenes with DMSO and Hydrogen Halide: A Mild Protocol for Late-Stage Functionalization. Org. Lett. 2015, 17, 2886–2889. [Google Scholar] [CrossRef]
- Chen, J.; Xiong, X.; Chen, Z.; Huang, J. Imidazolium Salt Catalyzed para-Selective Halogenation of Electron-Rich Arenes. Synlett 2015, 26, 2831–2834. [Google Scholar]
- Li, L.; Liu, W.; Zeng, H.; Mu, X.; Cosa, G.; Mi, Z.; Li, C.-J. Photo-induced Metal-Catalyst-Free Aromatic Finkelstein Reaction. J. Am. Chem. Soc. 2015, 137, 8328–8331. [Google Scholar] [CrossRef]
- Racys, D.T.; Warrilow, C.E.; Pimlott, S.L.; Sutherland, A. Highly Regioselective Iodination of Arenes via Iron(III)-Catalyzed Activation of N-Iodosuccinimide. Org. Lett. 2015, 17, 4782–4785. [Google Scholar]
- Racys, D.T.; Sharif, S.A.I.; Pimlott, S.L.; Sutherland, A. Silver(I)-Catalyzed Iodination of Arenes: Tuning the Lewis Acidity of N-Iodosuccinimide Activation. J. Org. Chem. 2016, 81, 772–780. [Google Scholar] [CrossRef]
- Fu, Z.; Li, Z.; Song, Y.; Yang, R.; Liu, Y.; Cai, H. Decarboxylative Halogenation and Cyanation of Electron-Deficient Aryl Carboxylic Acids via Cu Mediator as Well as Electron-Rich Ones through Pd Catalyst under Aerobic Conditions. J. Org. Chem. 2016, 81, 2794–2803. [Google Scholar] [CrossRef]
- Ajvazi, N.; Stavber, S. Electrophilic Iodination of Organic Compounds Using Elemental Iodine or Iodides: Recent Advances 2008–2021: Part I. Compounds 2022, 2, 3–24. [Google Scholar] [CrossRef]
- Brunei, Y.; Rousseau, G. Iodination of phenols and anilines with bis(sym-collidine)iodine(I) hexafluorophosphate. Tetrahedron Lett. 1995, 36, 8217–8220. [Google Scholar] [CrossRef]
- Homsi, F.; Robin, S.; Rousseau, G.; Patterson, S.; Hart, D.J. Preparation of Bis(2,4,6-Trimethylpyridine)Iodine(I) Hexafluorophosphate and Bis(2,4,6-Trimethylpyridine) Bromine(I) Hexafluorophosphate. Org. Synth. 2000, 77, 206–211. [Google Scholar]
- Rosenthaler, L.; Capuano, L. Process for preparing and therapeutical applications of the 2,4,6-triiodophenol. Pharm. Acta Helv. 1946, 21, 225–228. [Google Scholar] [PubMed]
- Emmanuvel, L.; Shukla, R.K.; Sudalai, A.; Gurunath, S.; Sivaram, S. NaIO4/KI/NaCl: A new reagent system for iodination of activated aromatics through in situ generation of iodine monochloride. Tetrahedron Lett. 2006, 47, 4793–4796. [Google Scholar] [CrossRef]
- Hauenschild, T.; Reichenwallner, J.; Enkelmann, V.; Hinderberger, D. Characterizing Active Pharmaceutical Ingredient Binding to Human Serum Albumin by Spin-Labeling and EPR Spectroscopy. Chem. Eur. J. 2016, 22, 12825–12838. [Google Scholar] [CrossRef]
- Sultani, H.N.; Haeri, H.H.; Hinderberger, D.; Westermann, B. Spin-labelled diketopiperazines and peptide–peptoid chimera by Ugi-multi-component-reactions. Org. Biomol. Chem. 2016, 14, 11336–11341. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Entry | Substrate | Reaction Conditions a (Reaction Time (min); eq. of FIC*17*) | Product | Yield b (%) |
|---|---|---|---|---|
| 1 |  | (5; 1) |  | 98 |
| 2 |  | (10; 1) |  | 99 |
| 3 |  | (5; 1) |  | 95 |
| 4 |  | (10; 1) |  | 96 |
| 5 |  | (5; 1) |  | 95 |
| 6 |  | (10; 1) |  | 95 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hauenschild, T.; Hinderberger, D. A New Rapid and Specific Iodination Reagent for Phenolic Compounds. Organics 2023, 4, 137-145. https://doi.org/10.3390/org4020011
Hauenschild T, Hinderberger D. A New Rapid and Specific Iodination Reagent for Phenolic Compounds. Organics. 2023; 4(2):137-145. https://doi.org/10.3390/org4020011
Chicago/Turabian StyleHauenschild, Till, and Dariush Hinderberger. 2023. "A New Rapid and Specific Iodination Reagent for Phenolic Compounds" Organics 4, no. 2: 137-145. https://doi.org/10.3390/org4020011
APA StyleHauenschild, T., & Hinderberger, D. (2023). A New Rapid and Specific Iodination Reagent for Phenolic Compounds. Organics, 4(2), 137-145. https://doi.org/10.3390/org4020011