
Extremely Halophilic Biohydrogen Producing Microbial Communities from High-Salinity Soil and Salt Evaporation Pond

 , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. DNA Extraction and Amplicon Sequencing
2.3. Data Analysis
3. Results
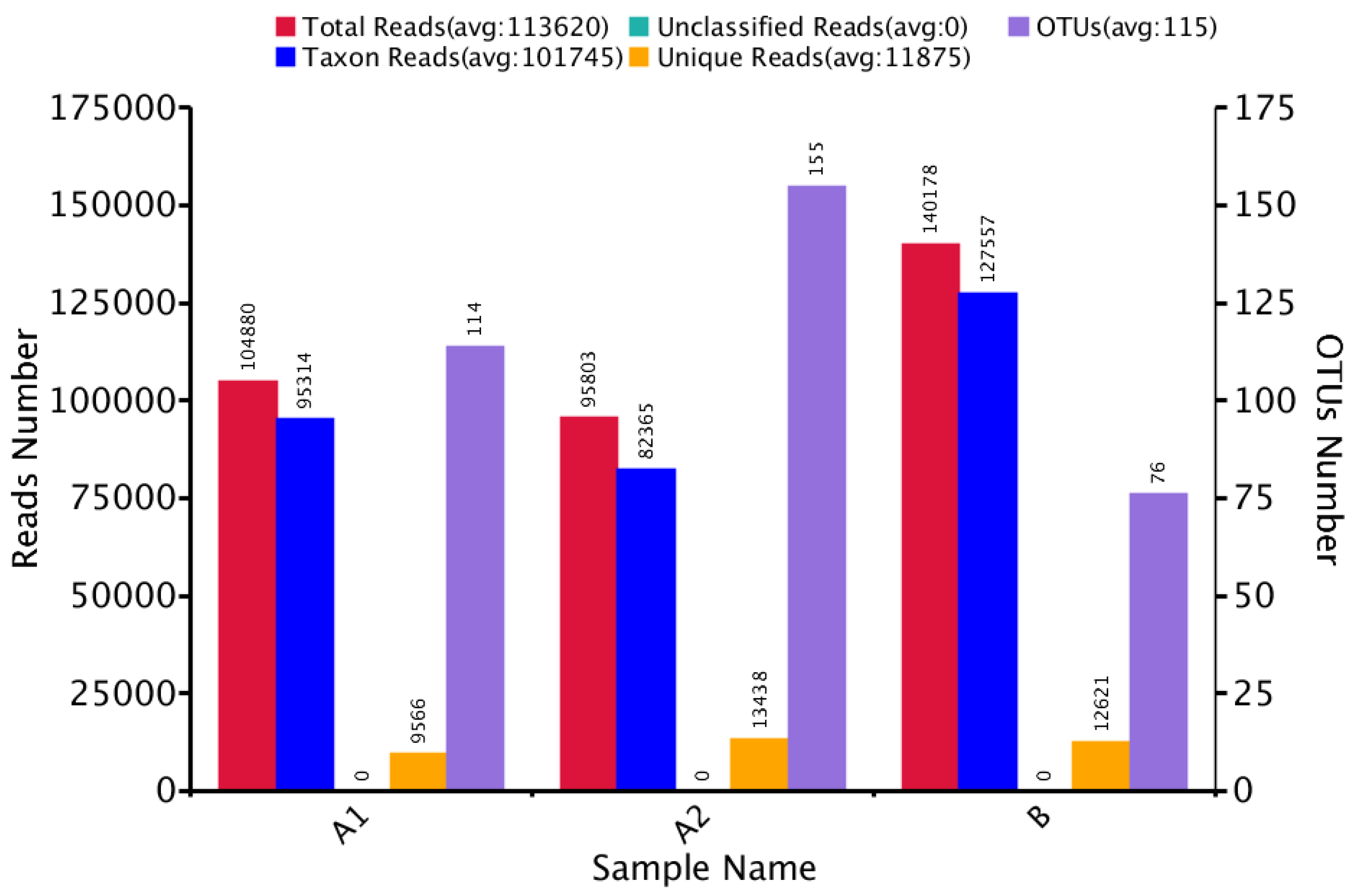
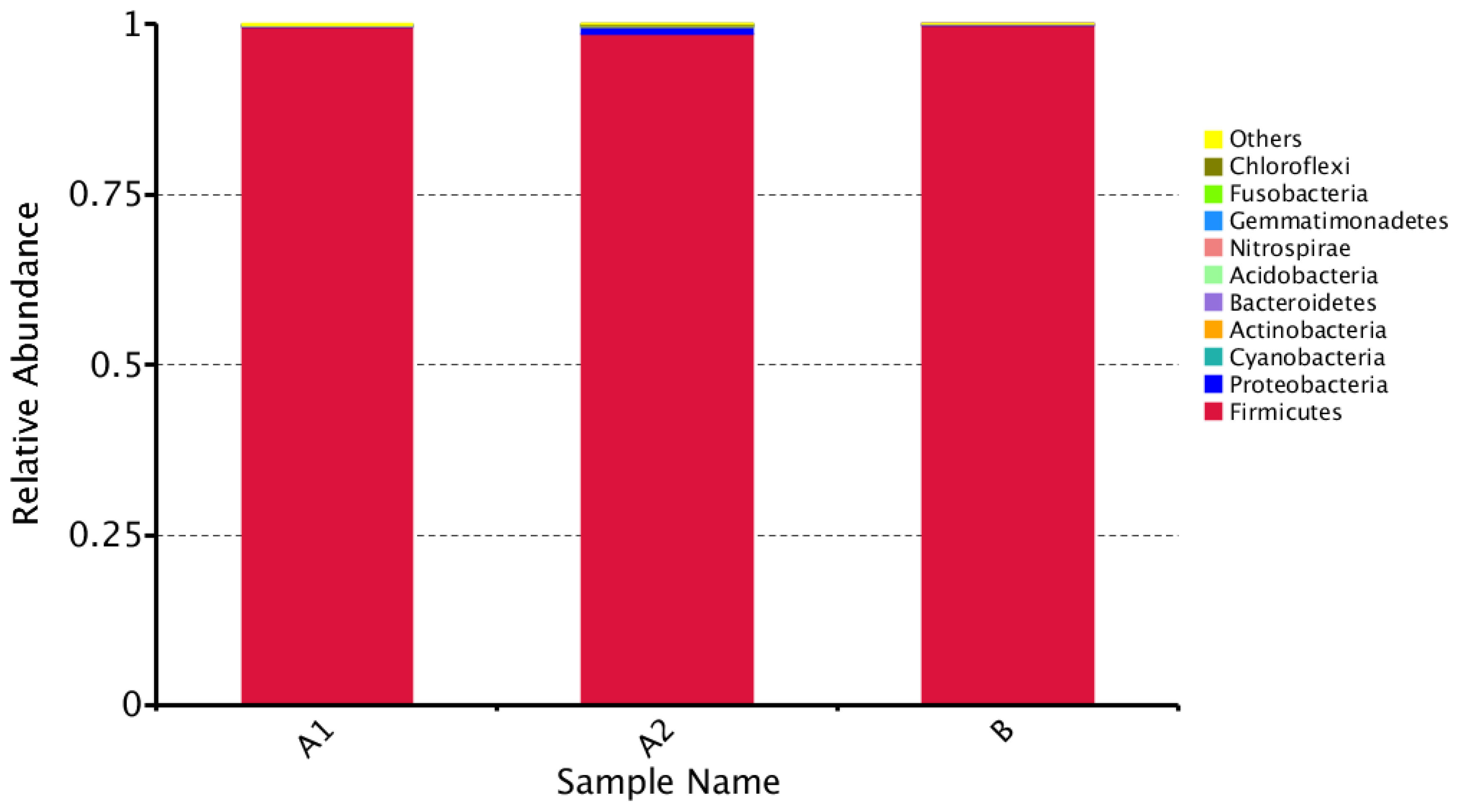
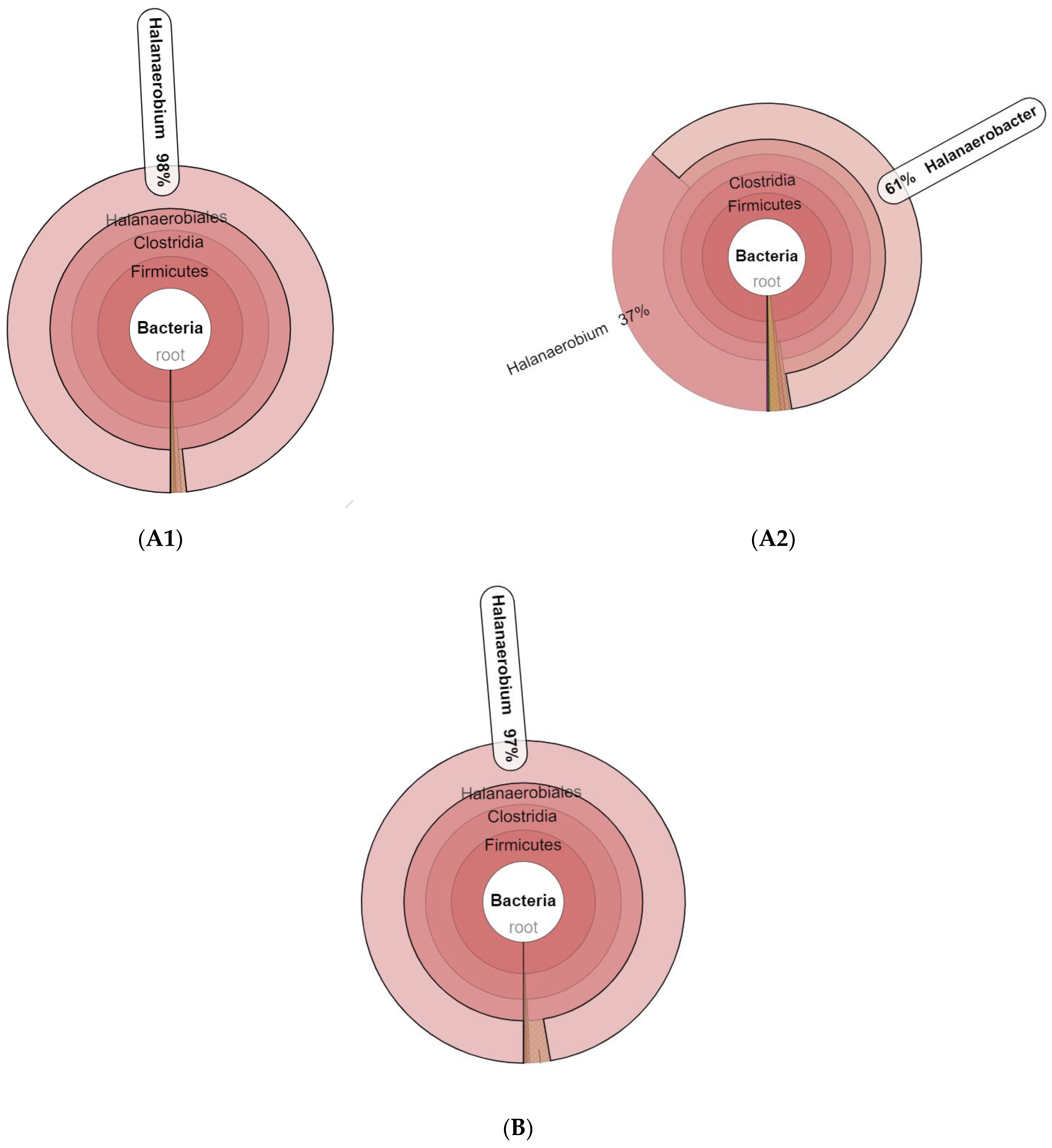
3.1. Microbial Community Amplicon Sequencing
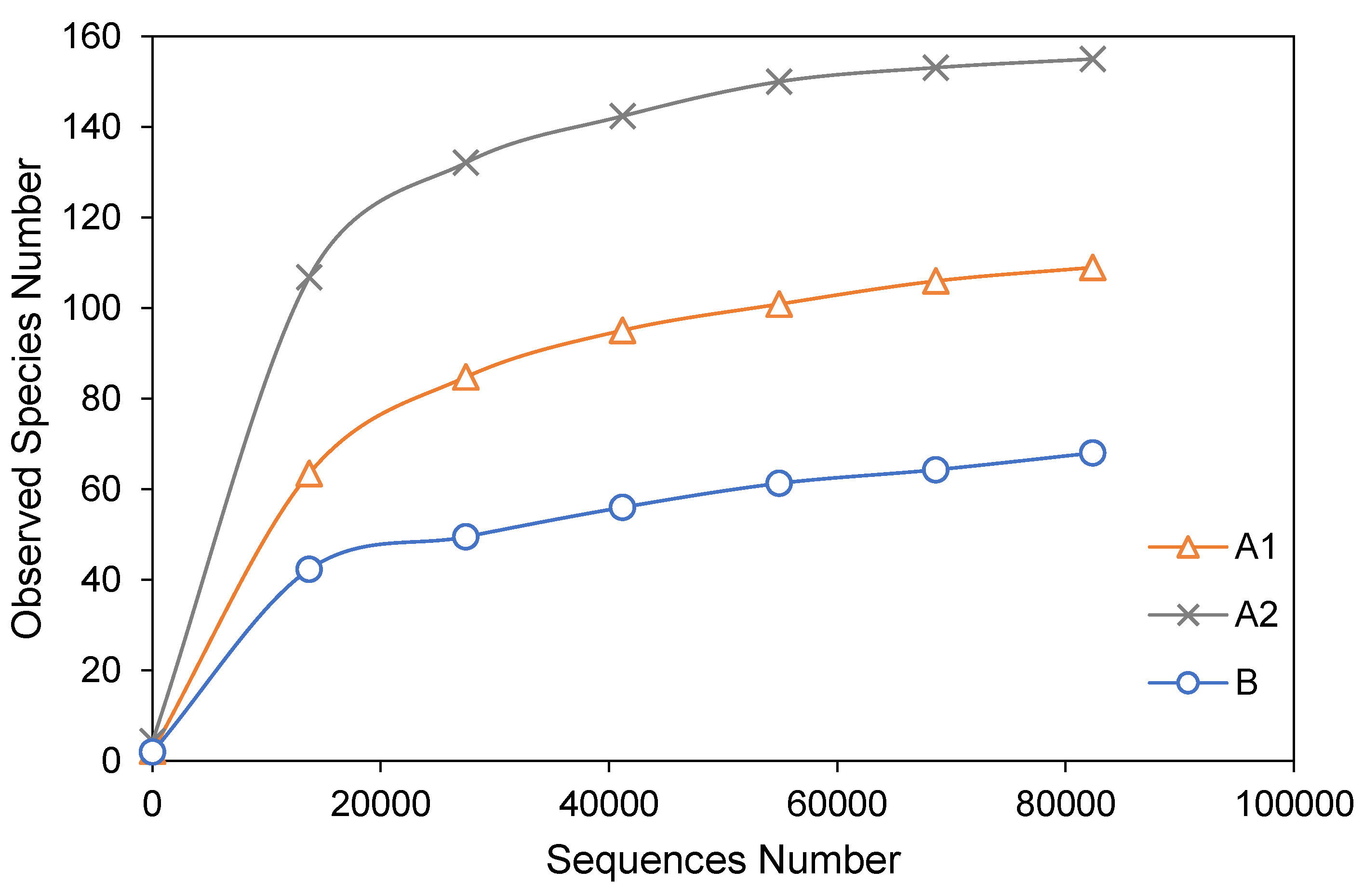
3.2. Alpha Diversity Analysis
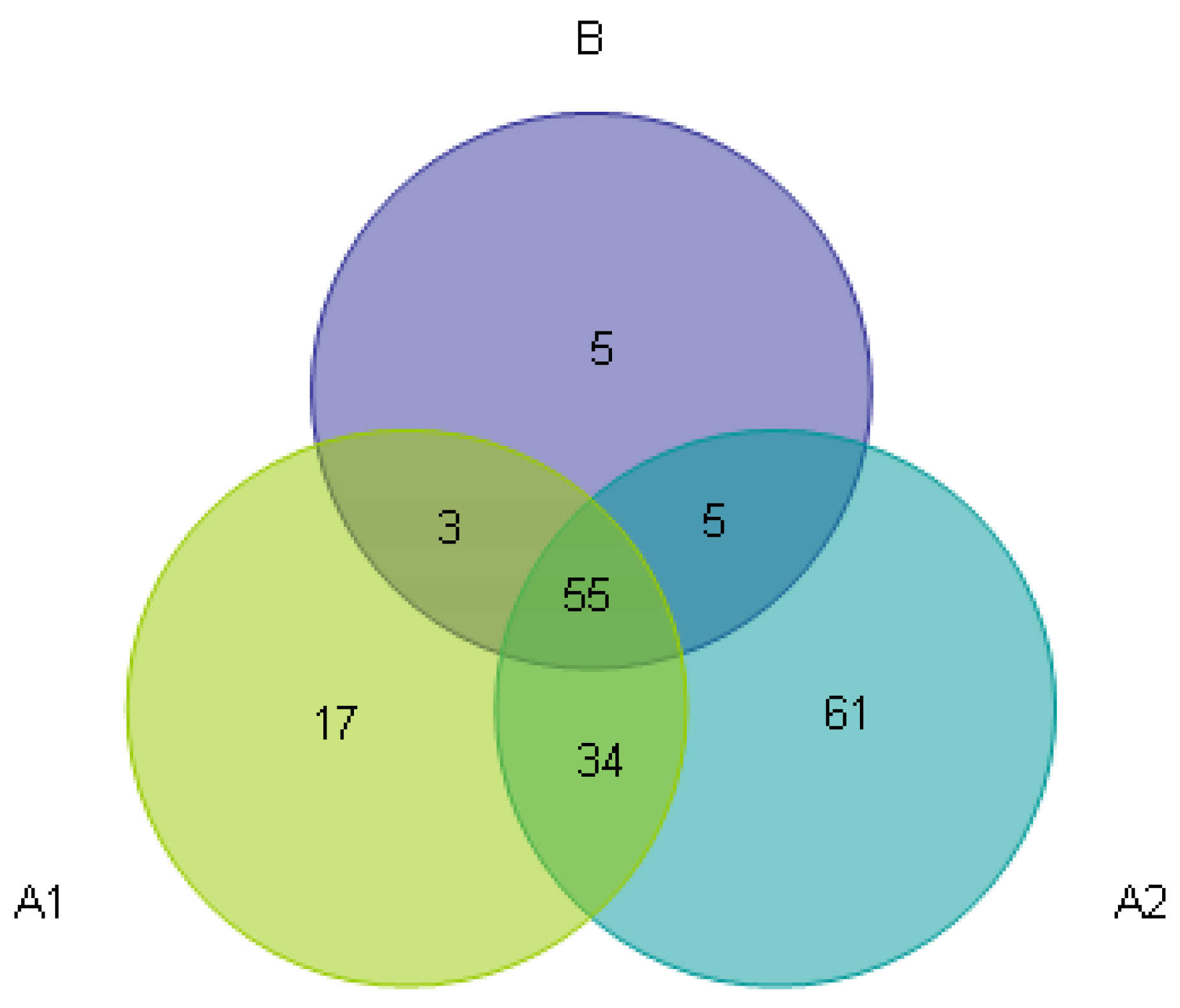
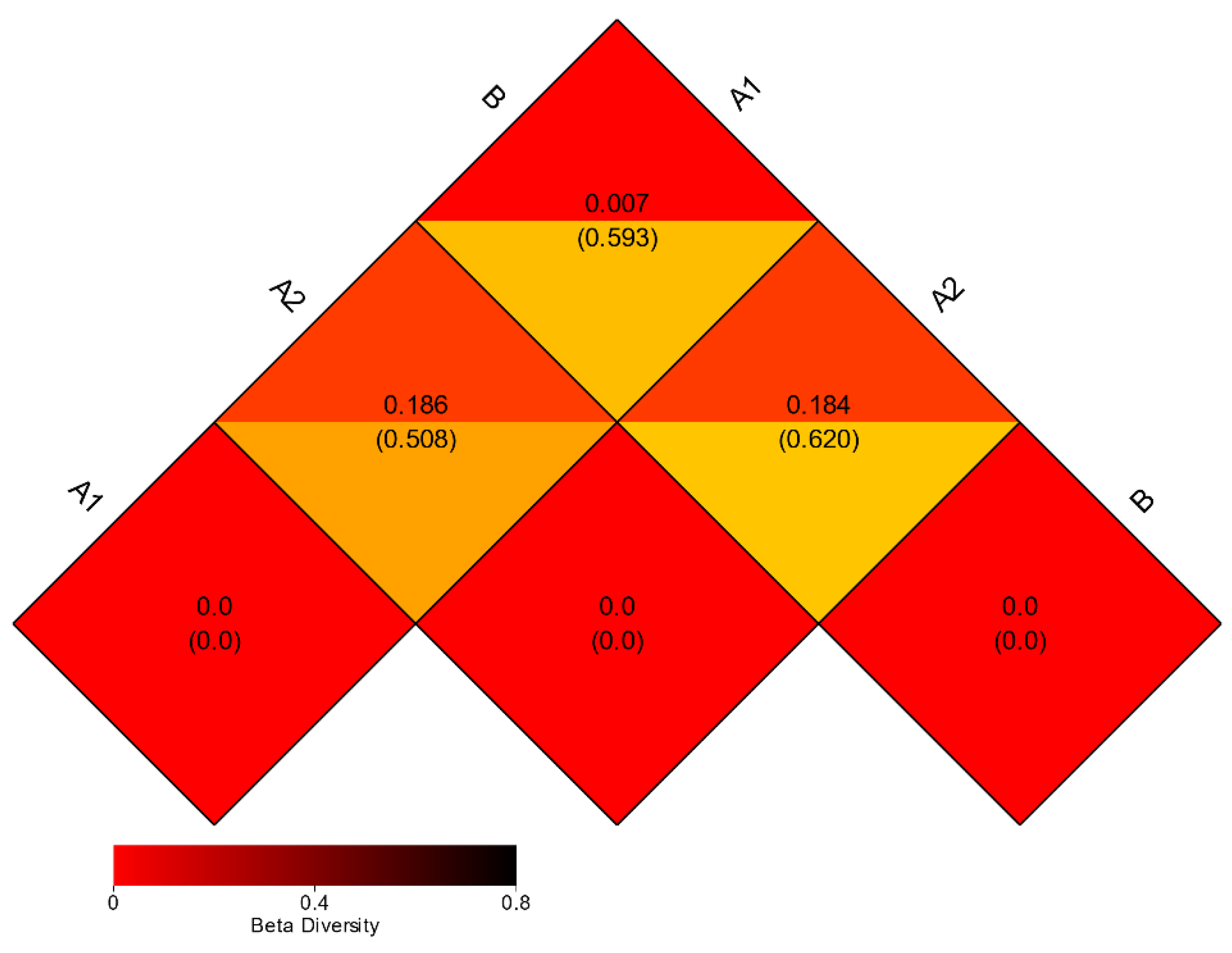
3.3. Beta Diversity Analysis
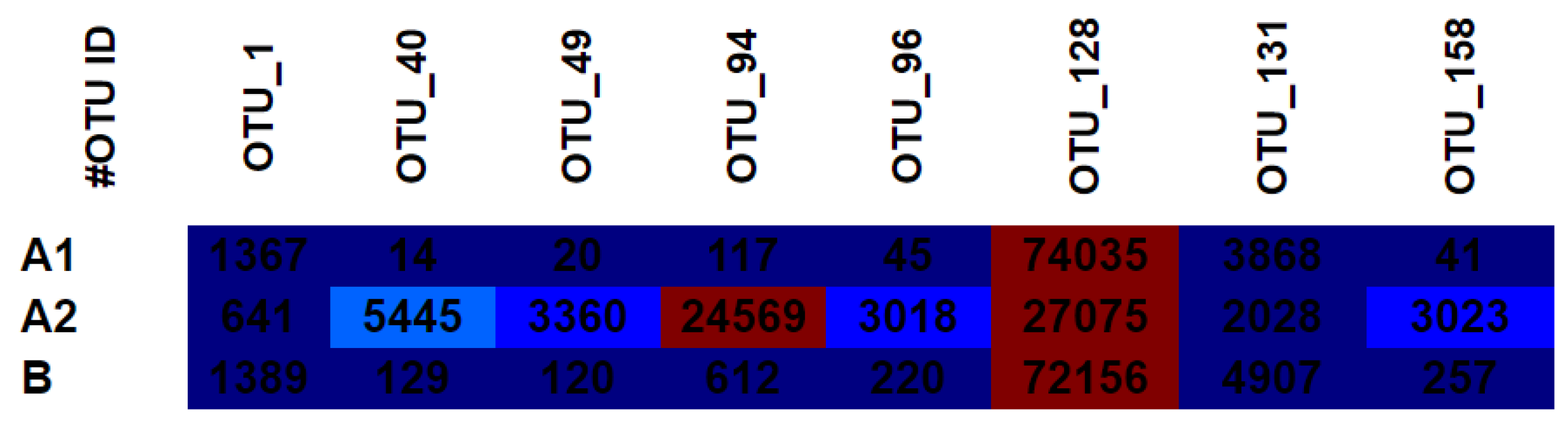
3.4. OTU Heat Map
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sarangi, P.K.; Nanda, S. Biohydrogen Production Through Dark Fermentation. Chem. Eng. Technol. 2020, 43, 601–612. [Google Scholar] [CrossRef]
- Soares, J.F.; Confortin, T.C.; Todero, I.; Mayer, F.D.; Mazutti, M.A. Dark Fermentative Biohydrogen Production from Lignocellulosic Biomass: Technological Challenges and Future Prospects. Renew. Sustain. Energy Rev. 2020, 117, 109484. [Google Scholar] [CrossRef]
- Pachapur, V.L.; Kutty, P.; Pachapur, P.; Brar, S.K.; Le Bihan, Y.; Galvez-Cloutier, R.; Buelna, G. Seed Pretreatment for Increased Hydrogen Production Using Mixed-Culture Systems with Advantages over Pure-Culture Systems. Energies 2019, 12, 530. [Google Scholar] [CrossRef]
- Sivagurunathan, P.; Kumar, G.; Mudhoo, A.; Rene, E.R.; Saratale, G.D.; Kobayashi, T.; Xu, K.; Kim, S.-H.; Kim, D.-H. Fermentative Hydrogen Production Using Lignocellulose Biomass: An Overview of Pre-Treatment Methods, Inhibitor Effects and Detoxification Experiences. Renew. Sustain. Energy Rev. 2017, 77, 28–42. [Google Scholar] [CrossRef]
- Oren, A. Anaerobic Degradation of Organic Compounds at High Salt Concentrations. Antonie Van Leeuwenhoek 1988, 54, 267–277. [Google Scholar] [CrossRef]
- Kivistö, A.; Santala, V.; Karp, M. Hydrogen Production from Glycerol Using Halophilic Fermentative Bacteria. Bioresour. Technol. 2010, 101, 8671–8677. [Google Scholar] [CrossRef]
- Pierra, M.; Trably, E.; Godon, J.J.; Bernet, N. Fermentative Hydrogen Production under Moderate Halophilic Conditions. Int. J. Hydrogen Energy 2013, 9, 1–10. [Google Scholar] [CrossRef]
- Begemann, M.; Mormile, M.; Sitton, O.; Wall, J.; Elias, D. A Streamlined Strategy for Biohydrogen Production with Halanaerobium Hydrogeniformans, an Alkaliphilic Bacterium. Front. Microbiol. 2012, 3, 93. [Google Scholar] [CrossRef]
- Ollivier, B.; Caumette, P.; Garcia, J.L.; Mah, R.A. Anaerobic Bacteria from Hypersaline Environments. Microbiol. Rev. 1994, 58, 27–38. [Google Scholar] [CrossRef]
- García-Depraect, O.; Valdez-Vázquez, I.; Rene, E.R.; Gómez-Romero, J.; López-López, A.; León-Becerril, E. Lactate- and Acetate-Based Biohydrogen Production through Dark Co-Fermentation of Tequila Vinasse and Nixtamalization Wastewater: Metabolic and Microbial Community Dynamics. Bioresour. Technol. 2019, 282, 236–244. [Google Scholar] [CrossRef]
- García-Depraect, O.; Diaz-Cruces, V.F.; Rene, E.R.; León-Becerril, E. Changes in Performance and Bacterial Communities in a Continuous Biohydrogen-Producing Reactor Subjected to Substrate- and PH-Induced Perturbations. Bioresour. Technol. 2020, 295, 122182. [Google Scholar] [CrossRef] [PubMed]
- Goud, R.K.; Arunasri, K.; Yeruva, D.K.; Krishna, K.V.; Dahiya, S.; Mohan, S.V. Impact of Selectively Enriched Microbial Communities on Long-Term Fermentative Biohydrogen Production. Bioresour. Technol. 2017, 242, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Muyzer, G. Genetic Fingerprinting of Microbial Communities–Present Status and Future Perspectives. In Proceedings of the 8th International Symposium on Microbial Ecology, Halifax, NS, Canada, 9–14 August 1998; Atlantic Canada Society for Microbial Ecology, 1999; Volume 1, Available online: http://socrates.acadiau.ca/isme/Symposium16/muyzer.PDF (accessed on 9 June 2021).
- Kumar, G.; Cho, S.-K.; Sivagurunathan, P.; Anburajan, P.; Mahapatra, D.M.; Park, J.-H.; Pugazhendhi, A. Insights into Evolutionary Trends in Molecular Biology Tools in Microbial Screening for Biohydrogen Production through Dark Fermentation. Int. J. Hydrogen Energy 2018, 43, 19885–19901. [Google Scholar] [CrossRef]
- Taroepratjeka, D.A.H.; Imai, T.; Chairattanamanokorn, P. Investigation of Hydrogen-Producing Ability of Extremely Halotolerant Bacteria from a Salt Pan and Salt-Damaged Soil in Thailand. Int. J. Hydrogen Energy 2019, 44, 3407–3413. [Google Scholar] [CrossRef]
- Taroepratjeka, D.A.H.; Imai, T.; Chairattanamanokorn, P.; Reungsang, A. Biohydrogen Production by Extremely Halophilic Bacteria from the Salt Pan of Samut Sakhon, Thailand. Chiang Mai J. Sci. 2020, 47, 378–390. [Google Scholar]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME Improves Sensitivity and Speed of Chimera Detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing Mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools|Nucleic Acids Research|Oxford Academic. Available online: https://academic.oup.com/nar/article/41/D1/D590/1069277?login=true (accessed on 31 January 2021).
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive Metagenomic Visualization in a Web Browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef]
- Chao, A.; Hwang, W.-H.; Chen, Y.-C.; Kuo, C.-Y. Estimating the Number of Shared Species in Two Communities. Stat. Sin. 2000, 10, 227–246. [Google Scholar]
- Faith, D.P. Conservation Evaluation and Phylogenetic Diversity. Biol. Conserv. 1992, 61, 1–10. [Google Scholar] [CrossRef]
- Kim, B.-R.; Shin, J.; Guevarra, R.; Lee, J.H.; Kim, D.W.; Seol, K.-H.; Lee, J.-H.; Kim, H.B.; Isaacson, R. Deciphering Diversity Indices for a Better Understanding of Microbial Communities. J. Microbiol. Biotechnol. 2017, 27, 2089–2093. [Google Scholar] [CrossRef]
- Nam, Y.-D.; Lee, S.-Y.; Lim, S.-I. Microbial Community Analysis of Korean Soybean Pastes by Next-Generation Sequencing. Int. J. Food Microbiol. 2012, 155, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Pylro, V.S.; Roesch, L.F.W.; Morais, D.K.; Clark, I.M.; Hirsch, P.R.; Tótola, M.R. Data Analysis for 16S Microbial Profiling from Different Benchtop Sequencing Platforms. J. Microbiol. Methods 2014, 107, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Taroepratjeka, D.A.H. A Study of Biohydrogen Production by Extremely Halophilic Bacterial Communities from a Salt Pan and Salt Damaged Soil. Ph.D. Thesis, Yamaguchi University, Yamaguchi, Japan, March 2020. [Google Scholar]
- Xia, Q.; Rufty, T.; Shi, W. Soil Microbial Diversity and Composition: Links to Soil Texture and Associated Properties. Soil Biol. Biochem. 2020, 149, 107953. [Google Scholar] [CrossRef]
- Vos, P.; Garrity, G.; Jones, D.; Krieg, N.R.; Ludwig, W.; Rainey, F.A.; Schleifer, K.-H.; Whitman, W.B. Bergey’s Manual of Systematic Bacteriology: Volume 3: The Firmicutes; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2011; ISBN 978-0-387-68489-5. [Google Scholar]
- Moune, S.; Manac’h, N.; Hirschler, A.; Caumette, P.; Willison, J.C.; Matheron, R. Haloanaerobacter Salinarius Sp. Nov., a Novel Halophilic Fermentative Bacterium That Reduces Glycine-Betaine to Trimethylamine with Hydrogen or Serine as Electron Donors; Emendation of the Genus Haloanaerobacter. Int. J. Syst. Bacteriol. 1999, 49, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Liaw, H.J.; Mah, R.A. Isolation and Characterization of Haloanaerobacter Chitinovorans Gen. Nov., Sp. Nov., a Halophilic, Anaerobic, Chitinolytic Bacterium from a Solar Saltern. Appl. Environ. Microbiol. 1992, 58, 260–266. [Google Scholar] [CrossRef]
- Kobayashi, T.; Kimura, B.; Fujii, T. Haloanaerobium Fermentans Sp. Nov., a Strictly Anaerobic, Fermentative Halophile Isolated from Fermented Puffer Fish Ovaries. Int. J. Syst. Evol. Microbiol. 2000, 50, 1621–1627. [Google Scholar] [CrossRef]
- Marui, J.; Boulom, S.; Panthavee, W.; Momma, M.; Kusumoto, K.-I.; Nakahara, K.; Saito, M. Culture-Independent Bacterial Community Analysis of the Salty-Fermented Fish Paste Products of Thailand and Laos. Biosci. Microbiota Food Health 2015, 2014–2018. [Google Scholar] [CrossRef]
- Kim, M.-S.; Park, E.-J. Bacterial Communities of Traditional Salted and Fermented Seafoods from Jeju Island of Korea Using 16S RRNA Gene Clone Library Analysis. J. Food Sci. 2014, 79, M927–M934. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, F.D.; Palmigiano, A.; Paciello, I.; Pallach, M.; Garozzo, D.; Bernardini, M.-L.; Cono, V.L.; Yakimov, M.M.; Molinaro, A.; Silipo, A. The Deep-Sea Polyextremophile Halobacteroides Lacunaris TB21 Rough-Type LPS: Structure and Inhibitory Activity towards Toxic LPS. Mar. Drugs 2017, 15, 201. [Google Scholar] [CrossRef] [PubMed]
- Mezghani, M.; Alazard, D.; Karray, F.; Cayol, J.-L.; Joseph, M.; Postec, A.; Fardeau, M.-L.; Tholozan, J.-L.; Sayadi, S. Halanaerobacter Jeridensis Sp. Nov., Isolated from a Hypersaline Lake. Int. J. Syst. Evol. Microbiol. 2012, 62, 1970–1973. [Google Scholar] [CrossRef] [PubMed]
- Varrella, S.; Tangherlini, M.; Corinaldesi, C. Deep Hypersaline Anoxic Basins as Untapped Reservoir of Polyextremophilic Prokaryotes of Biotechnological Interest. Mar. Drugs 2020, 18, 91. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Wang, J. Pretreatment of Grass Waste Using Combined Ionizing Radiation-Acid Treatment for Enhancing Fermentative Hydrogen Production. Bioresour. Technol. 2018, 255, 7–15. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Observed Species | Simpson | Shannon– Wiener | ACE | Chao1 | Good’s Coverage | PD Whole Tree |
|---|---|---|---|---|---|---|---|
| A1 | 109 | 0.189 | 0.777 | 117.204 | 118.231 | 1.000 | 13.401 |
| A2 | 155 | 0.791 | 3.152 | 156.894 | 155.652 | 1.000 | 14.512 |
| B | 68 | 0.228 | 0.904 | 96.987 | 99.667 | 1.000 | 6.243 |
| OTU Number | Sequence Length (bp) | Closest Relative (NCBI Accession Number) | Similarity |
|---|---|---|---|
| OTU_128 | 448 | Halanaerobium fermentans R-9 (NR 024715.1) | 99.55% |
| OTU_94 | 447 | Halanaerobacter lacunarum TB21 (KJ677978.1) | 97.45% |
| OTU_40 | 449 | Halanaerobacter lacunarum TB21 (KJ677978.1) | 97.51% |
| OTU_49 | 450 | Halanaerobacter lacunarum TB21 (KJ677978.1) | 99.03% |
| OTU_158 | 438 | Halanaerobacter lacunarum TB21 (KJ677978.1) | 96.79% |
| OTU_96 | 438 | Halanaerobacter lacunarum TB21 (KJ677978.1) | 96.80% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taroepratjeka, D.A.H.; Imai, T.; Chairattanamanokorn, P.; Reungsang, A. Extremely Halophilic Biohydrogen Producing Microbial Communities from High-Salinity Soil and Salt Evaporation Pond. Fuels 2021, 2, 241-252. https://doi.org/10.3390/fuels2020014
Taroepratjeka DAH, Imai T, Chairattanamanokorn P, Reungsang A. Extremely Halophilic Biohydrogen Producing Microbial Communities from High-Salinity Soil and Salt Evaporation Pond. Fuels. 2021; 2(2):241-252. https://doi.org/10.3390/fuels2020014
Chicago/Turabian StyleTaroepratjeka, Dyah Asri Handayani, Tsuyoshi Imai, Prapaipid Chairattanamanokorn, and Alissara Reungsang. 2021. "Extremely Halophilic Biohydrogen Producing Microbial Communities from High-Salinity Soil and Salt Evaporation Pond" Fuels 2, no. 2: 241-252. https://doi.org/10.3390/fuels2020014
APA StyleTaroepratjeka, D. A. H., Imai, T., Chairattanamanokorn, P., & Reungsang, A. (2021). Extremely Halophilic Biohydrogen Producing Microbial Communities from High-Salinity Soil and Salt Evaporation Pond. Fuels, 2(2), 241-252. https://doi.org/10.3390/fuels2020014