
Why Does Inflammation Result in Resorptive Bone Loss? What the Study of Burns Teaches Us


{kind=link}
Highlights
- Inflammation promotes bone resorption by triggering immune cells to release cytokines, which in turn stimulate osteoblasts and osteocytes to produce RANKL (receptor activator of nuclear factor kappa-B ligand), a key factor that enhances osteoclastogenesis.
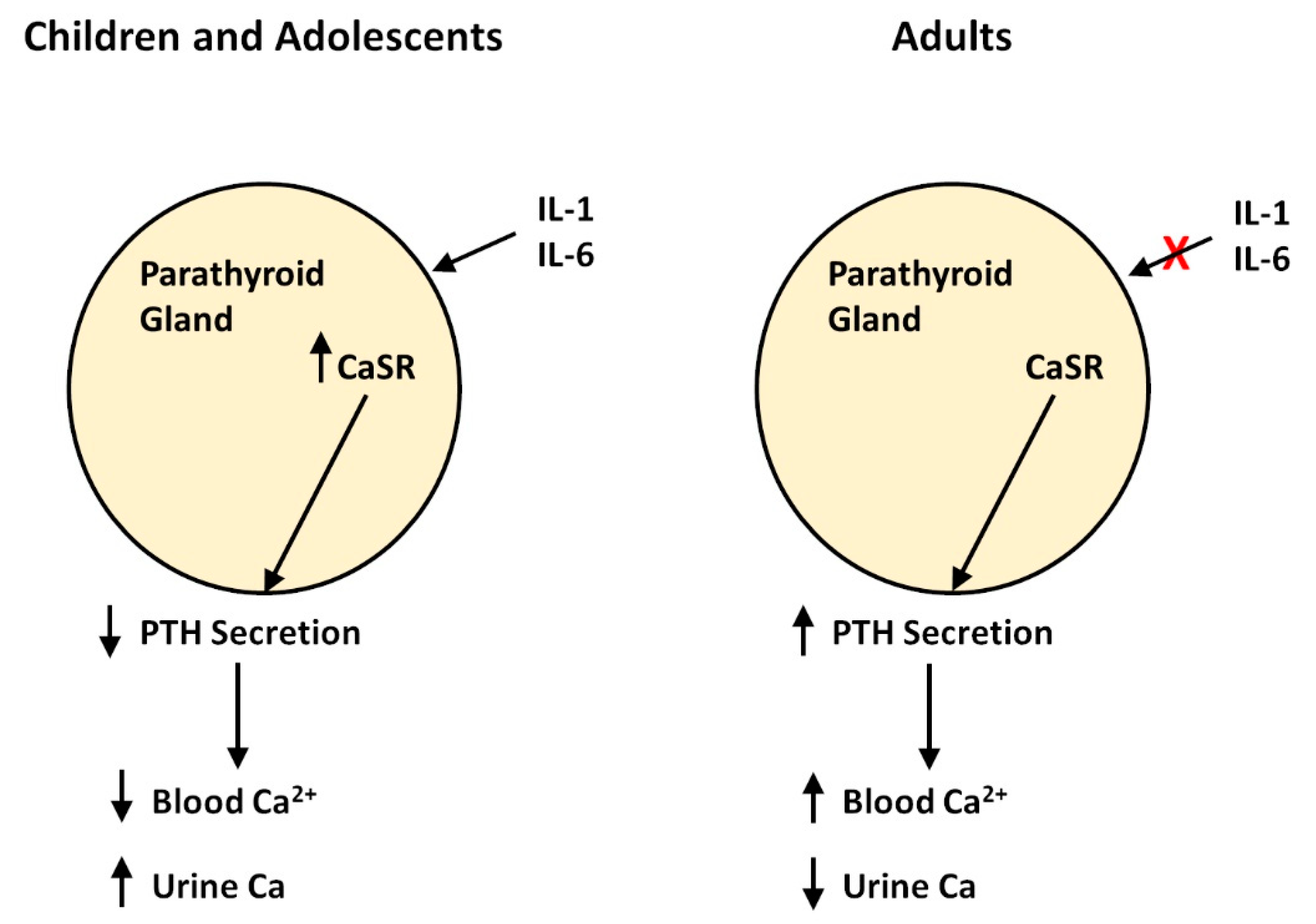
- Bone resorption releases calcium and phosphate into the bloodstream. The calcium-sensing receptor in the parathyroid detects the elevated calcium levels and signals the chief parathyroid cells to reduce the secretion of the parathyroid hormone. In children, this results in a hypoparathyroid state, which leads to increased urinary excretion of excess calcium.
- In adults, the calcium-sensing receptor on the parathyroid gland loses its ability to suppress parathyroid hormone secretion in response to excess calcium released by bone, resulting in the accumulation of calcium in the bloodstream. The cause of this dysfunction remains unclear.
- Extracellular calcium can stimulate peripheral blood mononuclear cells to produce chemokines and upregulate the NLRP3 inflammasome, thereby amplifying or sustaining the inflammatory response. This calcium, released from bones during resorption, acts as a fuel for inflammation.
Abstract
1. Introduction
2. Extracellular Calcium and Inflammation
3. Preservation of the Hypocalcemic Response in Small Burns
4. Developmental Disappearance of Calcium-Sensing Receptor Response to Inflammatory Cytokines
5. Potential Implications of the Disappearance of Calcium-Sensing Receptor Response to Inflammatory Cytokines
6. Other Clinical Conditions
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Klein, G.L.; Herndon, D.N.; Goodman, W.G.; Langman, C.B.; Phillips, W.A.; Dickson, I.R.; Eastell, R.; Naylor, K.; Maloney, N.; Desai, M.; et al. Histomorphometric and biochemical characterization of bone following acute severe burns in children. Bone 1995, 17, 455–460. [Google Scholar] [CrossRef]
- Klein, G.L.; Bi, L.X.; Sherrard, D.J.; Beavan, S.R.; Ireland, D.; Compston, J.E.; Williams, W.G.; Herndon, D.N. Evidence supporting a role of glucocorticoids in short-term bone loss in burned children. Osteoporos. Int. 2004, 15, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, P.K.; Rasmussen, A.K.; Butters, R.; Feldt-Rasmussen, U.; Bendtzen, K.; Diaz, R.; Brown, E.M.; Olgaard, K. Inhibition of PTH secretion by interleukin-1 beta in bovine parathyroid glands in vitro is associated with an up-regulation of the calcium-sensing receptor mRNA. Biochem. Biophys. Res. Commun. 1997, 238, 880–885. [Google Scholar] [CrossRef] [PubMed]
- Toribio, R.E.; Kohn, C.W.; Capen, C.C.; Rosol, T.J. Parathyroid hormone (PTH) secretion, PTH mRNA and calcium-sensing receptor mRNA expression in equine parathyroid cells and effects of interleukin (IL)-1, IL-6 and tumor necrosis factor alpha on equine parathyroid cell function. J. Mol. Endocrinol. 2003, 31, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Canaff, L.; Zhou, X.; Hendy, G.N. The proinflammatory cytokine, interleukin-6, up-regulates calcium-sensing receptor gene transcription via Stat1/3 and Sp 1/3. J. Biol. Chem. 2008, 283, 13586–13600. [Google Scholar] [CrossRef]
- Riccardi, D.; Brown, E.M. Physiology and pathophysiology of the calcium-sensing receptor in the kidney. Am. J. Physiol. Renal. Physiol. 2010, 298, F485–F499. [Google Scholar] [CrossRef]
- Goltzman, D.; Hendy, G.N. The calcium-sensing receptor in bone- mechanistic and therapeutic insights. Nat. Rev. Endocrinol. 2015, 11, 298–307. [Google Scholar] [CrossRef]
- Tang, L.; Cheng, C.Y.; Sun, X.; Pedicone, A.J.; Mohamadzadeh, M.; Cheng, S.X. The extracellular calcium-sensing receptor in the intestine: Evidence for regulation of colonic absorption, secretion, motility and immunity. Front. Physiol. 2016, 7, 245. [Google Scholar]
- Klein, G.L.; Enkhbaatar, P.; Traber, D.L.; Buja, M.; Jonkam, C.C.; Poindexter, B.J.; Bick, R.J. Cardiovascular distribution of the calcium-sensing receptor before and after burns. Burns 2008, 34, 370–375. [Google Scholar] [CrossRef]
- Schepelmann, M.; Yarova, P.L.; Lopez-Fernandez, I.; Davies, T.S.; Brennan, S.C.; Edwards, P.J.; Aggarwal, A.; Graça, J.; Rietdorf, K.; Matchkov, V.; et al. The vascular Ca2+-sensing receptor regulates blood vessel tone and blood pressure. Am. J. Physiol. Cell. Physiol. 2016, 310, C103–C204. [Google Scholar] [CrossRef]
- Murphey, E.D.; Chattopadhyay, N.; Bai, M.; Kifor, O.; Harper, D.; Traber, D.L.; Hawkins, H.K.; Brown, E.M.; Klein, G.L. Up-regulation of the parathyroid calcium-sensing receptor after burn injury in sheep: A potential contributory factor to postburn hypocalcemia. Crit. Care Med. 2000, 28, 3885–3890. [Google Scholar] [CrossRef] [PubMed]
- Klein, G.L.; Nicolai, M.; Langman, C.B.; Cuneo, B.F.; Sailer, D.E.; Herndon, D.N. Dysregulation of calcium homeostasis after burn injury in children: Possible role of magnesium depletion. J. Pediatr. 1997, 131, 246–251. [Google Scholar] [CrossRef]
- Klein, G.L.; Xie, Y.; Qin, Y.-X.; Lin, L.; Hu, M.; Enkhbaatar, P.; Bonewald, L.F. Preliminary evidence of bone resorption in a sheep model of acute burn injury: An observational study. J. Bone Miner. Metab. 2014, 32, 136–141. [Google Scholar] [CrossRef]
- Chattopadhyay, N.; Mithal, A.; Brown, E.M. The calcium-sensing receptor: A window into the physiology and pathophysiology of mineral ion metabolism. Endocr. Rev. 1996, 17, 289–307. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Klein, G.L.; Castro, S.M.; Garofalo, R.P. The calcium-sensing receptor as a mediator of inflammation. Semin. Cell Dev. Biol. 2016, 49, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Rossol, M.; Pierer, M.; Raulien, N.; Quandt, D.; Meusch, U.; Rothe, K.; Schubert, K.; Schöneberg, T.; Schaefer, M.; Krügel, U.; et al. Extracellular Ca2+ is a danger signal activating NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat. Commun. 2012, 3, 1329. [Google Scholar] [CrossRef] [PubMed]
- Jager, E.; Murthy, S.; Schmidt, C.; Hahn, M.; Strobel, S.; Peters, A.; Stäubert, C.; Sungur, P.; Venus, T.; Geisler, M.; et al. Calcium-sensing-receptor-mediated NLRP3 inflammasome response to calciprotein particles drives inflammation in rheumatoid arthritis. Nat. Commun. 2020, 11, 4243. [Google Scholar] [CrossRef] [PubMed]
- Olszak, I.T.; Poznansky, M.C.; Evans, R.H.; Olson, D.; Kos, C.; Pollak, M.R.; Brown, E.M.; Scadden, D.T. Extracellular calcium elicits a chemokinetic response from monocytes in vitro and in vivo. J. Clin. Investig. 2000, 105, 1299–1305. [Google Scholar] [CrossRef]
- Michalick, L.; Kuebler, W.M. TRPV4—A missing link between mechanosensation and immunity. Front. Immunol. 2020, 11, 413. [Google Scholar] [CrossRef]
- Gugala, Z.; Cacciani, N.; Klein, G.L.; Larsson, L. Acute and severe trabecular bone loss in a rat model of critical illness myopathy.J. Orthop. Res. 2022, 40, 1293–1300. [Google Scholar] [CrossRef]
- Klein, G.L.; Herndon, D.N.; Langman, C.B.; Rutan, T.C.; Young, W.E.; Pembleton, G.; Nusynowitz, M.; Barnett, J.L.; Broemeling, L.D.; Sailer, D.E.; et al. Long-term reduction in bone mass after severe burn injury in children. J. Pediatr. 1995, 126, 252–256. [Google Scholar] [CrossRef]
- Klein, G.L.; Wimalawansa, S.J.; Kulkarni, G.; Sherrard, D.J.; Sanford, A.P.; Herndon, D.N. The efficacy of the acute administration of pamidronate on the conservation of bone mass following severe burn injury in children: A double-blind, randomized controlled study. Osteoporos. Int. 2005, 16, 631–635. [Google Scholar] [CrossRef]
- Klein, G.L.; Herndon, D.N.; Rutan, T.C.; Sherrard, D.J.; Coburn, J.W.; Langman, C.B.; Thomas, M.L.; Haddad, J.G., Jr.; Cooper, C.W.; Miller, N.L.; et al. Bone disease in burn patients. J. Bone Miner. Res. 1993, 8, 337–345. [Google Scholar] [CrossRef] [PubMed]
- Klein, G.L.; Benjamin, D.A.; Herndon, D.N. Calcemic response to burns differs between adults and children: A review of the literature. Osteoporos. Sarcopenia 2017, 3, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, A.F.; Foidart-DeSalle, M.; Ledoux, D.; Remy, C.; Croisier, J.L.; Damas, P.; Cavalier, E. Effects of cholecalciferol supplementation and optimized calcium intakes on vitamin D status, muscle strength and bone health: A one-year pilot randomized controlled trial in adults with severe burns. Burns 2015, 41, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, M.G.; Pinto, P.; Kraft, R.; Nathens, A.B.; Finnerty, C.C.; Gamelli, R.L.; Gibran, N.S.; Klein, M.B.; Arnoldo, B.D.; Tompkins, R.G.; et al. Morbidity and survival probability in burn patients in modern burn care. Crit. Care Med. 2015, 43, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Hundeshagen, G.; Herndon, D.N.; Clayton, R.P.; Wurzer, P.; McQuitty, A.; Jennings, K.; Branski, L.K.; Collins, V.N.; Marques, N.R.; Finnerty, C.C.; et al. Long-term effect of critical illness after burn injury on cardiac function in adolescent survivors: An observational study. Lancet Child Adolesc. Health 2017, 1, 293–301. [Google Scholar] [CrossRef]
- Pin, F.; Bonetto, A.; Bonewald, L.F.; Klein, G.L. Molecular mechanisms responsible for the rescue effects of pamidronate on muscle atrophy in pediatric burn patients. Front. Endocrinol. 2019, 10, 543. [Google Scholar] [CrossRef]
- Borsheim, E.; Herndon, D.N.; Hawkins, H.K.; Suman, O.E.; Cotter, M.; Klein, G.L. Pamidronate attenuates muscle loss after pediatric burn injury. J. Bone Miner. Res. 2014, 29, 1369–1372. [Google Scholar] [CrossRef]
- Waning, D.L.; Mohammad, K.S.; Reiken, S.; Xie, W.; Andersson, D.C.; John, S.; Chiechi, A.; Wright, L.E.; Umanskaya, A.; Niewolna, M.; et al. Excess TGF-β mediates muscle weakness associated with bone metastases in mice. Nat. Med. 2015, 21, 1262–1271. [Google Scholar] [CrossRef]
- Abrigo, J.; Simon, F.; Cabrera, D.; Cordova, G.; Trollet, C.; Cabello-Verrugio, C. Central role of transforming growth factor-beta 1 in skeletal muscle dysfunctions: An update on therapeutic strategies. Curr. Protein Pept. Sci. 2018, 19, 1189–1200. [Google Scholar] [CrossRef] [PubMed]
- Alexander, W.; Schneider, H.-G.; Smith, C.; Cleland, H. The incidence and significance of raised troponin levels in acute burns. J. Burn Care Res. 2018, 39, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Campos-Obando, N.; Kavousi, M.; Roeters van Lennep, J.E.; Rivadeneira, F.; Hofman, A.; Uitterlinden, A.G.; Franco, O.; Zillikens, M.C. Bone health and coronary artery calcification: The Rotterdam Study. Atherosclerosis 2015, 241, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Bauman, W.A.; Cardozo, C. Bone and muscle loss after spinal cord injury: Organ interactions. Ann. N. Y. Acad. Sci. 2010, 1211, 66–84. [Google Scholar] [CrossRef]
- Peterson, M.D.; Berri, M.; Lin, P.; Kamdar, N.; Rodriguez, G.; Mahmoudi, E.; Tate, D. Cardiovascular and metabolic morbidity following spinal cord injury. Spine J. 2021, 21, 1520–1527. [Google Scholar] [CrossRef]
- Porter, C.; Sousse, L.E.; Irick, R.; Schryver, E.; Klein, G.L. Interactions of phosphate with serious injury including burns. JBMR Plus 2017, 1, 59–65. [Google Scholar] [CrossRef]
- Del Rivero, T.; Bethea, J. The effects of spinal cord injury on bone loss and dysregulation of the calcium/parathyroid hormone loop in mice. Osteoporos. Sarcopenia 2016, 2, 164–169. [Google Scholar] [CrossRef]
- Bauman, W.A.; Spungeon, A.M. Coronary artery disease in individuals with spinal cord injury: Assessment of risk factors. Spinal Cord 2008, 46, 466–476. [Google Scholar] [CrossRef]
- Lieberman, J.A.; Hammond, F.M.; Barringer, T.A.; Norton, H.J.; Goff, D.C., Jr.; Bockenek, W.L.; Scelza, W.M. Comparison of coronary artery calcification scores and National Cholesterol Education program guidelines for coronary heart disease assessment and treatment paradigms in individuals with chronic traumatic spinal cord injury. J. Spinal Cord Med. 2011, 34, 233–240. [Google Scholar] [CrossRef]
- Karpouzas, G.A.; Ormseth, S.R.; Hernandez, E.; Budoff, M.J. Impact of cumulative inflammation, cardiac risk factors and medication exposure on coronary atherosclerosis progression in rheumatoid arthritis. Arthritis Rheumatol. 2020, 72, 400–408. [Google Scholar] [CrossRef]
- Houri Levi, E.; Watad, A.; Whitby, A.; Tiosano, S.; Comaneshter, D.; Cohen, A.D.; Amital, H. Coexistence of ischemic heart disease and rheumatoid arthritis patients- A case controlled study. Autoimmun. Rev. 2016, 15, 393–396. [Google Scholar] [CrossRef]
- Macedo, M.B.; Ostrovski Sousa Santos, V.M.; Rodrigues Pereira, R.M.; Fuller, R. Association between osteoarthritis and atherosclerosis: A systematic review and meta-analysis. Exp. Gerontol. 2022, 161, 111734. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Bai, J.; He, B.; Hu, X.; Liu, D. Osteoarthritis and the risk of cardiovascular disease: A meta-analysis of observational studies. Sci. Rep. 2016, 6, 39672. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.J.; Stubbs, B.; Mamas, M.A.; Myint, P.K.; Smith, T.O. Association between osteoarthritis and cardiovascular disease: Systematic review and meta-analysis. Eur. J. Prev. Cardiol. 2016, 23, 938–946. [Google Scholar] [CrossRef]
- Suetomi, T.; Willeford, A.; Brand, C.S.; Cho, Y.; Ross, R.S.; Miyamoto, S.; Brown, J.H. Inflammation and NLRP3 inflammasome activation initiated in response to pressure overload by Ca2+/calmodulin-dependent protein kinase II δ signaling in cardiomyocytes are essential for adverse cardiac remodeling. Circulation 2018, 138, 2530–2544. [Google Scholar] [CrossRef]
- Yu, J.; Chen, Y.; Xu, M.; Sun, L.; Luo, H.; Bao, X.; Meng, G.; Zhang, W. Ca2+/Calmodulin-dependent protein kinase II regulation by Inhibitor 1 of Protein Phosphatase 1 protects against ischemia-reperfusion injury. J. Cardiovasc. Pharmacol. Ther. 2019, 24, 460–473. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klein, G.L. Why Does Inflammation Result in Resorptive Bone Loss? What the Study of Burns Teaches Us. Endocrines 2022, 3, 452-459. https://doi.org/10.3390/endocrines3030036
Klein GL. Why Does Inflammation Result in Resorptive Bone Loss? What the Study of Burns Teaches Us. Endocrines. 2022; 3(3):452-459. https://doi.org/10.3390/endocrines3030036
Chicago/Turabian StyleKlein, Gordon L. 2022. "Why Does Inflammation Result in Resorptive Bone Loss? What the Study of Burns Teaches Us" Endocrines 3, no. 3: 452-459. https://doi.org/10.3390/endocrines3030036
APA StyleKlein, G. L. (2022). Why Does Inflammation Result in Resorptive Bone Loss? What the Study of Burns Teaches Us. Endocrines, 3(3), 452-459. https://doi.org/10.3390/endocrines3030036