Abstract
Despite the improvements in diagnostic and therapeutic approaches, breast cancer still remains one of the world’s leading causes of death among women. Particularly, triple negative breast cancer (TNBC) is characterized by aggressiveness, metastatic spreading, drug resistance and a very high percentage of death in patients. Nowadays, identification of new targets in TNBC appears very compelling. TNBC are considered negative for the estrogen receptor alpha (ERα) expression. Nevertheless, they often express ERβ and its variants. As such, this TNBC subtype still responds to estrogens. While the ERβ1 variant seems to act as a tumor-suppressor, the two variants ERβ2 and 5 exhibit pro-oncogenic activities in TNBC. Thus, ERβ1 activation might be used to limit the growth and spreading as well as to increase the drug sensitivity of TNBC. In contrast, the pro-oncogenic properties of ERβ2 and ERβ5 suggest the possible development and clinical use of specific antagonists in TNBC treatment. Furthermore, the role of ERβ might be regarded in the context of the androgen receptor (AR) expression, which represents another key marker in TNBC. The relationship between AR and ERβ as well as the ability to modulate the receptor-mediated effects through agonists/antagonists represent a challenge to develop more appropriate therapies in clinical management of TNBC patients. In this review, we will discuss the most recent data in the field. Therapeutic implications of these findings are also presented in the light of the discovery of specific ERβ modulators.
1. Introduction
Breast cancer (BC) represents the second most diagnosed malignancy and the fifth commonest cause of cancer-related death worldwide [1]. The BC incidence is higher in economically developed countries, probably because the disease’s onset is linked to risk factors, such as obesity, sedentary lifestyle, smoking, alcohol drinking, high consumption of red meat reach in hormones, use of oral contraceptives. Additionally, BC mortality is higher in countries with a low Human Development Index (HDI) [2,3].
Despite the advancements in methods for early detection and novel treatment options, BC often shows drug-resistance, likely due to its wide heterogeneity. BC is, indeed, characterized by different molecular signatures responsible for the disparate response to therapeutics and differences in patients’ long-term survival [4]. To date, on the basis of the expression of the classical BC markers, estrogen receptor alpha (ERα), progesterone receptor (PR) and the human epidermal growth factor receptor II (Her2), the molecular classification divides BC in five subtypes. The luminal A and luminal B, which are both characterized by ERα expression, while differing from each other in Her2 overexpression in luminal B; the Her2-enriched subtype; the basal like subtype, lacking the expression of ERα and PR and the amplification of the Her2 gene, ERBB2; the normal like subtype, with molecular characteristics similar to normal breast epithelium. Among them, the basal like subtype is also known as triple-negative breast cancer (TNBC), lacking expression of the three most important BC markers [5]. TNBC are currently treated with systemic chemotherapy. However, patients have a poor prognosis, poor recurrence-free and overall survival outcomes [4,5,6]. These considerations highlight the need for new molecular targets in TNBC.
Estrogen receptor β (ERβ) is a sex steroid receptor and a transcription factor expressed in different cancers, such as prostate [7,8], colon [9] and breast [10]. ERβ has been detected in 30% of BC patients [11]. As such, many reports in literature aim to determine the prognostic role of ERβ in TNBC, with conflicting results, likely because of the lack of specific antibodies and immunohistochemistry (IHC) approaches [12].
Nowadays, it is accepted that TNBC cells express different ERβ variants. ERβ1, which contains the entire predicted sequence, seems to play an inhibitory effect on TNBC growth and metastasis, while the truncated variants ERβ2 and 5 trigger proliferation and migration of TNBC. Therefore, the different ERβ variants represent promising molecular targets to develop precision strategies in TNBC clinical approaches [13].
In this manuscript, we discuss the role of ERβ in TNBC. An overview of the pathways controlled by ERβ through genomic and non-genomic mechanisms is also presented. In this scenario, the complexity of ERβ interaction with other partners, mainly the androgen receptor (AR), is underscored. These findings might pave the way for new and unexpected options for therapeutic clinical management of patients.
2. Estrogen Action in Target Cells
Estrogens play a crucial role in physiological and pathologic processes, such as development and maintenance of reproductive organs, cell proliferation, metabolism, programmed cell death, homeostasis, differentiation and cancer progression. They are also involved in neurodegenerative and cardio-vascular diseases [14]. Estrogens commonly bind two receptor isoforms (ERα or β). Once activated by estrogens, the receptors might act through two types of mechanisms. The first is also called nuclear or genomic mechanism, the second is defined extra-nuclear or non-genomic mechanism. According to the genomic mechanism, estrogens diffuse across cell membranes and bind to either, or both, of their intra-nuclear or cytoplasmic receptor, which undergoes dimerization. The receptor(s) thus bind estrogen responsive elements (ERE) sequences in the promoter region of target genes involved in cell proliferation, differentiation and metabolism [15]. In contrast, extra-nuclear/cytoplasmic receptors rapidly activate the non-transcriptional or non-genomic pathway upon estrogen binding. Since the studies from Szego and Davis reported a rapid increase in uterine cAMP within 15 s after treatment with physiological doses of estradiol [16], data collected over the last decade have shown that extra-nuclear ERs undergo post-transcriptional modifications in cytoplasm of target cells and rapidly trigger activation of signaling effectors, causing different hormonal effects upon ligand binding [17,18,19,20]. We appreciate, however, that an interconnection between genomic and non-genomic pathways occurs in target cells [21]. The balance between the different mechanisms (transcriptional versus non transcriptional) often impinges on cell outcome and pathophysiological processes [22]. In different hormone-responsive BC cells, estrogens can also mediate non genomic signalling activating the G protein coupled receptor (GPCR), known as GPR30. The G-protein-coupled receptor (GPR)-30 is a seven transmembrane domain protein, working as an alternative estrogen receptor and showing a structure distinct from the ER α and β. It specifically binds estrogens and cross-reacts with different cell signaling pathways such as the epidermal growth factor receptor (EGFR), the mitogen-activated protein kinase (MAPK) and the Notch pathways, thus controlling BC cell proliferation, migration and invasion and the tamoxifen-resistance [23]. As discussed in a subsequent section of this review, ERβ mediates both genomic and non-genomic actions in TNBC.
3. ERβ: Structure and Function
ERβ, a ligand-regulated transcription factor, was discovered and cloned from rat prostate in 1996 [24]. It has received increasing attention over the last 20 years, because of its involvement in many physiological and pathological processes [12]. The ESR2 gene is located on chromosome 14 and encodes for the full-length ERβ protein, consisting of 530 amino-acids (59 kDa). The receptor is arranged in six functional domains. The N-terminal domain A/B (named AF-1) responsible for the receptor transcriptional activity, the C domain responsible for the binding to the DNA, the E-domain responsible for the ligand binding and containing a second functional region, named AF-2. Finally, the D domain links the C and E domains, while the F domain represents the C-terminal end of the receptor (Figure 1).
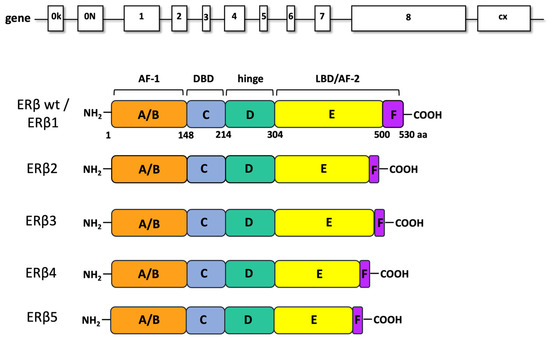
Figure 1.
ERβ gene and isoforms. In ERβ gene representation, introns (lanes) and exons (boxes) are indicated. ERβ wt (ERβ1 or ERβ) contains eight exons. The ERβ splice variants differ for the ligand binding domain (LBD) and contains six exons. AF1: activation function 1; DBD: DNA-binding domain; AF2: activation function 2.
Because of the high homology shared in C and D domains (96 and 60% homology, respectively) with the corresponding domains of ERα, the two receptor isoforms, α and β, also share various targets and functions. Moreover, they bind common ligands, such as estrogens and selective estrogen receptor modulators (SERMs; 24). ERβ is expressed in a higher number of BCs than ERα and in different cells of the cancer microenvironment such as fibroblasts, endothelial cells and BC infiltrating lymphocytes [9]. Beyond the firstly cloned full-length variant, named ERβ1, four more variants of ERβ have been identified [25,26]. They result from alternative splicing of exon 8 [25,26,27] and are named ERβ2, ERβ3, ERβ4 and ERβ5. These variants have different functions and tissue distribution, as they can be detected both in normal as well as in cancer tissues [12]. ERβ tissue localization is, indeed, wider than that of ERα [28] and includes the gastrointestinal tract, lungs, brain, luminal and myoepithelial cells of normal breast, as well as prostate, testis, uterus and ovary. Breast tissues mainly express ERβ1, ERβ2 and ERβ5, while ERβ4 is less present. ERβ3 is typical of prostate tissues [29]. ERβ1 is able to form functional homodimers and heterodimers with the other isoforms and with ERα thus inhibiting their signaling pathways [29,30]. In breast cancer, ERβ2 isoform is the best known; this variant shows an undetectable affinity for 17-β-estradiol and is related to the ERα negative phenotype. ERβ2 forms heterodimers with ERα thus inducing its degradation, inhibiting its recruitment to the EREs and leading to the suppression of its transcriptional activity [29]. In ERα positive patients, ERβ2 is related to an excellent response to tamoxifen therapy. Otherwise, in a limited number of samples with a low PR expression and in ERα negative BC, the ERβ2 correlates to a poor prognosis [31,32]. In literature there is no information about ERβ3, probably because scientists have found this isoform neither in BC cell lines nor in BC samples [33]. ERβ4 and 5 isoforms are truncated transcripts unable to bind ligands [25]; both can heterodimerize with ERβ1 and enhance its transactivation in a ligand-dependent manner [27]. ERβ4 can transform normal mammary epithelial cells, ERβ5 and ERβ2 sustain the tumor growth in a hypoxic environment and increase the c-Myc expression that correlates to bad prognosis in BC [34].
As far as cellular localization is concerned, available information indicates that ERβ mainly localizes to cytoplasm and the nucleus, but it can also be found inside mitochondria and at the peri-membrane [35]. Unliganded ERβ is inactive and it is bound to cytoplasmic chaperones, such as heat shock protein 90 (Hsp90). Only upon ligand binding does the receptor become activated and exert its effects, leading to different cell outcome [29].
4. ERβ in TNBC
TNBC is difficult to treat, since it is often resistant to chemotherapeutical treatments. Therefore, the discovery of new molecular targets to develop more effective therapies has received great attention in the last years by molecular and clinical oncologists. The study of ERβ is a recent and emerging field in TNBC. The receptor is expressed in TNBC lacking ERα and this appears to be a peculiar and intriguing situation. In the next paragraphs, we will present an overview about the role of ERβ in TNBC, distinguishing between genomic and non-genomic actions controlled by the receptor. A specific section will be dedicated to the relationship between ERβ and AR in TNBC.
4.1. Genomic Action of ERβ in TNBC
The discovery of ERβ was enthusiastically received by endocrinologists and oncologists, since it suggested that the pleiotropic effects of estrogen can be mediated through ERβ and its isoforms, other than the well-known ERα. Recent advances in molecular analysis of pathways activated by ERs have allowed for identification of EREs in the promoters of numerous genes and to characterize changes in the gene expression profile upon estradiol treatment of TNBC cells. Since these TNBC cells do not express ERα, these findings have suggested that a different isoform of ER is present and functionally active in TNBC. Consistent with these hypotheses, many findings have reported an oncosuppressor role for ERβ in TNBC. Inducible expression of full-length ERβ in MDA-MB-468 cells and treatment with estradiol or the selective ERβ ligand, ERB-041, induces a G1 cell-cycle arrest, blocks the colony formation and reduces the tumor size in xenografted mice. The antagonists, ICI 182,780 or 4-hydroxytamoxifen, restore cell growth. RNA sequencing showed that most (about 80%) of the target genes regulated by ERβ are ligand-dependent, while only 20% are ligand-independent. The ligand-mediated growth-inhibitory effects of ERβ are due to regulation of target genes involved in the Wnt/β-catenin pathway and G1/S cell cycle checkpoint control, two critical steps in cancer cell proliferation [36]. Notably, ERβ expression and activation by estradiol upregulates both the gene encoding the cyclin dependent kinase inhibitor p21 and CDKN1A, as well as the noncanonical Wnt ligand, WNT4, and the β-catenin interacting protein, CDH1. Downregulation of the Wnt inhibitor, DKK1, can also be observed [36]. Consistent with these studies, the ligand-mediated ERβ activation by estrogens or the ERβ selective agonist, LY500307, decreases cell proliferation and blocks the cell cycle in doxycycline (Dox)-inducible ERβ expressing MDA-MB-231 cells. Microarray data and qPCR analysis showed that activation of ERβ suppresses the cell cycle-related genes, such as cyclin-dependent kinase 1 (CDK1), cyclin B and cyclin H [37]. These findings are consistent with previous studies indicating that the ligand-mediated activation of ERβ suppresses proliferation in other cancer cell lines [36,37,38].
An antimetastatic role for ERβ has also been proposed in TNBC. Treatment with estrogen or LY500307 induces changes in gene expression profiles of ERβ-expressing TNBC cells, with several (almost 976) differently regulated genes. Among them, some genes coding for interleukins and other inflammation-related factors are significantly inhibited by estrogen. In contrast, four members of a superfamily of cystatins (cystatins 1, 2, 4 and 5) are upregulated by the ligand. Because of the inhibition of the TGF-β/SMAD pathway, high cystatin 1, 2, 4 and 5 expression levels have been associated with improved relapse-free survival and decreased metastatic potential in TNBC patients [39]. Consistent with this study, ERβ knockdown leads to the improper activation of TGF-β signalling pathway in TNBC models, thereby inducing the transcription of genes involved with either assembly, organization or in combination, of the extracellular matrix as well as the migration/invasion potential. ERβ agonists, such as ERB-041, WAY2000070, 3βA-diol and liquiritigenin, result in a significant decrease of TNBC cell invasiveness [40]. By repressing epidermal growth factor receptor (EGFR) transcription, ERβ and its ligand 3βA-diol suppress insulin-like growth factor II (IGF-II) mRNA binding protein 3 (IMP3) in MDA-MB-231 and MDA-MB-468 cells. In this way, the receptor likely inhibits cell invasiveness. The specific ERβ antagonist, PHTPP (4-[2-Phenyl-5,7-bis(trifluoromethyl) pyrazolo [1,5-a] pyrimidin-3-y1] phenol), restores IMP3 and EGFR expression [41]. A novel mechanism through which ERβ1 might inhibit invasiveness of TNBC cells has also been described. Most TNBC cells harbor a mutant version of p53 showing oncogenic functions, including the ability to promote metastasis. In MDA-MB-231 cells, ERβ upregulates SHARP-1 and CCNG2, which inhibit the metastatic events, while downregulating the pro-metastatic factor, follistatin. Thus, ERβ counteracts the oncogenic functions mediated by a p53 mutant and exerts its antimetastatic properties through a transcriptional mechanism [42]. Further, ERβ1 inhibits migration and invasiveness of MDA-MB-231 and Hs578T cells by regulating the expression level of epithelial-mesenchymal transition (EMT) markers. The ability of ERβ to reduce tumor metastases has been further corroborated by findings showing that hyperexpression of ERβ1 in MDA MB231 and Hs578T cells induces a low recurrence of lung metastases in xenografted mice [43].
Finally, given the presence of ERβ variants in target cells [44], many reports have investigated their specific role in TNBC cells. MDA-MB-468 and BT-549 cells, for instance, express very low levels of ERβ1, which exerts antioncogenic properties. In contrast, the most abundant isoforms, ERβ2 and ERβ5, exhibit pro-oncogenic activities by acting on cell proliferation, migration and invasion [13]. Thus, it might be argued that the final outcome of ERβ activation depends on expression and content of the receptor variants in TNBC.
Nowadays, it is largely recognized that genomic effects mediated by ERβ exert a potential anti-oncogenic role in TNBC. Specific targeting of ERβ might represent an interesting pharmacological option in TNBC patients who often exhibit or develop drug-resistance.
4.2. Non-Genomic Actions of ERβ in TNBC
Aside from the aforementioned genomic actions, cytoplasmic ERβ can also work in a non-genomic way upon ligand binding. These actions are much faster than the genomic ones, taking place in cytoplasm within seconds to minutes, and involving generation of second messengers, such as calcium [45] and cAMP [46] as well as activation of the MAPK pathway [47] or interaction with components of the proteasome degradation pathway [48]. These actions escape the transcription and translation inhibition.
The PI3K/AKT pathway is crucial for growth, proliferation, angiogenesis and migration of BC [49,50] and is upregulated in aggressive TNBCs [51]. A loss of function of the PI3-K inhibitor, PTEN, is often associated with a worse prognosis and outcome for BC patients. In TNBCs, increased ERβ1 expression correlates with downregulation of pAKT, which represents a favorable prognostic marker for the overall and disease-free-survival [11]. In TNBC cells, high levels of ERβ are associated with increased sensitivity to doxorubicin, which is controlled by the activation of the PI3K/AKT/mTOR pathway [52]. Triggering MDA-MB-231 and BT549 cell lines, with a specific agonist (liquiritigenin) for ERβ, increases the sensitivity to doxorubicin, a chemotherapeutic agent. In both TNBC cell lines, combinatorial treatment with liquiritigenin and doxorubicin showed a stronger effect than that exerted by each drug in monotherapy. Analysis of the molecular mechanisms has shown that combinatorial treatment causes a strong inhibition of the PI3K/AKT/mTOR pathway and this mechanism is correlated to ERβ expression [52].
Other studies have analyzed the role of ERβ in mediating the action of commonly used adjuvant drugs (such as tamoxifen or raloxifene) in TNBC cell lines. In MDA-MB-231 cells, ERβ expression fuels their overall effect or increases TNBC sensitivity to them. Since the mechanism of action of these drugs involves regulation of multiple signaling pathways, including EGFR, MAPK and PI3K, it has been reported that ERβ expression influences the effect of the aforementioned drugs, likely acting through a non-genomic way [53]. Again, by acting through a non-genomic action, ERβ decreases cell survival in TNBC. In MDA-MB231 cells, ERβ stimulation or upregulation reduces cell survival and enhances apoptosis by activating the stress-regulated cell-death pathway at endoplasmic reticulum [54]. Notably, by activating rapid actions, ERβ is also able to control the epithelial to mesenchymal transition (EMT) in TNBC. By overexpressing or downregulating ERβ in basal-like breast cancer cells (MDA-MB231 and Hs857T cells), the stabilization of an epithelial phenotype occurs, followed by inhibition of cell motility and invasion. These findings have been attributed to the ERβ-mediated upregulation of miR-200a/b/429, as well as the transcriptional repression of ZEB1 and SIP1, accounting for the increase in E-cadherin expression and inhibition of EMT. The direct correlation between ERβ and E-cadherin expression has been further confirmed in BC specimens. In the same experimental setting, Thomas and colleagues [55] also showed that by reinforcing the interaction between EGFR and the ubiquitin ligase Cbl, ERβ increases the EGFR degradation and lowers the EGFR-mediated downstream signaling. In this way, ERβ further impairs EMT in TNBC. These findings were confirmed by in vivo experiments in zebrafish [55].
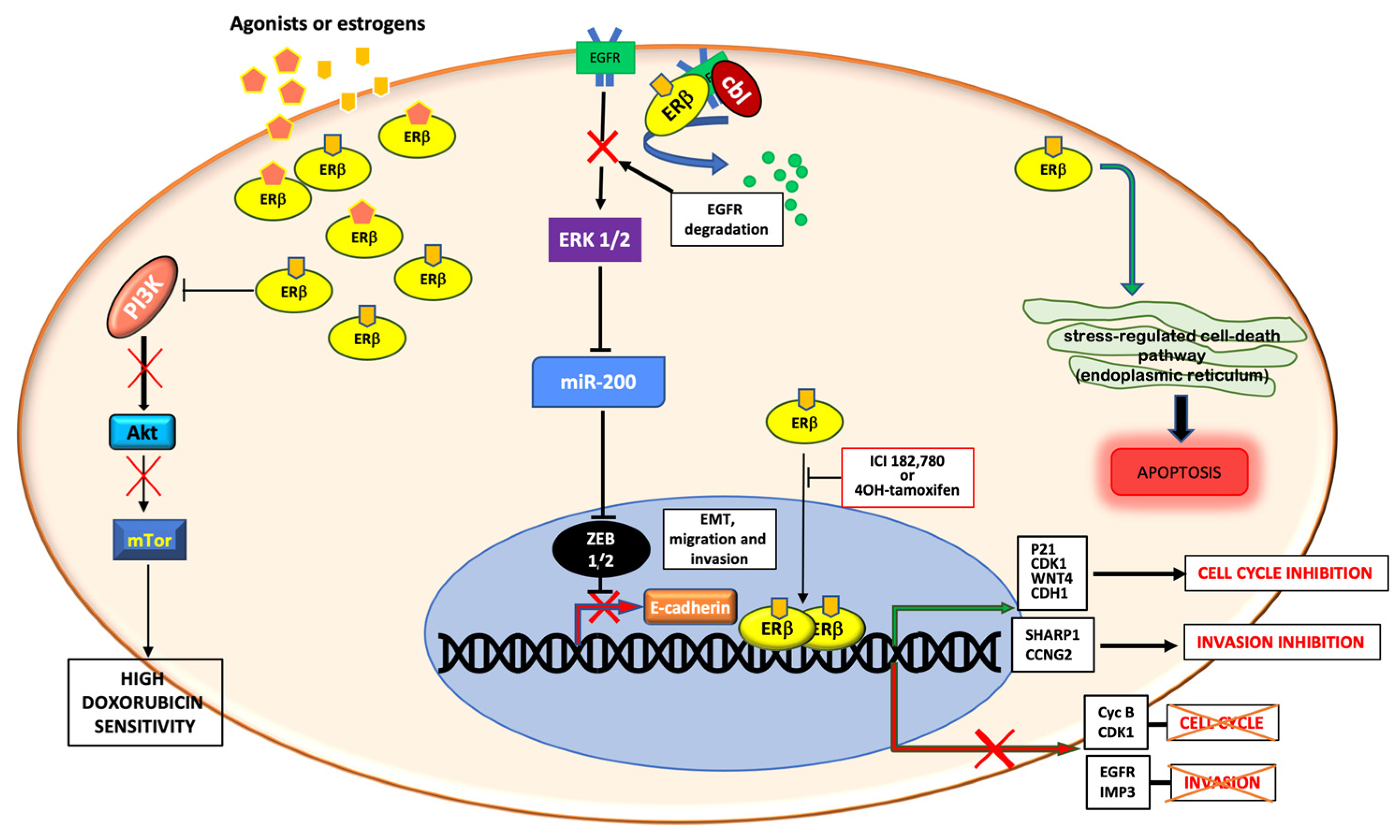
Taken together, the data so far discussed underscore the relevance of ERβ2, ERβ5 and ERβ1 in TNBC. These studies also imply that quantification of the amount or relative ratios of the different ERβ isoforms might have prognostic and therapeutic relevance in TNBC. These considerations would help us for a better stratification of patients, although the question of ERβ ligand efficacy in TNBC still remains pending and agonism of ERβ in patients with advanced TNBC has shown a limited efficacy in phase 2 study [31]. Figure 2 summarizes the aforementioned genomic and non-genomic pathways controlled by ERβ in TNBC.
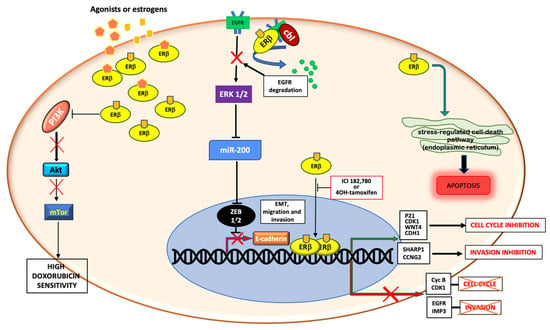
Figure 2.
Schematic representation of genomic and non-genomic actions of ERβ in TNBC. ERβ controls genomic and non-genomic actions upon ligand binding. It is able to control cell cycle and cell migration and invasion by upregulating or downregulating the transcription of different genes. Cytoplasmic ERβ rapidly activates the stress-regulated cell death pathway or inhibits the PI3K/Akt/mTor pathway or stimulates the EGFR degradation, thus promoting apoptosis, increasing the doxorubicin sensitivity and inhibiting the EMT transition and cell invasion, respectively.
4.3. ERβ and Androgen Receptor Interaction in TNBCs
Several findings have recently highlighted the role of androgen receptor (AR) in TNBC. The concept that this steroid receptor modulates the growth and progression of BC is currently undeniable [56]. AR is able to work in both transcriptional and non-transcriptional ways, thus influencing the behavior of TNBC. In particular, AR controls growth [57] and invasiveness [58] of TNBC. Indeed, AR inhibitors seem to promise a new opportunity to cure AR-positive TNBC. The relevance of AR expression in BC is underscored by the worst prognosis of quadruple negative breast cancer (QNBC), a TNBC subtype also lacking AR [59]. Considering that ERβ and AR belong to the same genic superfamily and share similar biological actions, it is conceivable that the two receptors act in cooperation to modulate the functions of TNBC cells.
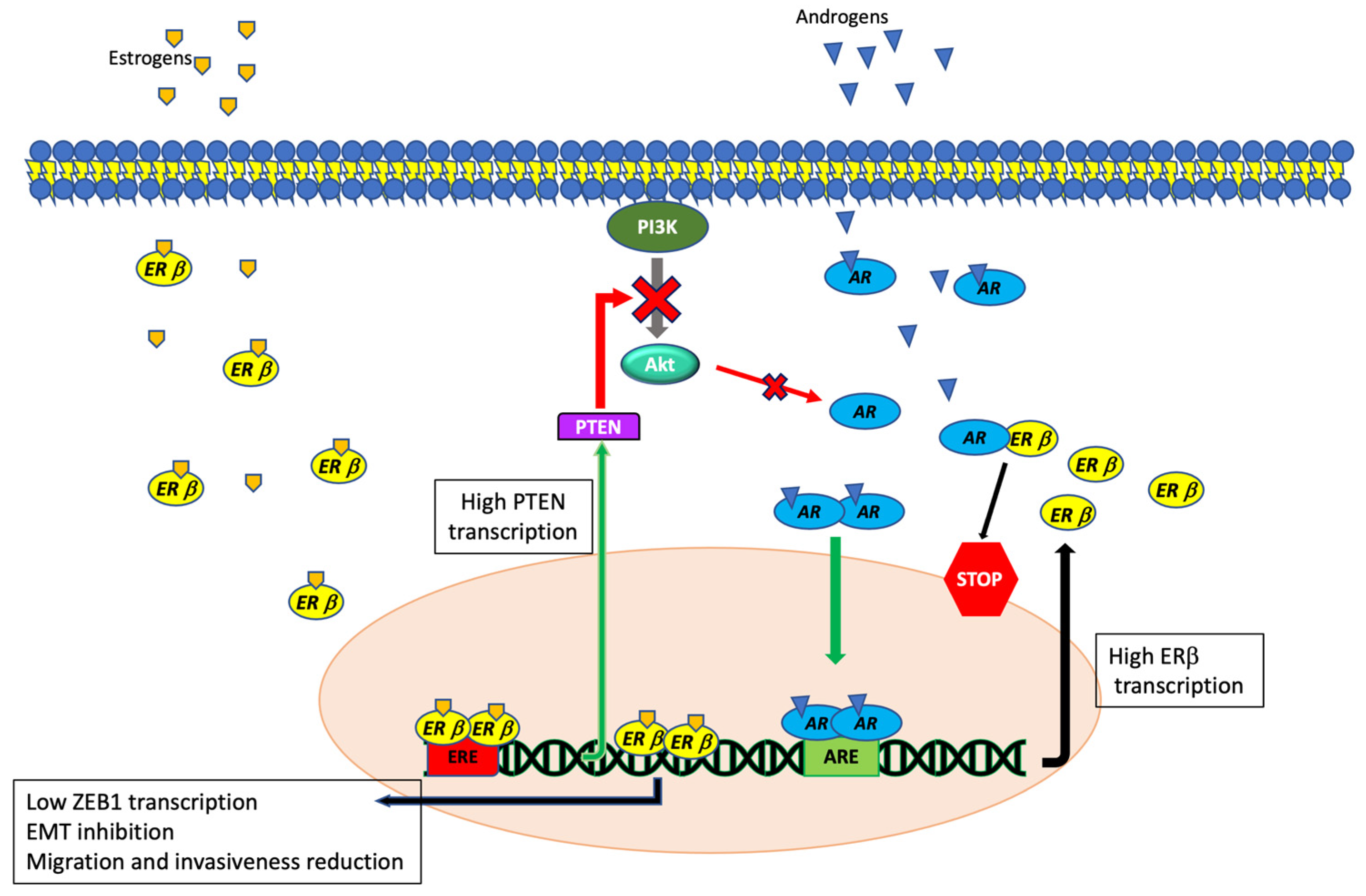
In AR+ TNBC cell lines (MB-231 and Hs578T), ERβ1 inhibits migration and invasion by regulating, through ZEB1, the expression level of EMT markers. Androgen stimulation of AR-positive TNBC cells reinforces this effect. Notably, AR controls the ERβ-mediated transcription by binding androgen responsive elements (ARE) localized into the ERβ promoter. After DHT stimulation, AR stimulates the ERβ expression, thus enhancing the anti-metastatic effect of ERβ in TNBC [43]. Additionally, ERβ might influence BC cell responsiveness to classical chemotherapeutic agents by acting in an indirect and in a direct way. On one hand, transient expression of ERβ in AR+ MDA-MB-453 cells positively regulates the transcription of PTEN, a known inhibitor of PI3-K, thus reducing the Akt phosphorylation and the consequent AR activation. The indirect inhibition of AR signaling causes the reduction of cell proliferation, survival and metastatic potential in TNBC cells [60]. Contrarily, anti-androgen treatment of MDA-MB-453 cells transiently transfected with ERβ fosters the cell necrosis/apoptosis and inhibits cell migration. Moreover, ERβ impairs the androgen-induced AR nuclear translocation, just as the antiandrogens, enzalutamide or bicalutamide are able to do. When ERβ is overexpressed, AR is, indeed, mainly localized in cytoplasm, where it likely interacts with ERβ. This process might be due to the presence of AR:ERβ heterodimers in TNBC cells overexpressing ERβ. As such, androgen addition displaces this interaction, promoting AR:AR homodimerization, while enzalutamide fosters AR:ERβ heterodimer formation, thereby increasing the oncosuppressor functions of ERβ [60]. Figure 3 summarizes the known interactions between AR and ERβ in TNBC.
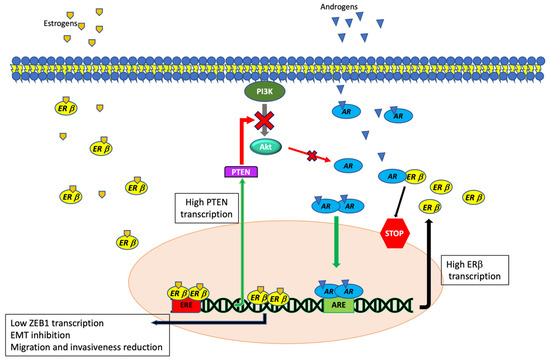
Figure 3.
Schematic representation of ERβ activated pathways and their interactions with AR-dependent signaling in TNBC. By reducing ZEB1 levels, ERβ negatively controls cell migration and EMT in AR-positive TNBC. AR controls the ERβ-mediated transcription by binding androgen responsive elements (ARE) localized into the ERβ promoter, while ERβ inhibits AR by increasing the PTEN protein level and inhibiting the AR nuclear translocation.
In summary, despite the scant number of studies in literature, a tight correlation between AR and ERβ occurs in TNBC. A deeper knowledge of the mechanisms regulating the connection between these two receptors might open new perspectives for better knowledge of TNBC biology.
5. Concluding Remarks
Many studies have described the clinical significance of ERβ in TNBC. Only recently, however, has the overall picture seemed clearer. This is probably due to better knowledge of ERβ and its isoforms and, not less important, to the existence of validated tools to recognize this receptor and its variants. The findings reported in this review depict a new and fascinating role of ERβ in TNBC. In particular, ERβ1 seems to work as a tumor suppressor in TNBC, while the other two isoforms most commonly revealed in this BC subtype, ERβ2 and ERβ5, exhibit pro-oncogenic activities. Fortunately, ERβ1 is more frequently expressed than the other two isoforms and its activation could be used to limit growth and spread as well as to increase the drug-sensitivity of TNBC.
The pro-oncogenic properties of ERβ 2 and ERβ 5 suggest the possible development and clinical use of specific antagonists in treating TNBC. Lastly, the tight relationship between AR and ERβ in TNBC and the ability of ERβ to potentiate the antiandrogen effect is another aspect to be considered when developing therapeutic strategies in TNBC. Overall, the results collected and described in this review underscore the importance to ameliorate the detection methods for revealing ERβ. Only in this way, might we select the best and more tailored treatment for TNBC patients.
Author Contributions
Conceptualization, P.G. and G.C.; writing—original draft preparation, P.G., G.C., A.M. (Alessandra Monac) and F.L.; writing—review and editing, A.M. (Alessandra Monac), F.L., M.D.G., G.G., M.D.D., P.G., A.M. (Antimo Migliaccio) and G.C.; visualization, P.G. and A.M. (Antimo Migliaccio); supervision, A.M. (Antimo Migliaccio) and G.C.; funding acquisition, A.M. (Antimo Migliaccio), G.C., M.D.D., P.G., G.G. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the Italian Ministry of University and Scientific Research (P.R.I.N. 2017EKMFTN_002 to G.C.); VALERE Program (“Desire” Vanvitelli per la Ricerca Program to P.G.; GoMAGIC to A.M.; AdipCARE to G.C.; Campania to P.G.). Marzia Di Donato is supported by iCURE Project (B21C17000030007- Regione Campania). Pia Giovannelli and Giovanni Galasso are supported by VALERE (Vanvitelli per la Ricerca) Program.
Institutional Review Board Statement
Not Applicable.
Informed Consent Statement
Not Applicable.
Data Availability Statement
Not Applicable.
Conflicts of Interest
The Authors declare that they have no financial or non-financial competing interests.
Abbreviations
| AR | androgen receptor |
| BC | breast cancer |
| EnR | endoplasmic reticulum |
| ER | estradiol receptor |
| TNBC | triple negative breast cancer |
References
- Sharma, R. Global, regional, national burden of breast cancer in 185 countries: Evidence from GLOBOCAN 2018. Breast Cancer Res. Treat. 2021, 187, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Salvi, S.; Bonafè, M.; Bravaccini, S. Androgen receptor in breast cancer: A wolf in sheep’s clothing? A lesson from prostate cancer. Semin. Cancer Biol. 2020, 60, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chan, P.S.; Lok, V.; Chen, X.; Ding, H.; Jin, Y.; Yuan, J.; Lao, X.-Q.; Zheng, Z.-J.; Wong, M.C. Global incidence and mortality of breast cancer: A trend analysis. Aging 2021, 13, 5748–5803. [Google Scholar] [CrossRef] [PubMed]
- Rivenbark, A.G.; O’Connor, S.M.; Coleman, W.B. Molecular and cellular heterogeneity in breast cancer: Challenges for personalized medicine. Am. J. Pathol. 2013, 183, 1113–1124. [Google Scholar] [CrossRef] [Green Version]
- Giovannelli, P.; Di Donato, M.; Galasso, G.; di Zazzo, E.; Bilancio, A.; Migliaccio, A. The Androgen Receptor in Breast Cancer. Front. Endocrinol. 2018, 9, 492. [Google Scholar] [CrossRef] [Green Version]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-Negative Breast Cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [Green Version]
- Migliaccio, A.; Di Domenico, M.; Castoria, G.; Nanayakkara, M.; Lombardi, M.; De Falco, A.; Bilancio, A.; Varricchio, L.; Ciociola, A.; Auricchio, F. Steroid Receptor Regulation of Epidermal Growth Factor Signaling through Src in Breast and Prostate Cancer Cells: Steroid Antagonist Action. Cancer Res. 2005, 65, 10585–10593. [Google Scholar] [CrossRef] [Green Version]
- di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Castoria, G. Estrogens and Their Receptors in Prostate Cancer: Therapeutic Implications. Front. Oncol. 2018, 8, 2. [Google Scholar] [CrossRef] [Green Version]
- Hartman, J.; Edvardsson, K.; Lindberg, K.; Zhao, C.; Williams, C.; Ström, A.; Gustafsson, J.-Å. Tumor Repressive Functions of Estrogen Receptor β in SW480 Colon Cancer Cells. Cancer Res. 2009, 69, 6100–6106. [Google Scholar] [CrossRef] [Green Version]
- Paruthiyil, S.; Parmar, H.; Kerekatte, V.; Cunha, G.R.; Firestone, G.L.; Leitman, D.C. Estrogen Receptor β Inhibits Human Breast Cancer Cell Proliferation and Tumor Formation by Causing a G2 Cell Cycle Arrest. Cancer Res. 2004, 64, 423–428. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Zhang, C.; Chen, K.; Tang, H.; Tang, J.; Song, C.; Xie, X. ERβ1 inversely correlates with PTEN/PI3K/AKT pathway and predicts a favorable prognosis in triple-negative breast cancer. Breast Cancer Res. Treat. 2015, 152, 255–269. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, J.-A.; Strom, A.; Warner, M. Update on ERbeta. J. Steroid Biochem. Mol. Biol. 2019, 191, 105312. [Google Scholar] [CrossRef]
- Yan, S.; Dey, P.; Ziegler, Y.; Jiao, X.; Kim, S.H.; Katzenellenbogen, J.A.; Katzenellenbogen, B.S. Contrasting activities of estrogen receptor beta isoforms in triple negative breast cancer. Breast Cancer Res. Treat. 2021, 185, 281–292. [Google Scholar] [CrossRef]
- Hamilton, K.J.; Hewitt, S.C.; Arao, Y.; Korach, K.S. Estrogen Hormone Biology. Curr. Top. Dev. Biol. 2017, 125, 109–146. [Google Scholar] [CrossRef]
- Di Croce, L.; Okret, S.; Kersten, S.; Gustafsson, J.-Å.; Parker, M.; Wahli, W.; Beato, M. Steroid and nuclear receptors. Villefranche-sur-Mer, France, May 25–27, 1999. EMBO J. 1999, 18, 6201–6210. [Google Scholar] [CrossRef] [Green Version]
- Szego, C.M.; Davis, J.S. Adenosine 3′,5′-monophosphate in rat uterus: Acute elevation by estrogen. Proc. Natl. Acad. Sci. USA 1967, 58, 1711–1718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Romancer, M.; Poulard, C.; Cohen, P.; Sentis, S.; Renoir, J.-M.; Corbo, L. Cracking the Estrogen Receptor’s Posttranslational Code in Breast Tumors. Endocr. Rev. 2011, 32, 597–622. [Google Scholar] [CrossRef] [Green Version]
- Giovannelli, P. Targeting rapid action of sex-steroid receptors in breast and prostate cancers. Front. Biosci. 2012, E4, 453–461. [Google Scholar] [CrossRef]
- Migliaccio, A.; Castoria, G.; Auricchio, F. Src-dependent signalling pathway regulation by sex-steroid hormones: Therapeutic implications. Int. J. Biochem. Cell Biol. 2007, 39, 1343–1348. [Google Scholar] [CrossRef]
- Levin, E.R.; Hammes, S.R. Nuclear receptors outside the nucleus: Extranuclear signalling by steroid receptors. Nat. Rev. Mol. Cell Biol. 2016, 17, 783–797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicent, G.P.; Zaurin, R.; Nacht, A.S.; Font-Mateu, J.; Le Dily, F.; Beato, M. Nuclear Factor 1 Synergizes with Progesterone Receptor on the Mouse Mammary Tumor Virus Promoter Wrapped around a Histone H3/H4 Tetramer by Facilitating Access to the Central Hormone-responsive Elements. J. Biol. Chem. 2010, 285, 2622–2631. [Google Scholar] [CrossRef] [Green Version]
- Castoria, G.; Auricchio, F.; Migliaccio, A. Extranuclear partners of androgen receptor: At the crossroads of proliferation, migration, and neuritogenesis. FASEB J. 2017, 31, 1289–1300. [Google Scholar] [CrossRef] [Green Version]
- Elappano, R.; Episano, A.; Maggiolini, M. GPER Function in Breast Cancer: An Overview. Front. Endocrinol. 2014, 5, 66. [Google Scholar] [CrossRef] [Green Version]
- Kuiper, G.G.; Shughrue, P.J.; Merchenthaler, I.; Gustafsson, J.A. The estrogen receptor beta subtype: A novel mediator of estrogen action in neuroendocrine systems. Front. Neuroendocrinol. 1998, 19, 253–286. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.T.; McKee, D.D.; Slentz-Kesler, K.; Moore, L.B.; Jones, S.A.; Horne, E.L.; Su, J.-L.; Kliewer, S.A.; Lehmann, J.M.; Willson, T.M. Cloning and Characterization of Human Estrogen Receptor β Isoforms. Biochem. Biophys. Res. Commun. 1998, 247, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Leygue, E.; Murphy, L.C. A bi-faceted role of estrogen receptor β in breast cancer. Endocrine-Relat. Cancer 2013, 20, R127–R139. [Google Scholar] [CrossRef] [PubMed]
- Leung, Y.-K.; Mak, P.; Hassan, S.; Ho, S.-M. Estrogen receptor (ER)-beta isoforms: A key to understanding ER-beta signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 13162–13167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuiper, G.; Enmark, E.; PeltoHuikko, M.; Nilsson, S.; Gustafsson, J.A. Cloning of a novel estrogen receptor expressed in rat prostate and ovary. Proc. Natl. Acad. Sci. USA 1996, 93, 5925–5930. [Google Scholar] [CrossRef] [Green Version]
- Božović, A.; Mandušić, V.; Todorović, L.; Krajnović, M. Estrogen Receptor Beta: The Promising Biomarker and Potential Target in Metastases. Int. J. Mol. Sci. 2021, 22, 1656. [Google Scholar] [CrossRef] [PubMed]
- Warner, M.; Gustafsson, J.A. The role of estrogen receptor beta (ERbeta) in malignant diseases—A new potential target for antipro-liferative drugs in prevention and treatment of cancer. Biochem. Biophys. Res. Commun. 2010, 396, 63–66. [Google Scholar] [CrossRef]
- Wisinski, K.B.; Xu, W.; Tevaarwerk, A.J.; Saha, S.; Kim, K.; Traynor, A.; Dietrich, L.; Hegeman, R.; Patel, D.; Blank, J.; et al. Targeting Estrogen Receptor Beta in a Phase 2 Study of High-Dose Estradiol in Metastatic Triple-Negative Breast Cancer: A Wisconsin Oncology Network Study. Clin. Breast Cancer 2016, 16, 256–261. [Google Scholar] [CrossRef] [Green Version]
- Chantzi, Ν.Ι.; Tiniakos, D.G.; Palaiologou, M.; Goutas, N.; Filippidis, T.; Vassilaros, S.D.; Dhimolea, E.; Mitsiou, D.J.; Alexis, Μ.N. Estrogen receptor beta 2 is associated with poor prognosis in estrogen receptor alpha-negative breast carcinoma. J. Cancer Res. Clin. Oncol. 2013, 139, 1489–1498. [Google Scholar] [CrossRef]
- Tong, D.; Schuster, E.; Seifert, M.; Czerwenka, K.; Leodolter, S.; Zeillinger, R. Expression of estrogen receptor beta isoforms in human breast cancer tissues and cell lines. Breast Cancer Res. Treat. 2002, 71, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Faria, M.; Karami, S.; Granados-Principal, S.; Dey, P.; Verma, A.; Choi, D.S.; Elemento, O.; Bawa-Khalfe, T.; Chang, J.C.; Strom, A.M.; et al. The ERβ4 variant induces transformation of the normal breast mammary epithelial cell line MCF-10A; the ERβ variants ERβ2 and ERβ5 increase aggressiveness of TNBC by regulation of hypoxic signaling. Oncotarget 2018, 9, 12201–12211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yaşar, P.; Ayaz, G.; User, S.D.; Güpür, G.; Muyan, M. Molecular mechanism of estrogen-estrogen receptor signaling. Reprod. Med. Biol. 2017, 16, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Shanle, E.K.; Zhao, Z.; Hawse, J.; Wisinski, K.; Keles, S.; Yuan, M.; Xu, W. Research Resource: Global Identification of Estrogen Receptor β Target Genes in Triple Negative Breast Cancer Cells. Mol. Endocrinol. 2013, 27, 1762–1775. [Google Scholar] [CrossRef] [Green Version]
- Reese, J.M.; Bruinsma, E.S.; Monroe, D.G.; Negron, V.; Suman, V.J.; Ingle, J.N.; Goetz, M.P.; Hawse, J.R. ERβ inhibits cyclin dependent kinases 1 and 7 in triple negative breast cancer. Oncotarget 2017, 8, 96506–96521. [Google Scholar] [CrossRef] [Green Version]
- Lazennec, G.; Bresson, D.; Lucas, A.; Chauveau, C.; Vignon, F. ERβ Inhibits Proliferation and Invasion of Breast Cancer Cells. Endocrinology 2001, 142, 4120–4130. [Google Scholar] [CrossRef]
- Reese, J.M.; Bruinsma, E.S.; Nelson, A.W.; Chernukhin, I.; Carroll, J.S.; Li, Y.; Subramaniam, M.; Suman, V.J.; Negron, V.; Monroe, D.G.; et al. ERβ-mediated induction of cystatins results in suppression of TGFβ signaling and inhibition of triple-negative breast cancer metastasis. Proc. Natl. Acad. Sci. USA 2018, 115, E9580–E9589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schüler-Toprak, S.; Häring, J.; Inwald, E.C.; Moehle, C.; Ortmann, O.; Treeck, O. Agonists and knockdown of estrogen receptor β differentially affect invasion of triple-negative breast cancer cells in vitro. BMC Cancer 2016, 16, 951. [Google Scholar] [CrossRef] [Green Version]
- Samanta, S.; Sharma, V.M.; Khan, A.; Mercurio, A.M. Regulation of IMP3 by EGFR signaling and repression by ERβ: Implications for triple-negative breast cancer. Oncogene 2012, 31, 4689–4697. [Google Scholar] [CrossRef] [PubMed]
- Bado, I.; Nikolos, F.; Rajapaksa, G.; Gustafsson, J.-Å.; Thomas, C. ERβ decreases the invasiveness of triple-negative breast cancer cells by regulating mutant p53 oncogenic function. Oncotarget 2016, 7, 13599–13611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.; Tang, L.; Xu, Y.; Sun, Q.; Yang, F.; Guan, X. ERβ1 inhibits metastasis of androgen receptor-positive triple-negative breast cancer by suppressing ZEB1. J. Exp. Clin. Cancer Res. 2017, 36, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, V.; Di Zazzo, E.; Galasso, G.; De Rosa, C.; Abbondanza, C.; Sinisi, A.A.; Altucci, L.; Migliaccio, A.; Castoria, G. Estrogens Modulate Somatostatin Receptors Expression and Synergize with the Somatostatin Analog Pasireotide in Prostate Cells. Front. Pharmacol. 2019, 10, 28. [Google Scholar] [CrossRef] [Green Version]
- Improta-Brears, T.; Whorton, A.R.; Codazzi, F.; York, J.D.; Meyer, T.; McDonnell, D.P. Estrogen-induced activation of mitogen-activated protein kinase requires mobilization of intracellular calcium. Proc. Natl. Acad. Sci. USA 1999, 96, 4686–4691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aronica, S.M.; Kraus, W.L.; Katzenellenbogen, B.S. Estrogen action via the cAMP signaling pathway: Stimulation of adenylate cyclase and cAMP-regulated gene transcription. Proc. Natl. Acad. Sci. USA 1994, 91, 8517–8521. [Google Scholar] [CrossRef] [Green Version]
- Migliaccio, A.; Castoria, G.; Di Domenico, M.; De Falco, A.; Bilancio, A.; Lombardi, M.; Barone, M.V.; Ametrano, D.; Zannini, M.S.; Abbondanza, C.; et al. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J. 2000, 19, 5406–5417. [Google Scholar] [CrossRef] [Green Version]
- Stellato, C.; Nassa, G.; Tarallo, R.; Giurato, G.; Ravo, M.; Rizzo, F.; Marchese, G.; Alexandrova, E.; Cordella, A.; Baumann, M.; et al. Identification of cytoplasmic proteins interacting with unliganded estrogen receptor α and β in human breast cancer cells. Proteomics 2015, 15, 1801–1807. [Google Scholar] [CrossRef]
- Castoria, G.; Migliaccio, A.; Bilancio, A.; Di Domenico, M.; De Falco, A.; Lombardi, M.; Fiorentino, R.; Varricchio, L.; Barone, M.V.; Auricchio, F. PI3-kinase in concert with Src promotes the S-phase entry of oestradiol-stimulated MCF-7 cells. EMBO J. 2001, 20, 6050–6059. [Google Scholar] [CrossRef] [Green Version]
- Le Romancer, M.; Treilleux, I.; Leconte, N.; Robin-Lespinasse, Y.; Sentis, S.; Bouchekioua-Bouzaghou, K.; Goddard, S.; Gobert-Gosse, S.; Corbo, L. Regulation of Estrogen Rapid Signaling through Arginine Methylation by PRMT1. Mol. Cell 2008, 31, 212–221. [Google Scholar] [CrossRef]
- Dillon, R.L.; White, D.E.; Muller, W.J. The phosphatidyl inositol 3-kinase signaling network: Implications for human breast cancer. Oncogene 2007, 26, 1338–1345. [Google Scholar] [CrossRef]
- Lei, S.; Fan, P.; Wang, M.; Zhang, C.; Jiang, Y.; Huang, S.; Fang, M.; He, Z.; Wu, A. Elevated estrogen receptor β expression in triple negative breast cancer cells is associated with sensitivity to doxorubicin by inhibiting the PI3K/AKT/mTOR signaling pathway. Exp. Ther. Med. 2020, 20, 1630–1636. [Google Scholar] [CrossRef]
- Greish, K.; Nehoff, H.; Bahman, F.; Pritchard, T.; Taurin, S. Raloxifene nano-micelles effect on triple-negative breast cancer is mediated through estrogen receptor-β and epidermal growth factor receptor. J. Drug Target. 2019, 27, 903–916. [Google Scholar] [CrossRef] [PubMed]
- Rajapaksa, G.; Nikolos, F.; Bado, I.; Clarke, R.; Gustafsson, J.-Å.; Thomas, C. ERβ decreases breast cancer cell survival by regulating the IRE1/XBP-1 pathway. Oncogene 2015, 34, 4130–4141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, C.; Rajapaksa, G.; Nikolos, F.; Hao, R.; Katchy, A.; Mccollum, C.W.; Bondesson, M.; Quinlan, P.; Thompson, A.; Krishnamurthy, S.; et al. ERβ1 represses basal-like breast cancer epithelial to mesenchymal transition by destabilizing EGFR. Breast Cancer Res. 2012, 14, R148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesei, A.; Castoria, G. Editorial: The Androgen Receptor in Breast Cancer. Front. Endocrinol. 2021, 11, 636480. [Google Scholar] [CrossRef] [PubMed]
- Traina, T.A.; Miller, K.; Yardley, D.A.; Eakle, J.; Schwartzberg, L.S.; O’Shaughnessy, J.; Gradishar, W.; Schmid, P.; Winer, E.; Kelly, C.; et al. Enzalutamide for the Treatment of Androgen Receptor–Expressing Triple-Negative Breast Cancer. J. Clin. Oncol. 2018, 36, 884–890. [Google Scholar] [CrossRef]
- Giovannelli, P.; Di Donato, M.; Auricchio, F.; Castoria, G.; Migliaccio, A. Androgens Induce Invasiveness of Triple Negative Breast Cancer Cells Through AR/Src/PI3-K Complex Assembly. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, M.; Wu, J.; Ling, R.; Li, N. Quadruple negative breast cancer. Breast Cancer 2020, 27, 527–533. [Google Scholar] [CrossRef]
- Anestis, A.; Sarantis, P.; Theocharis, S.; Zoi, I.; Tryfonopoulos, D.; Korogiannos, A.; Koumarianou, A.; Xingi, E.; Thomaidou, D.; Kontos, M.; et al. Estrogen receptor beta increases sensitivity to enzalutamide in androgen receptor-positive triple-negative breast cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 1221–1233. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).