

The Human Early Maternal–Embryonic Interactome

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Bulk RNA-Seq Data
2.2. ScRNA-Seq Data
2.3. In Silico Modelling of the TE-EEC Interface
2.4. Hypernetworks
2.5. Gene Network Construction and Visualisation
2.6. Enrichment Analysis
2.7. Data Availability and Materials
3. Results
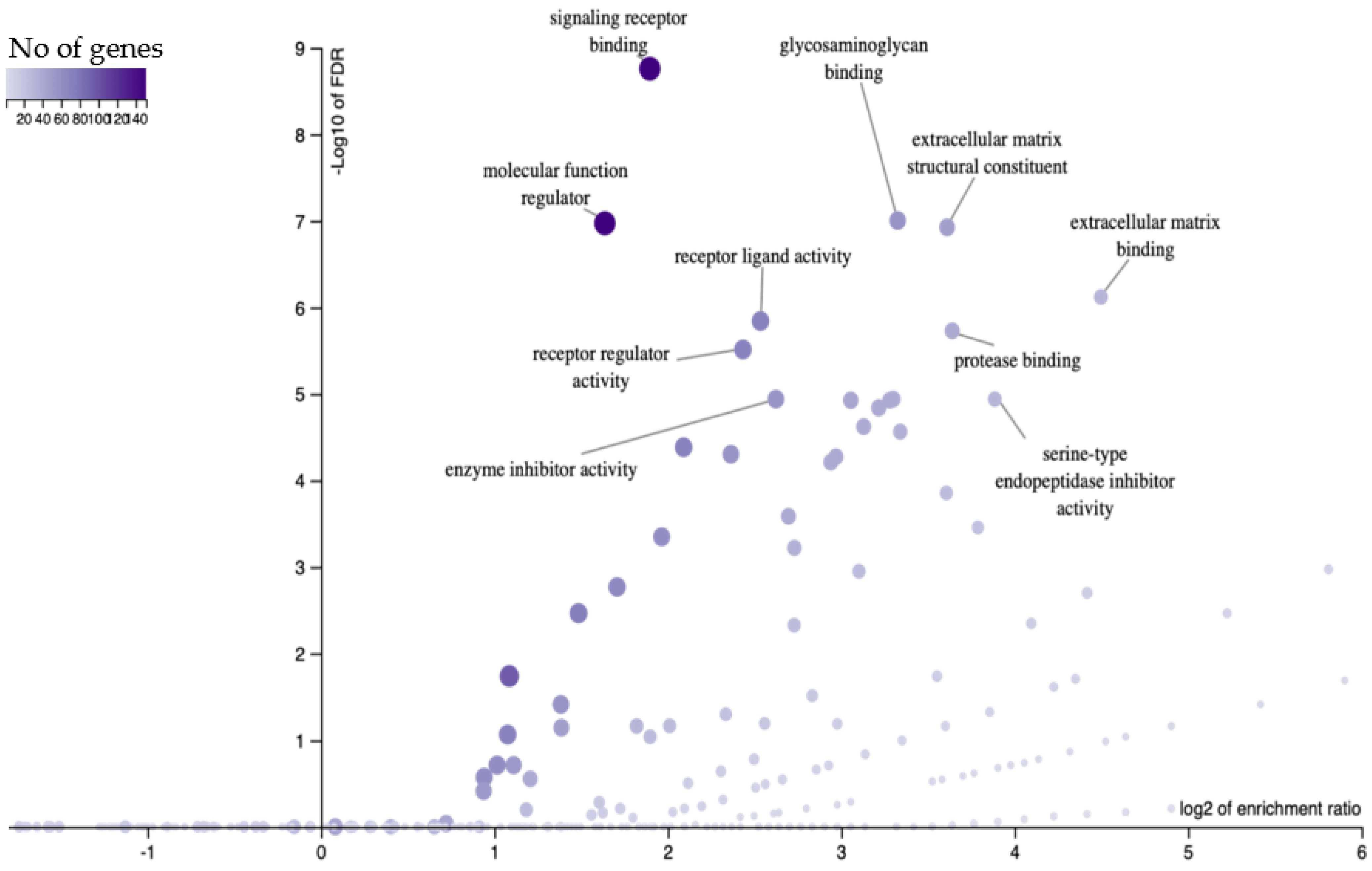
3.1. Secretory Phase EEC Genes Used to Model the Maternal-TE Interface
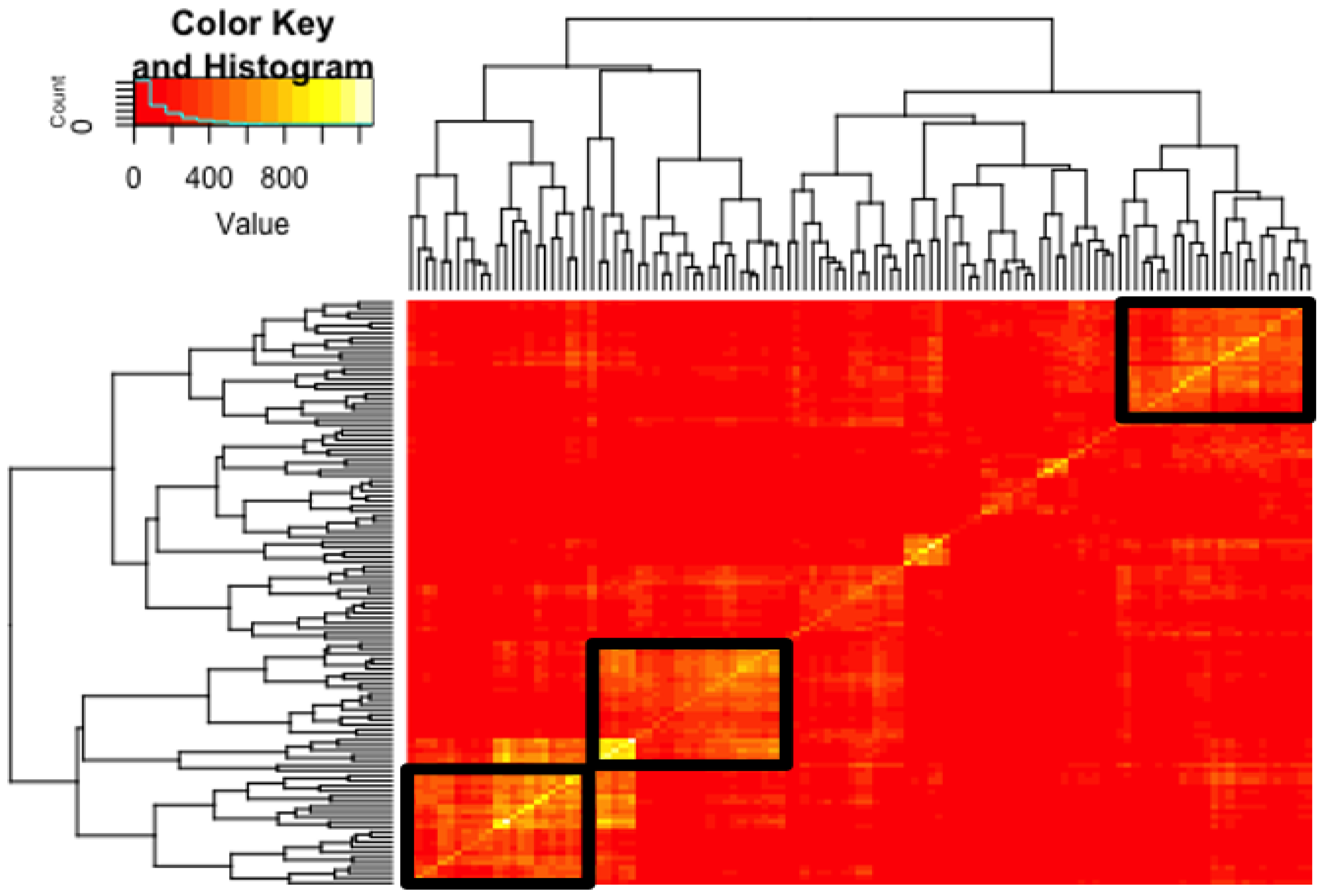
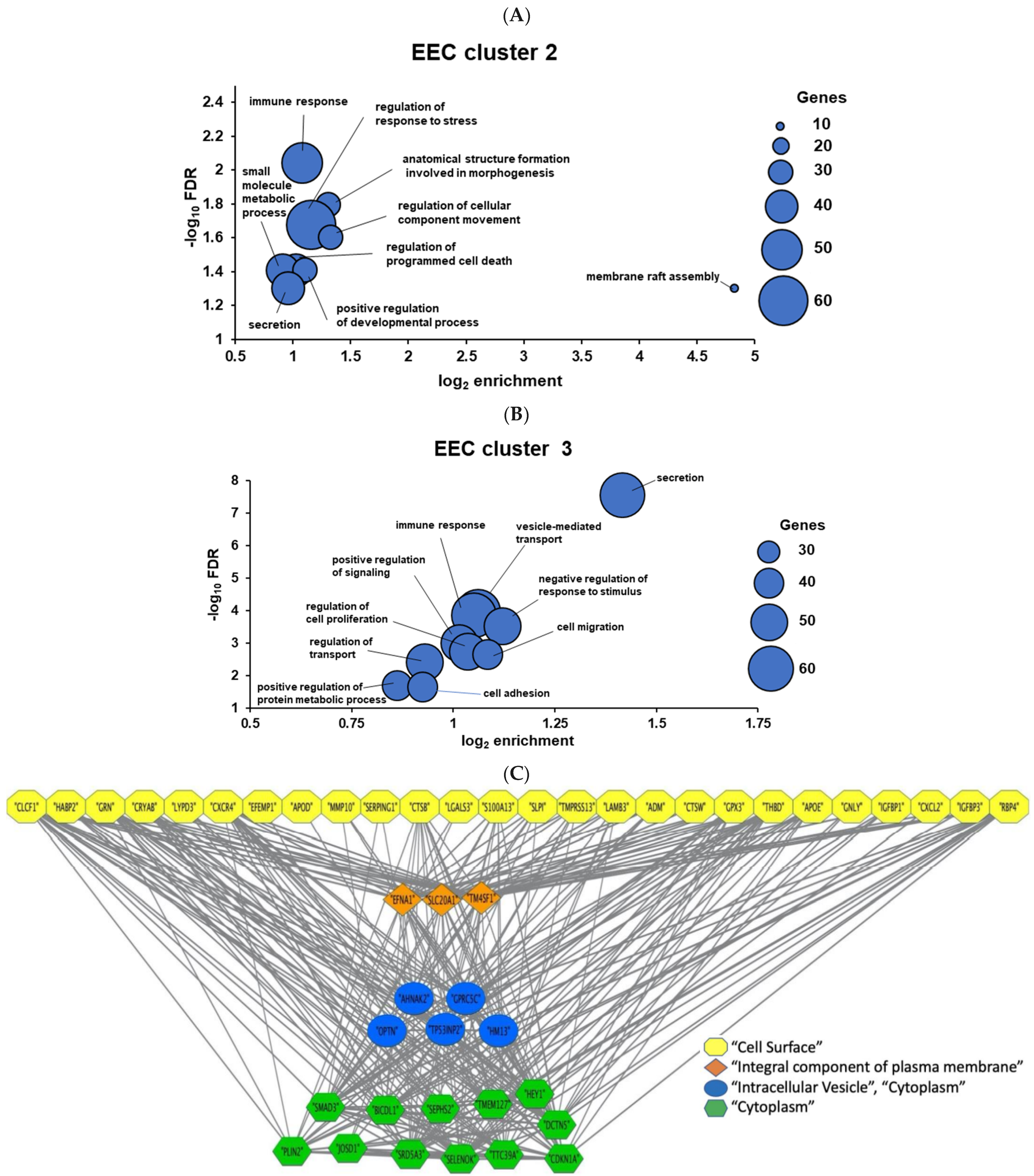
3.2. Networks Functioning at the EEC-TE Interface Examined Using Hypernetwork Analysis
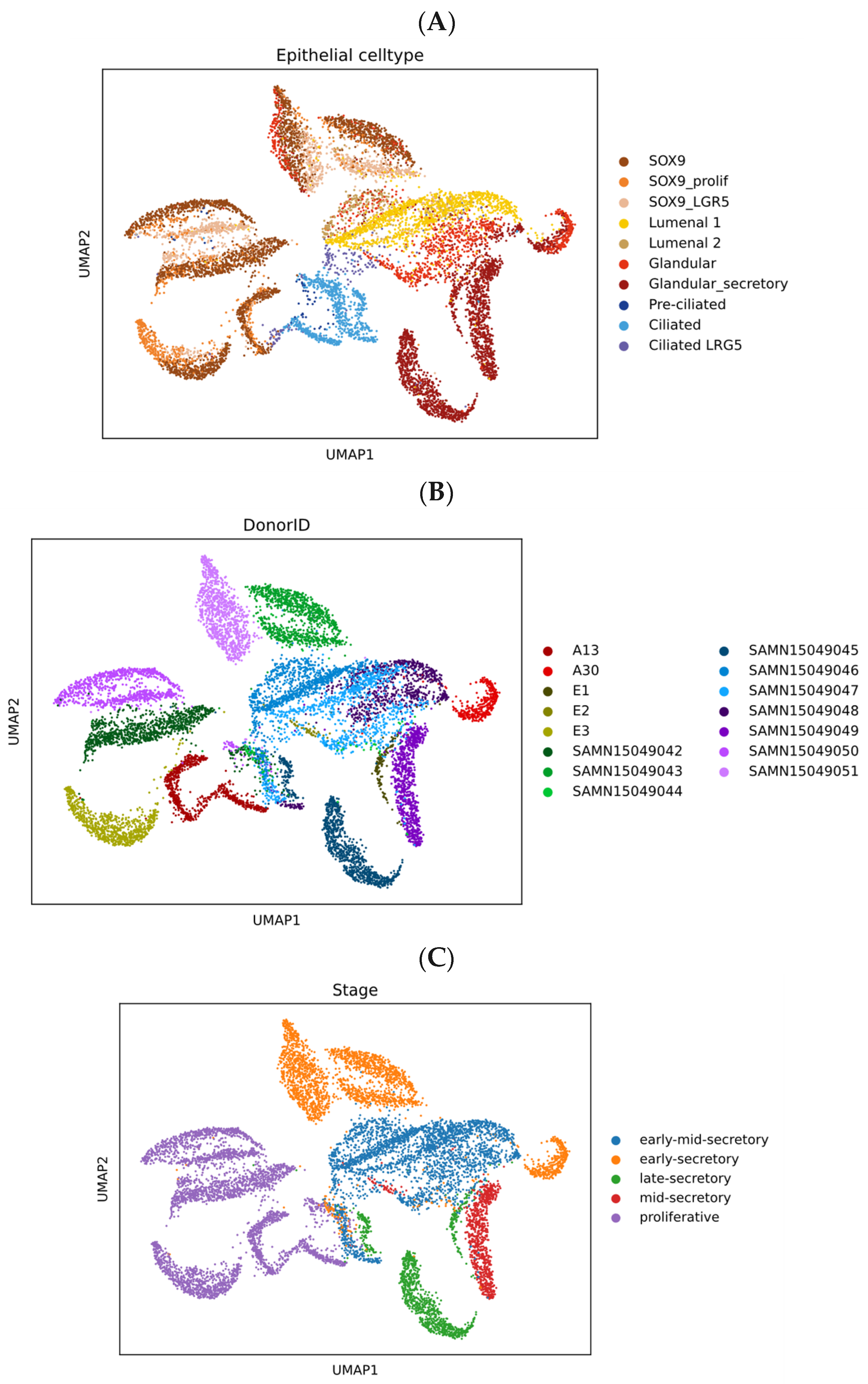
3.3. Epithelial Heterogeneity
3.4. Trophectoderm-Luminal Epithelial Gene Networks
3.5. Trophectoderm-Ciliated Epithelium and Trophectoderm-Glandular Epithelium Gene Networks
3.6. Comparison of Maternal Gene Networks at the Different TE Interface Models
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Aplin, J.D.; Ruane, P.T. Embryo-epithelium interactions during implantation at a glance. J. Cell Sci. 2017, 130, 15–22. [Google Scholar] [CrossRef]
- Meseguer, M.; Aplin, J.D.; Caballero-Campo, P.; O’Connor, J.E.; Martín, J.C.; Remohí, J.; Pellicer, A.; Simón, C. Human endometrial mucin MUC1 is up-regulated by progesterone and down-regulated in vitro by the human blastocyst. Biol. Reprod. 2001, 64, 590–601. [Google Scholar] [CrossRef]
- Aplin, J.D.; Meseguer, M.; Simón, C.; Ortíz, M.E.; Croxatto, H.; Jones, C.J. MUC1, glycans and the cell-surface barrier to embryo implantation. Biochem. Soc. Trans. 2001, 29 Pt 2, 153–156. [Google Scholar] [CrossRef]
- Ruane, P.T.; Berneau, S.C.; Koeck, R.; Watts, J.; Kimber, S.J.; Brison, D.R.; Westwood, M.; Aplin, J.D. Apposition to endometrial epithelial cells activates mouse blastocysts for implantation. Mol. Hum. Reprod. 2017, 23, 617–627. [Google Scholar] [CrossRef] [PubMed]
- Ruane, P.T.; Garner, T.; Parsons, L.; Babbington, P.A.; Wangsaputra, I.; Kimber, S.J.; Stevens, A.; Westwood, M.; Brison, D.R.; Aplin, J.D. Trophectoderm differentiation to invasive syncytiotrophoblast is promoted by endometrial epithelial cells during human embryo implantation. Hum. Reprod. 2022, 37, 777–792. [Google Scholar] [CrossRef]
- Savio Figueira Rde, C.; Setti, A.S.; Braga, D.P.A.F.; Iaconelli, A., Jr.; Borges, E., Jr. Blastocyst Morphology Holds Clues Concerning The Chromosomal Status of The Embryo. Int. J. Fertil. Steril. 2015, 9, 215–220. [Google Scholar] [PubMed]
- Wilcox, A.J.; Weinberg, C.R.; O’Connor, J.F.; Baird, D.D.; Schlatterer, J.P.; Canfield, R.E.; Armstrong, E.G.; Nisula, B.C. Incidence of early loss of pregnancy. N. Engl. J. Med. 1988, 319, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, C.; Wang, L.; Chen, D.; Guang, W.; French, J. Conception, early pregnancy loss, and time to clinical pregnancy: A population-based prospective study. Fertil. Steril. 2003, 79, 577–584. [Google Scholar] [CrossRef]
- Foo, L.; Johnson, S.; Marriott, L.; Bourne, T.; Bennett, P.; Lees, C. Peri-implantation urinary hormone monitoring distinguishes between types of first-trimester spontaneous pregnancy loss. Paediatr. Perinat. Epidemiol. 2020, 34, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Franasiak, J.M.; Alecsandru, D.; Forman, E.J.; Gemmell, L.C.; Goldberg, J.M.; Llarena, N.; Margolis, C.; Laven, J.; Schoenmakers, S.; Seli, E. A review of the pathophysiology of recurrent implantation failure. Fertil. Steril. 2021, 116, 1436–1448. [Google Scholar] [CrossRef]
- Pirtea, P.; De Ziegler, D.; Tao, X.; Sun, L.; Zhan, Y.; Ayoubi, J.M.; Seli, E.; Franasiak, J.M.; Scott, R.T. Rate of true recurrent implantation failure is low: Results of three successive frozen euploid single embryo transfers. Fertil. Steril. 2021, 115, 45–53. [Google Scholar] [CrossRef]
- Díaz-Gimeno, P.; Ruiz-Alonso, M.; Sebastian-Leon, P.; Pellicer, A.; Valbuena, D.; Simón, C. Window of implantation transcriptomic stratification reveals different endometrial subsignatures associated with live birth and biochemical pregnancy. Fertil. Steril. 2017, 108, 703–710.e3. [Google Scholar] [CrossRef]
- Ben Rafael, Z. Endometrial Receptivity Analysis (ERA) test: An unproven technology. Hum. Reprod. Open 2021, 2021, hoab010. [Google Scholar] [CrossRef]
- Aplin, J.D.; Stevens, A. Use of ’omics for endometrial timing: The cycle moves on. Hum. Reprod. 2022, 37, 644–650. [Google Scholar] [CrossRef]
- Battiston, F.; Amico, E.; Barrat, A.; Bianconi, G.; de Arruda, G.F.; Franceschiello, B.; Iacopini, I.; Kéfi, S.; Latora, V.; Moreno, Y.; et al. The physics of higher-order interactions in complex systems. Nat. Phys. 2021, 17, 1093–1098. [Google Scholar] [CrossRef]
- Murgas, K.A.; Saucan, E.; Sandhu, R. Hypergraph geometry reflects higher-order dynamics in protein interaction networks. Sci. Rep. 2022, 12, 20879. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, H.; Javali, A.; Khoei, H.H.; Sommer, T.M.; Sestini, G.; Novatchkova, M.; Reimer, Y.S.O.; Castel, G.; Bruneau, A.; Maenhoudt, N.; et al. Human blastoids model blastocyst development and implantation. Nature 2021, 601, 600–605. [Google Scholar] [CrossRef]
- Chi, R.-P.A.; Wang, T.; Adams, N.; Wu, S.-P.; Young, S.L.; Spencer, T.E.; DeMayo, F. Human Endometrial Transcriptome and Progesterone Receptor Cistrome Reveal Important Pathways and Epithelial Regulators. J. Clin. Endocrinol. Metab. 2020, 105, e1419-39. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple hypothesis testing. J. R Stat. Soc. B. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Garcia-Alonso, L.; Handfield, L.-F.; Roberts, K.; Nikolakopoulou, K.; Fernando, R.C.; Gardner, L.; Woodhams, B.; Arutyunyan, A.; Polanski, K.; Hoo, R.; et al. Mapping the temporal and spatial dynamics of the human endometrium in vivo and in vitro. Nat. Genet. 2021, 53, 1698–1711. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Hao, S.; Andersen-Nissen, E.; Mauck, W.M., 3rd; Zheng, S.; Butler, A.; Lee, M.J.; Wilk, A.J.; Darby, C.; Zager, M.; et al. Integrated analysis of multimodal single-cell data. Cell 2021, 184, 3573–3587 e29. [Google Scholar] [CrossRef]
- Petropoulos, S.; Edsgärd, D.; Reinius, B.; Deng, Q.; Panula, S.P.; Codeluppi, S.; Reyes, A.P.; Linnarsson, S.; Sandberg, R.; Lanner, F. Single-Cell RNA-Seq Reveals Lineage and X Chromosome Dynamics in Human Preimplantation Embryos. Cell 2016, 165, 1012–1026. [Google Scholar] [CrossRef]
- Starostik, M.R.; Sosina, O.A.; McCoy, R.C. Single-cell analysis of human embryos reveals diverse patterns of aneuploidy and mosaicism. Genome Res. 2020, 30, 814–825. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Zheng, X.; Yang, J.; Imamichi, T.; Stephens, R.; Lempicki, R.A. Extracting biological meaning from large gene lists with DAVID. Curr. Protoc. Bioinform. 2009, 27, 13.11.1–13.11.13. [Google Scholar] [CrossRef]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [PubMed]
- Smoot, M.E.; Ono, K.; Ruscheinski, J.; Wang, P.-L.; Ideker, T. Cytoscape 2.8: New features for data integration and network visualization. Bioinformatics 2011, 27, 431–432. [Google Scholar] [CrossRef] [PubMed]
- Settle, B.; Otasek, D.; Morris, J.H.; Demchak, B. aMatReader: Importing adjacency matrices via Cytoscape Automation. F1000Research 2018, 7, 823. [Google Scholar] [CrossRef]
- Liao, Y.; Wang, J.; Jaehnig, E.J.; Shi, Z.; Zhang, B. WebGestalt 2019: Gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. 2019, 47, W199–W205. [Google Scholar] [CrossRef]
- Wang, W.; Vilella, F.; Alama, P.; Moreno, I.; Mignardi, M.; Isakova, A.; Pan, W.; Simon, C.; Quake, S.R. Single-cell transcriptomic atlas of the human endometrium during the menstrual cycle. Nat. Med. 2020, 26, 1644–1653. [Google Scholar] [CrossRef]
- Deng, W.; Wang, H. Efficient cell chatting between embryo and uterus ensures embryo implantationdagger. Biol. Reprod. 2022, 107, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Robertson, S.A.; Moldenhauer, L.M.; Green, E.S.; Care, A.S.; Hull, M.L. Immune determinants of endometrial receptivity: A biological perspective. Fertil. Steril. 2022, 117, 1107–1120. [Google Scholar] [CrossRef]
- Aplin, J. Uterus-Endometrium, in Encyclopedia of Reproduction; Skinner, M.K., Ed.; Academic Press: Cambridge, MA, USA; Elsevier: Amsterdam, The Netherlands, 2018; pp. 326–332. [Google Scholar]
- Haller-Kikkatalo, K.; Tagoma, A.; Uibo, R.; Salumets, A.; Altmäe, S. Autoimmune activation toward embryo implantation is rare in immune-privileged human endometrium. Semin. Reprod. Med. 2014, 32, 376–384. [Google Scholar] [CrossRef] [PubMed]
- Fragouli, E.; Alfarawati, S.; Spath, K.; Babariya, D.; Tarozzi, N.; Borini, A.; Wells, D. Analysis of implantation and ongoing pregnancy rates following the transfer of mosaic diploid-aneuploid blastocysts. Hum. Genet. 2017, 136, 805–819. [Google Scholar] [CrossRef]
- Liu, X.; Tan, J.P.; Schröder, J.; Aberkane, A.; Ouyang, J.F.; Mohenska, M.; Lim, S.M.; Sun, Y.B.Y.; Chen, J.; Sun, G.; et al. Modelling human blastocysts by reprogramming fibroblasts into iBlastoids. Nature 2021, 591, 627–632. [Google Scholar] [CrossRef]
- Yang, M.; Rito, T.; Metzger, J.; Naftaly, J.; Soman, R.; Hu, J.; Albertini, D.F.; Barad, D.H.; Brivanlou, A.H.; Gleicher, N. Depletion of aneuploid cells in human embryos and gastruloids. Nat. Cell Biol. 2021, 23, 314–321. [Google Scholar] [CrossRef]
- Moser, G.; Windsperger, K.; Pollheimer, J.; de Sousa Lopes, S.C.; Huppertz, B. Human trophoblast invasion: New and unexpected routes and functions. Histochem. Cell Biol. 2018, 150, 361–370. [Google Scholar] [CrossRef]
- Hjortebjerg, R. IGFBP-4 and PAPP-A in normal physiology and disease. Growth Horm. IGF Res. 2018, 41, 7–22. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Shiojima, I.; Ito, Y.; Li, Z.; Ikeda, H.; Yoshida, M.; Naito, A.T.; Nishi, J.-I.; Ueno, H.; Umezawa, A.; et al. IGFBP-4 is an inhibitor of canonical Wnt signalling required for cardiogenesis. Nature 2008, 454, 345–349. [Google Scholar] [CrossRef] [PubMed]
- Allègre, N.; Chauveau, S.; Dennis, C.; Renaud, Y.; Meistermann, D.; Estrella, L.V.; Pouchin, P.; Cohen-Tannoudji, M.; David, L.; Chazaud, C. NANOG initiates epiblast fate through the coordination of pluripotency genes expression. Nat. Commun. 2022, 13, 3550. [Google Scholar] [CrossRef]
- Lipecki, J.; Mitchell, A.; Muter, J.; Lucas, E.; Makwana, K.; Fishwick, K.; Odendaal, J.; Hawkes, A.; Vrljicak, P.; Brosens, J.; et al. EndoTime: Non-categorical timing estimates for luteal endometrium. Hum. Reprod. 2022, 37, 747–761. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, T.M.; Makwana, K.; Taylor, D.M.; Molè, M.A.; Fishwick, K.J.; Tryfonos, M.; Odendaal, J.; Hawkes, A.; Zernicka-Goetz, M.; Hartshorne, G.M.; et al. Modelling the impact of decidual senescence on embryo implantation in human endometrial assembloids. Elife 2021, 10, e69603. [Google Scholar] [CrossRef] [PubMed]
- Vento-Tormo, R.; Efremova, M.; Botting, R.A.; Turco, M.Y.; Vento-Tormo, M.; Meyer, K.B.; Park, J.E.; Stephenson, E.; Polański, K.; Goncalves, A.; et al. Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 2018, 563, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Ferenczy, A.; Richart, R.M.; Agate, F.J.; Purkerson, M.L.; Dempsey, E.W. Scanning electron microscopy of the human endometrial surface epithelium. Fertil. Steril. 1972, 23, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Haider, S.; Gamperl, M.; Burkard, T.R.; Kunihs, V.; Kaindl, U.; Junttila, S.; Fiala, C.; Schmidt, K.; Mendjan, S.; Knöfler, M.; et al. Estrogen Signaling Drives Ciliogenesis in Human Endometrial Organoids. Endocrinology 2019, 160, 2282–2297. [Google Scholar] [CrossRef]
- Ruane, P.T.; Tan, C.M.J.; Adlam, D.J.; Kimber, S.J.; Brison, D.R.; Aplin, J.D.; Westwood, M. Protein O-GlcNAcylation Promotes Trophoblast Differentiation at Implantation. Cells 2020, 9, 2246. [Google Scholar] [CrossRef]
- Ruane, P.T.; Koeck, R.; Berneau, S.; Kimber, S.J.; Westwood, M.; Brison, D.R.; Aplin, J. Osmotic stress induces JNK-dependent embryo invasion in a model of implantation. Reproduction 2018, 156, 421–428. [Google Scholar] [CrossRef]
- Fleming, T.P.; Eckert, J.J.; Denisenko, O. The Role of Maternal Nutrition During the Periconceptional Period and Its Effect on Offspring Phenotype. Adv. Exp. Med. Biol. 2017, 1014, 87–105. [Google Scholar]
- Sharma, N.; Kaur, J.; Xu, H.; Nieden, N.Z.; Rancourt, D. Characterization of secretory leukocyte protease inhibitor as an inhibitor of implantation serine proteinases. Mol. Reprod. Dev. 2008, 75, 1136–1142. [Google Scholar] [CrossRef]
- Tang, Q.; Chen, J.; Di, Z.; Yuan, W.; Zhou, Z.; Liu, Z.; Han, S.; Liu, Y.; Ying, G.; Shu, X.; et al. TM4SF1 promotes EMT and cancer stemness via the Wnt/beta-catenin/SOX2 pathway in colorectal cancer. J. Exp. Clin. Cancer Res. 2020, 39, 232. [Google Scholar] [CrossRef]
- Xu, H.; Li, J.; Jin, L.; Zhang, D.; Chen, B.; Liu, X.; Lin, X.; Huang, Y.; Ke, Z.; Liu, J.; et al. Intrauterine hyperglycemia impairs endometrial receptivity via up-regulating SGK1 in diabetes. Sci. China Life Sci. 2022, 65, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Dong, X.-Y.; Yang, P.-W.; Yang, S.-L.; Hu, D.; Zhang, H.-W.; Sui, C. Activation of Uterine Smad3 Pathway Is Crucial for Embryo Implantation. Curr. Med. Sci. 2019, 39, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Kriseman, M.; Monsivais, D.; Agno, J.; Masand, R.P.; Creighton, C.J.; Matzuk, M.M. Uterine double-conditional inactivation of Smad2 and Smad3 in mice causes endometrial dysregulation, infertility, and uterine cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 3873–3882. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stevens, A.; Khashkhusha, T.; Sharps, M.; Garner, T.; Ruane, P.T.; Aplin, J.D. The Human Early Maternal–Embryonic Interactome. Reprod. Med. 2023, 4, 40-56. https://doi.org/10.3390/reprodmed4010006
Stevens A, Khashkhusha T, Sharps M, Garner T, Ruane PT, Aplin JD. The Human Early Maternal–Embryonic Interactome. Reproductive Medicine. 2023; 4(1):40-56. https://doi.org/10.3390/reprodmed4010006
Chicago/Turabian StyleStevens, Adam, Taqua Khashkhusha, Megan Sharps, Terence Garner, Peter T. Ruane, and John D. Aplin. 2023. "The Human Early Maternal–Embryonic Interactome" Reproductive Medicine 4, no. 1: 40-56. https://doi.org/10.3390/reprodmed4010006
APA StyleStevens, A., Khashkhusha, T., Sharps, M., Garner, T., Ruane, P. T., & Aplin, J. D. (2023). The Human Early Maternal–Embryonic Interactome. Reproductive Medicine, 4(1), 40-56. https://doi.org/10.3390/reprodmed4010006