Next-Generation Sequencing Reveals Downregulation of the Wnt Signaling Pathway in Human Dysmature Cumulus Cells as a Hallmark for Evaluating Oocyte Quality

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
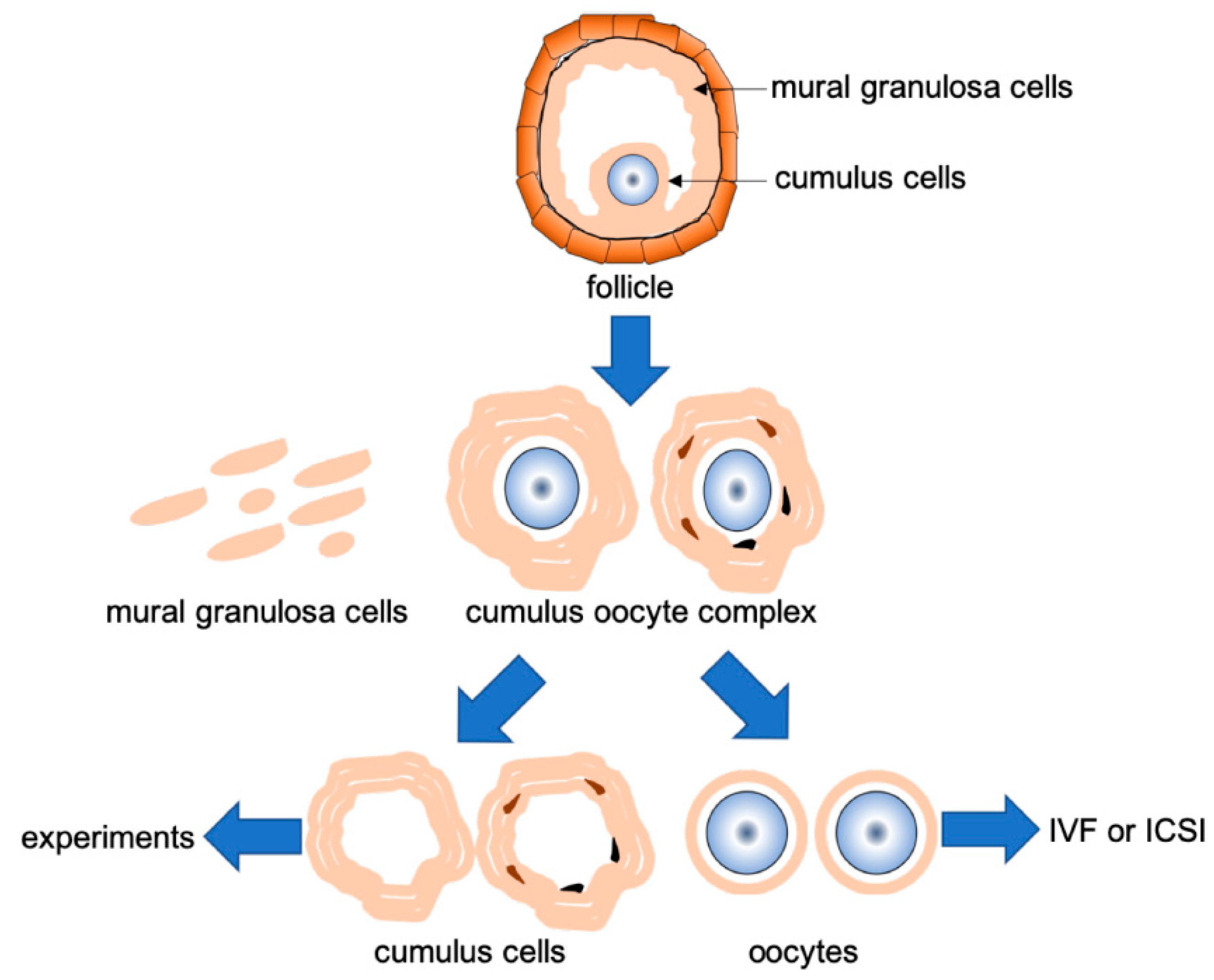
2.1. Patients
2.2. RNA Extraction and NGS
2.3. Gene Expression Profiling Analysis
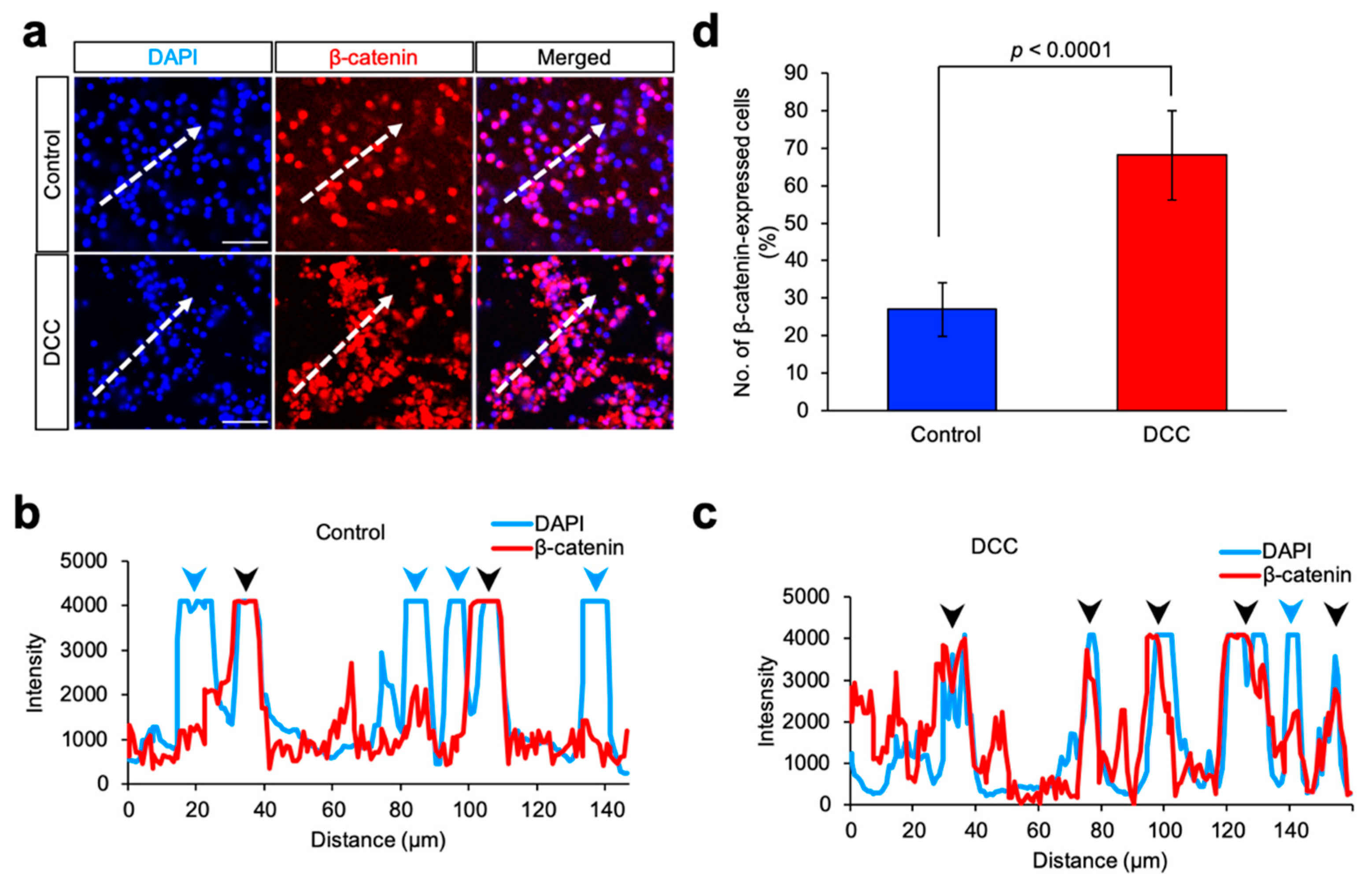
2.4. Immunofluorescence
3. Results
3.1. Sequencing and Mapping Summary
3.2. Differentially Expressed Genes
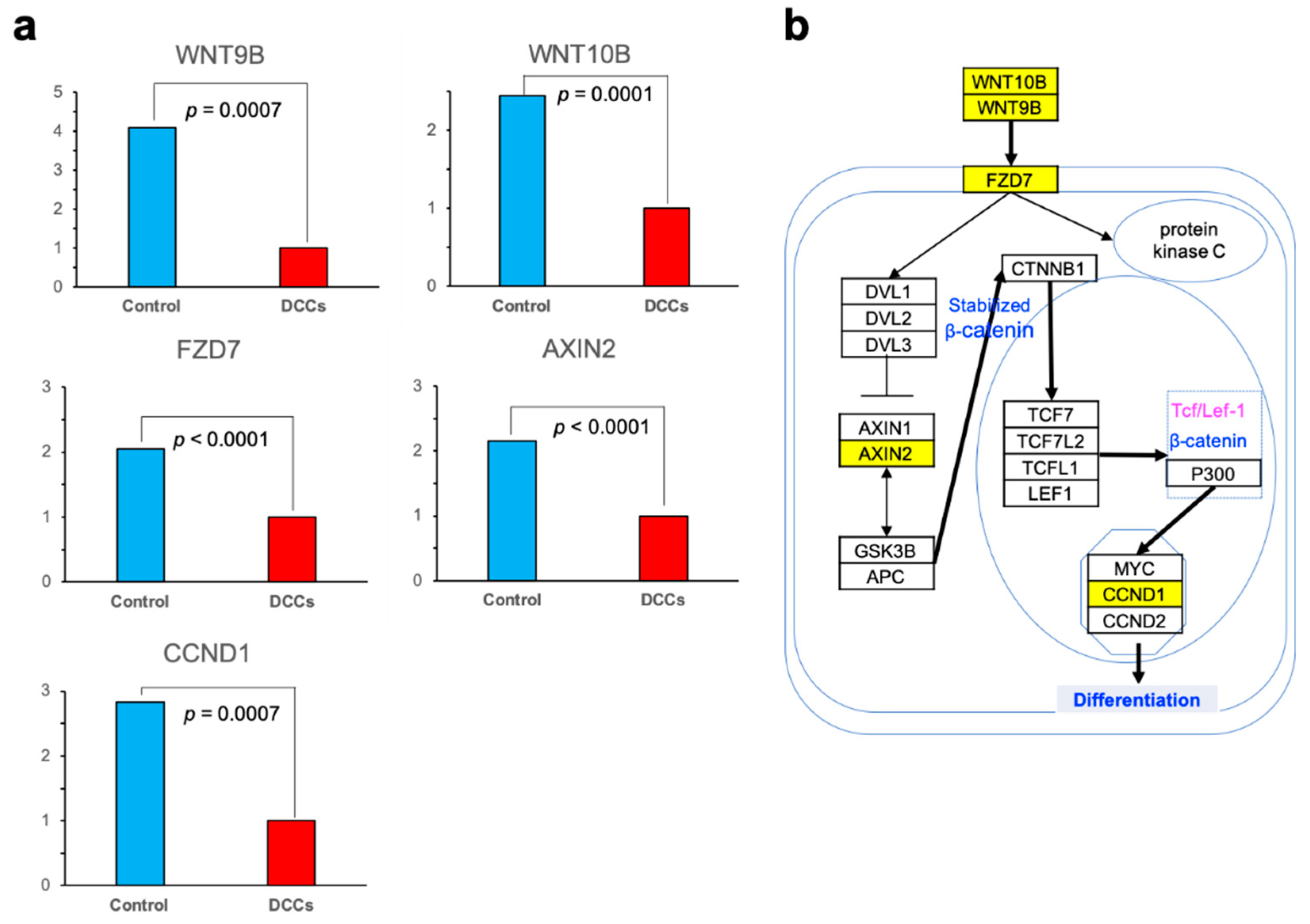
3.3. GO and Pathway Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, X.; Jafari, N.; Barnes, R.B.; Confino, E.; Milad, M.; Kazer, R.R. Studies of gene expression in human cumulus cells indicate pentraxin 3 as a possible marker for oocyte quality. Fertil. Steril. 2005, 83 (Suppl. 1), 1169–1179. [Google Scholar] [CrossRef] [PubMed]
- Heller, D.T.; Schultz, R.M. Ribonucleoside metabolism by mouse oocytes: Metabolic cooperativity between the fully grown oocyte and cumulus cells. J. Exp. Zool. 1980, 214, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Brower, P.T.; Schultz, R.M. Intercellular communication between granulosa cells and mouse oocytes: Existence and possible nutritional role during oocyte growth. Dev. Biol. 1982, 90, 144–153. [Google Scholar] [CrossRef]
- Larsen, W.J.; Wert, S.E.; Brunner, G.D. A dramatic loss of cumulus cell gap junctions is correlated with germinal vesicle breakdown in rat oocytes. Dev. Biol. 1986, 113, 517–521. [Google Scholar] [CrossRef]
- Dekel, N.; Beers, W.H. Development of the rat oocyte in vitro: Inhibition and induction of maturation in the presence or absence of the cumulus oophorus. Dev. Biol. 1980, 75, 247–254. [Google Scholar] [CrossRef]
- Lobo, R.A.; di Zerega, G.S.; Marrs, R.P. Follicular fluid steroid levels in dysmature and mature follicles from spontaneous and hyperstimulated cycles in normal and anovulatory women. J. Clin. Endocrinol. Metab. 1985, 60, 81–87. [Google Scholar] [CrossRef]
- Tsuiki, A.; Preyer, J.; Hung, T.T. Fibronectin and glycosaminoglycans in human preovulatory follicular fluid and their correlation to follicular maturation. Hum. Reprod. 1988, 3, 425–429. [Google Scholar] [CrossRef]
- Lefèvre, B.; Gougeon, A.; Nomé, F.; Testart, J. Effect of a gonadotropin-releasing hormone agonist and gonadotropins on ovarian follicles in cynomolgus monkey: A model for human ovarian hyperstimulation. Fertil. Steril. 1991, 56, 119–125. [Google Scholar] [CrossRef]
- Morgan, P.M.; Boatman, D.E.; Bavister, B.D. Relationships between follicular fluid steroid hormone concentrations, oocyte maturity, in vitro fertilization and embryonic development in the rhesus monkey. Mol. Reprod. Dev. 1990, 27, 145–151. [Google Scholar] [CrossRef]
- Ou, Y.C.; Lan, K.C.; Chang, S.Y.; Kung, F.T.; Huang, F.J. Increased progesterone/estradiol ratio on the day of HCG administration adversely affects success of in vitro fertilization-embryo transfer in patients stimulated with gonadotropin-releasing hormone agonist and recombinant follicle-stimulating hormone. Taiwan J. Obstet. Gynecol. 2008, 47, 168–174. [Google Scholar] [CrossRef]
- Gougeon, A. Dynamics of follicular growth in the human: A model from preliminary results. Hum. Reprod. 1986, 1, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.; Ishida, E.; Yamatoya, K.; Nakamura, A.; Miyado, M.; Miyamoto, Y.; Iwai, M.; Tatsumi, K.; Saito, T.; Saito, K.; et al. Autophagy-disrupted LC3 abundance leads to death of supporting cells of human oocytes. Biochem. Biophys. Rep. 2018, 15, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Muraki, M.; Takahashi, Y.; Imai, M.; Tsukui, T.; Yamakawa, N.; Nakagawa, K.; Ohgi, S.; Horikawa, T.; Iwasaki, W.; et al. Glutathione S-transferase theta 1 expressed in granulosa cells as a biomarker for oocyte quality in age-related infertility. Fertil. Steril. 2008, 90, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.-H.; Zheng, S.-X.; Yin, H.; Zhu, X.-Y.; Lu, F.-T.; Tong, X.-H.; Liu, Y.-S.; Zhang, Y.-W.; Xu, B. Identification of microRNAs that Regulate the MAPK Pathway in Human Cumulus Cells from PCOS Women with Insulin Resistance. Reprod. Sci. 2020, 27, 833–844. [Google Scholar] [CrossRef]
- Xu, Y.; Zhou, T.; Shao, L.; Zhang, B.; Liu, K.; Gao, C.; Gao, L.; Liu, J.; Cui, Y.; Chian, R.-C.; et al. Gene expression profiles in mouse cumulus cells derived from in vitro matured oocytes with and without blastocyst formation. Gene Expr. Patterns 2017, 25–26, 46–58. [Google Scholar] [CrossRef]
- Weizman, N.F.; Wyse, B.A.; Gat, I.; Balakier, H.; Sangaralingam, M.; Caballero, J.; Kenigsberg, S.; Librach, C.L. Triggering method in assisted reproduction alters the cumulus cell transcriptome. Reprod. Biomed. Online 2019, 39, 211–224. [Google Scholar] [CrossRef]
- Xu, B.; Zhang, Y.W.; Tong, X.H.; Liu, Y.S. Characterization of microRNA profile in human cumulus granulosa cells: Identification of microRNAs that regulate Notch signaling and are associated with PCOS. Mol. Cell Endocrinol. 2015, 404, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Chronowska, E. High-throughput analysis of ovarian granulosa cell transcriptome. BioMed Res. Int. 2014, 2014, 213570. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Wang, H.X.; Tekpetey, F.R.; Kidder, G.M. Identification of WNT/beta-CATENIN signaling pathway components in human cumulus cells. Mol. Hum. Reprod. 2009, 15, 11–17. [Google Scholar] [CrossRef]
- Harwood, B.N.; Cross, S.K.; Radford, E.E.; Haac, B.E.; De Vries, W.N. Members of the WNT signaling pathways are widely expressed in mouse ovaries, oocytes, and cleavage stage embryos. Dev. Dyn. 2008, 237, 1099–1111. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, M.; Johnson, M.A.; Greenberg, N.M.; Richards, J.S. Regulated expression of Wnts and Frizzleds at specific stages of follicular development in the rodent ovary. Endocrinology 2002, 143, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Hernandez Gifford, J.A. The role of WNT signaling in adult ovarian folliculogenesis. Reproduction 2015, 150, R137–R148. [Google Scholar] [CrossRef]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Julius, M.A.; Giarre, M.; Zheng, Z.; Brown, A.M.; Kitajewski, J. Transformation by Wnt family proteins correlates with regulation of beta-catenin. Cell Growth Differ. 1997, 8, 1349–1358. [Google Scholar]
- Bjarnadottir, T.K.; Gloriam, D.E.; Hellstrand, S.H.; Kristiansson, H.; Fredriksson, R.; Schioth, H.B. Comprehensive repertoire and phylogenetic analysis of the G protein-coupled receptors in human and mouse. Genomics 2006, 88, 263–273. [Google Scholar] [CrossRef]
- Nieto Gutierrez, A.; McDonald, P.H. GPCRs: Emerging anti-cancer drug targets. Cell Signal. 2018, 41, 65–74. [Google Scholar] [CrossRef]
- Lynch, J.R.; Wang, J.Y. G Protein-Coupled Receptor Signaling in Stem Cells and Cancer. Int. J. Mol. Sci. 2016, 17, 707. [Google Scholar] [CrossRef]
- Tepavčević, V.; Bs, C.K.; Bs, M.S.A.; Meppiel, E.; Ms, S.M.; Ms, R.A.; Ravassard, P.; Kennedy, T.E.; Nait-Oumesmar, B.; Lubetzki, C. Early netrin-1 expression impairs central nervous system remyelination. Ann. Neurol. 2014, 76, 252–268. [Google Scholar] [CrossRef]
- Gao, K.; Niu, J.; Dang, X. Neuroprotection of netrin-1 on neurological recovery via Wnt/β-catenin signaling pathway after spinal cord injury. Neuroreport 2020, 31, 537–543. [Google Scholar] [CrossRef]
- Li, Y.; Li, L.; Zhang, G.; Wang, Y.; Chen, H.; Kong, R.; Pan, S.; Sun, B. Crucial microRNAs and genes in metformin’s anti-pancreatic cancer effect explored by microRNA-mRNA integrated analysis. Investig. New Drugs 2018, 36, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Eisinger, F.; Patzelt, J.; Langer, H.F. The Platelet Response to Tissue Injury. Front. Med. 2018, 5, 317. [Google Scholar] [CrossRef] [PubMed]
- Bijak, M.; Saluk-Bijak, J. Flavonolignans inhibit the arachidonic acid pathway in blood platelets. BMC Complement. Altern. Med. 2017, 17, 396. [Google Scholar] [CrossRef] [PubMed]
- Vakifahmetoglu-Norberg, H.; Ouchida, A.T.; Norberg, E. The role of mitochondria in metabolism and cell death. Biochem. Biophys. Res. Commun. 2017, 482, 426–431. [Google Scholar] [CrossRef]
- Susin, S.A.; Lorenzo, H.K.; Zamzami, N.; Marzo, I.; Snow, B.E.; Brothers, G.M.; Mangion, J.; Jacotot, E.; Costantini, P.; Loeffler, M.; et al. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature 1999, 397, 441–446. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway | p-Value | Matched Entities | Pathway Entities of Experiment Type |
|---|---|---|---|
| Hs_Wnt_Signaling_Pathway_and_Pluripotency_ WP399_74897 | 0.0000586 | 5 | 100 |
| Hs_Integrated_Pancreatic_Cancer_Pathway_ WP2377_71228 | 0.009160941 | 4 | 200 |
| Hs_Platelet_homeostasis_WP1885_77031 | 0.005834554 | 2 | 31 |
| Hs_Arachidonic_acid_metabolism_ WP2650_76814 | 0.000182 | 3 | 27 |
| Hs_Netrin-1_signaling_WP1868_76847 | 0.000635 | 3 | 42 |
| Hs_Integration_of_energy_metabolism_ WP1831_77011 | 0.004354545 | 3 | 81 |
| Hs_GPCR_downstream_signaling_ WP1824_76910 | 0.005903522 | 6 | 406 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akino, R.; Matsui, D.; Kawahara-Miki, R.; Amita, M.; Tatsumi, K.; Ishida, E.; Kang, W.; Takada, S.; Miyado, K.; Sekizawa, A.; et al. Next-Generation Sequencing Reveals Downregulation of the Wnt Signaling Pathway in Human Dysmature Cumulus Cells as a Hallmark for Evaluating Oocyte Quality. Reprod. Med. 2020, 1, 205-215. https://doi.org/10.3390/reprodmed1030016
Akino R, Matsui D, Kawahara-Miki R, Amita M, Tatsumi K, Ishida E, Kang W, Takada S, Miyado K, Sekizawa A, et al. Next-Generation Sequencing Reveals Downregulation of the Wnt Signaling Pathway in Human Dysmature Cumulus Cells as a Hallmark for Evaluating Oocyte Quality. Reproductive Medicine. 2020; 1(3):205-215. https://doi.org/10.3390/reprodmed1030016
Chicago/Turabian StyleAkino, Ryosuke, Daisuke Matsui, Ryouka Kawahara-Miki, Mitsuyoshi Amita, Kuniko Tatsumi, Eri Ishida, Woojin Kang, Shuji Takada, Kenji Miyado, Akihiko Sekizawa, and et al. 2020. "Next-Generation Sequencing Reveals Downregulation of the Wnt Signaling Pathway in Human Dysmature Cumulus Cells as a Hallmark for Evaluating Oocyte Quality" Reproductive Medicine 1, no. 3: 205-215. https://doi.org/10.3390/reprodmed1030016
APA StyleAkino, R., Matsui, D., Kawahara-Miki, R., Amita, M., Tatsumi, K., Ishida, E., Kang, W., Takada, S., Miyado, K., Sekizawa, A., Saito, T., Kono, T., & Saito, H. (2020). Next-Generation Sequencing Reveals Downregulation of the Wnt Signaling Pathway in Human Dysmature Cumulus Cells as a Hallmark for Evaluating Oocyte Quality. Reproductive Medicine, 1(3), 205-215. https://doi.org/10.3390/reprodmed1030016