
Churg-Strauss Syndrome or Eosinophilic Granulomatosis with Polyangiitis

Abstract
:1. Introduction
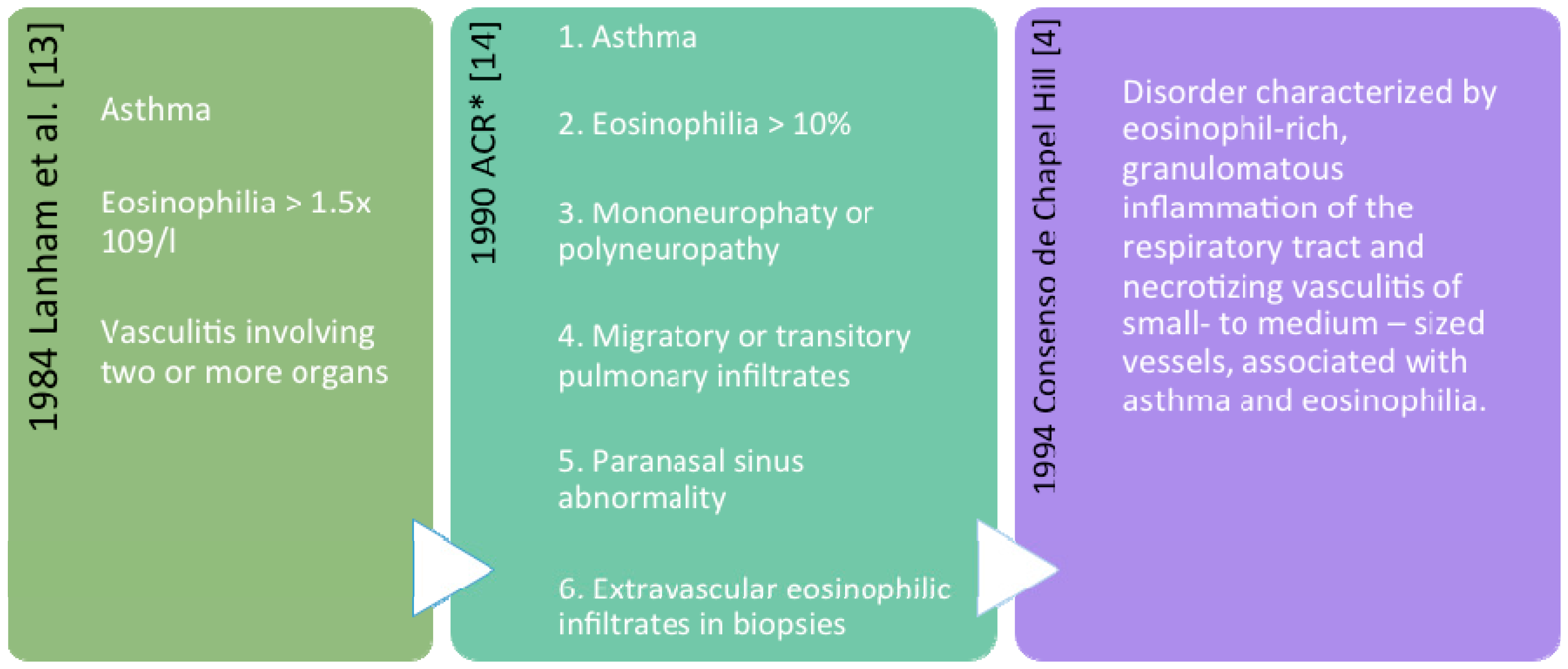
2. Definition and Classification
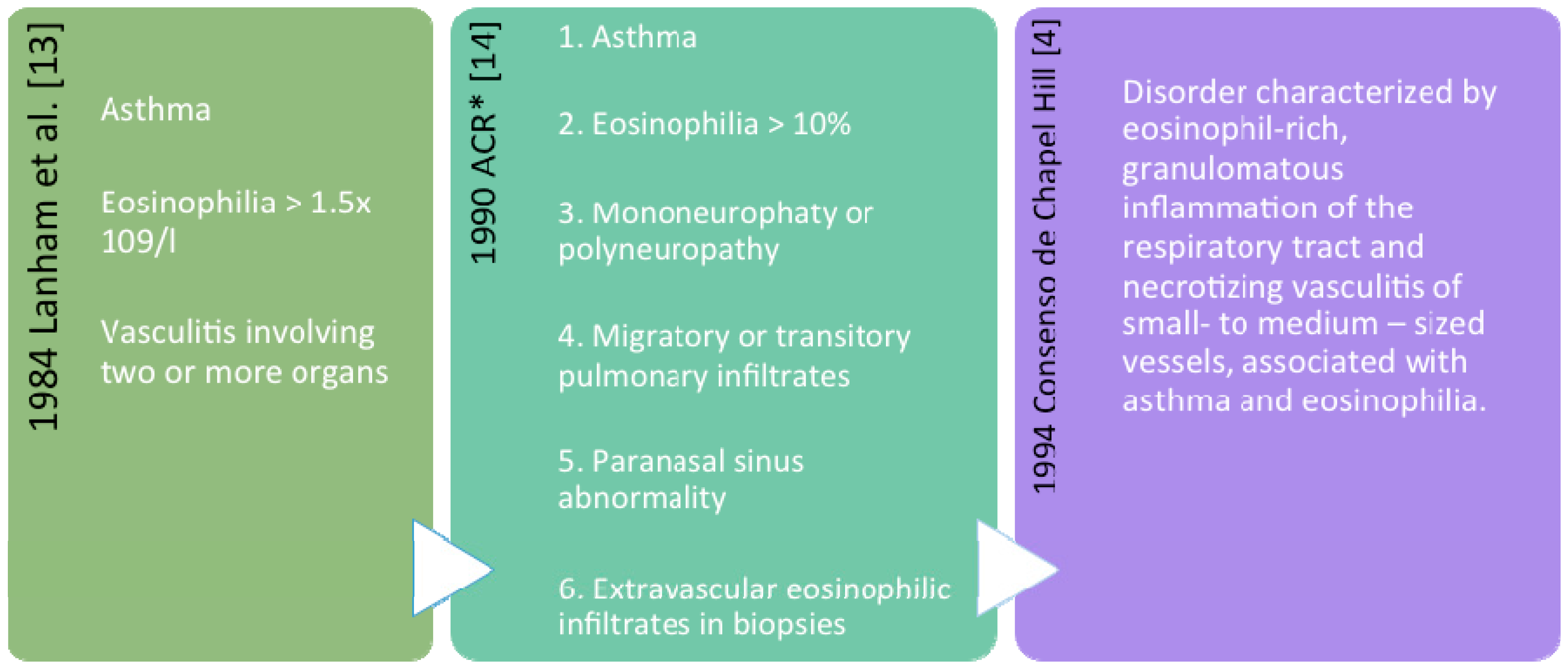
Nomenclature
3. Epidemiology
4. Genetic and Environmental Factors
5. Clinical Features
6. Specific Organs
6.1. Upper Airways
6.2. Lower Airways
6.3. Other Organs and Systems
7. ANCA Phenotypes

{kind=link}
| Sets of Clinical Manifestations | Subsets of Clinical Manifestations | ||||
|---|---|---|---|---|---|
| Allergic Phase | Eosinophilic Phase | Vasculitic Phase | ANCAs− [55] | ANCAs+ [49] | |
| Upper Airway | Allergic rhinitis | General symptoms (fever, weight loss, fatigue) Peripheral neuropathy (mononeuritis multiplex) Purpura Affected skin (livedo, ulcers) | Eosinophilic | Vasculitic | |
| Rhinosinusitis | Pulmonary infiltrates | Purpura | |||
| Nasal polyps | Endocardiomyopathy | Glomerulonephritis | |||
| Hearing loss | Peripheral neuropathy | ||||
| Lower Airway | Asthma | Pulmonary parenchymal involvement | Renal manifestations (glomerulonephritis) | ||
| Other Organ and Systems | Cardiac involvement | Cardiac involvement | Neuropathic pain and sensory deficits | ||
| Gastrointestinal involvement | |||||
| Thromboembolic events | Cardiac involvement | ||||
8. Diagnosis
8.1. Blood Cells and Biomarkers
8.2. Histopathology
8.3. Imaging
8.3.1. Chest Radiography and CT Scan
8.3.2. Sinonasal CT Scan
8.3.3. Sinunasal Plain X-rays
8.4. Lung Function
8.5. Bronchoscopy
9. Differential Diagnosis
9.1. Hypereosinophilic Syndrome (HES)
9.2. Allergic Bronchopulmonary Aspergillosis (ABPA)
9.3. Acute Eosinophilic Pneumonia
9.4. Other Vasculitis
9.5. IgG4-Related Disease
10. Therapeutical Options
10.1. General Treatment
- Cardiomyopathy
- Gastrointestinal involvement
- Central nervous system involvement
- Proteinuria (>1 g/24 h)
- Serum creatinine (>150 mmol/L)
10.2. Glucocorticoids and Immunosupressants
10.3. Biological Therapies (Monoclonal Antibodies)
10.4. Chronic Rhinosinusitis with Nasal Polyps
11. Prognosis
12. Conclusions
Author Contributions
Conflicts of Interest
References
- Churg, J.; Strauss, L. Allergic granulomatosis, allergic angiitis, and periarteritis nodosa. Am. J. Pathol. 1951, 27, 277–301. [Google Scholar] [PubMed]
- Jennette, J.; Falk, R.; Bacon, P.; Basu, N.; Cid, M.; Ferrario, F.; Flores-Suarez, L.F.; Gross, W.L.; Guillevin, L.; Hagen, E.C.; et al. 2012 Revised international chapel hill consensus conference nomenclature of vasculitides. Arthritis Rheum. 2013, 65, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Sinico, R.A.; di Toma, L.; Maggiore, U.; Tosoni, C.; Bottero, P.; Sabadini, E.; Giammarresi, G.; Tumiati, B.; Gregorini, G.; Pesci, A.; et al. Renal involvement in Churg-Strauss syndrome. Am. J. Kidney Dis. 2006, 47, 770–779. [Google Scholar] [CrossRef] [PubMed]
- Jennette, J.C.; Falk, R.J.; Andrassy, K.; Bacon, P.A.; Churg, J.; Gross, W.L.; Hagen, E.C.; Hoffman, G.S.; Hunder, G.G.; Kallenberg, C.G.; et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 1994, 37, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Dallos, T.; Heiland, G.R.; Strehl, J.; Karonitsch, T.; Gross, W.L.; Moosig, F.; Holl-Ulrich, C.; Distler, J.H.; Manger, B.; Schett, G.; et al. CCL17/thymus and activation-related chemokine in Churg-Strauss syndrome. Arthritis Rheum. 2010, 62, 3496–3503. [Google Scholar] [CrossRef] [PubMed]
- Terrier, B.; Bieche, I.; Maisonobe, T.; Laurendeau, I.; Rosenzwajg, M.; Kahn, J.E.; Diemert, M.C.; Musset, L.; Vidaud, M.; Sène, D.; et al. Interleukin-25: A cytokine linking eosinophils and adaptive immunity in Churg–Strauss syndrome. Blood 2010, 116, 4523–4531. [Google Scholar] [CrossRef] [PubMed]
- Vaglio, A.; Martorana, D.; Maggiore, U.; Grasselli, C.; Zanetti, A.; Pesci, A.; Garini, G.; Manganelli, P.; Bottero, P.; Tumiati, B.; et al. HLADRB4 as a genetic risk factor for Churg-Strauss syndrome. Arthritis Rheum. 2007, 56, 3159–3166. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, S.; Hellmich, B.; Gross, W.L.; Epplen, J.T. Associations of Churg-Strauss síndrome with the HLA-DRB1 locus, and relationship to the genetics of antineutrophil cytoplasmic antibody-associated vasculitides: Comment on the article by Vaglio et al. Arthritis Rheum. 2008, 58, 329–330. [Google Scholar] [CrossRef] [PubMed]
- Buzio, C.; Oliva, E. Diagnosis of Churg-Strauss syndrome: Eotaxin-3 makes it easier. Rheumatology 2011, 50, 1737–1738. [Google Scholar] [CrossRef] [PubMed]
- Guillevin, L.; Pagnoux, C.; Seror, R.; Mahr, A.; Mouthon, L.; le Toumelin, P. The five-factor score revisited: Assessment of prognoses of systemic necrotizing vasculitides based on the French Vasculitis Study Group (FVSG) cohort. Medicine 2011, 90, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Zwerina, J.; Bach, C.; Martorana, D.; Jatzwauk, M.; Hegasy, G.; Moosig, F.; Bremer, J.; Wieczorek, S.; Moschen, A.; Tilg, H.; et al. Eotaxin-3 in Churg-Strauss syndrome: A clinical and immunogenetic study. Rheumatology 2011, 50, 1823–1827. [Google Scholar] [CrossRef] [PubMed]
- Koukoulaki, M.; Smith, K.G.; Jayne, D.R. Rituximab in Churg-Strauss syndrome. Ann. Rheum. Dis. 2006, 65, 557–559. [Google Scholar] [CrossRef] [PubMed]
- Lanham, J.G.; Elkon, K.B.; Pusey, C.D.; Hughes, G.R. Systemic vasculitis with asthma and eosinophilia: A clinical approach to the Churg-Strauss syndrome. Medicine 1984, 63, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Masi, A.T.; Hunder, G.G.; Lie, J.T.; Michel, B.A.; Bloch, D.A.; Arend, W.P.; Calabrese, L.H.; Edworthy, S.M.; Fauci, A.S.; Leavitt, R.Y.; et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss síndrome (allergic granulomatosis and angiitis). Arthritis Rheum. 1990, 33, 1094–1100. [Google Scholar] [CrossRef] [PubMed]
- Watts, R.A.; Lane, S.; Scott, D.G. What is known about the epidemiology of the vasculitides? Best Pract. Res. Clin. Rheumatol. 2005, 19, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Herlyn, K.; Hellmich, B.; Gross, W.L.; Reinhold-Keller, E. Stable incidence of systemic vasculitides in schleswig-holstein, Germany. Dtsch. Arztebl. Int. 2008, 105, 355–361. [Google Scholar] [PubMed]
- Mahr, A.; Guillevin, L.; Poissonnet, M.; Ayme, S. Prevalences of polyarteritis nodosa, microscopic polyangiitis, Wegener’s granulomatosis, and Churg-Strauss syndrome in a French urban multiethnic population in 2000: A capture-recapture estimate. Arthritis Rheum 2004, 51, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, A.J.; Jacobsson, L.T.; Mahr, A.D.; Sturfelt, G.; Segelmark, M. Prevalence of Wegener’s granulomatosis, microscopic polyangiitis, polyarteritis nodosa and Churg-Strauss syndrome within a defined population in southern Sweden. Rheumatology 2007, 46, 1329–1337. [Google Scholar] [CrossRef] [PubMed]
- Moosig, F.; Bremer, J.P.; Hellmich, B.; Holle, J.U.; Holl-Ulrich, K.; Laudien, M.; Matthis, C.; Metzler, C.; Nölle, B.; Richardt, G.; et al. A vasculitis centre based management strategy leads to improved outcome in eosinophilic granulomatosis and polyangiitis (Churg-Strauss, EGPA): Monocentric experiences in 150 patients. Ann. Rheum. Dis. 2013, 72, 1011–1017. [Google Scholar] [CrossRef] [PubMed]
- Zwerina, J.; Eger, G.; Englbrecht, M.; Manger, B.; Schett, G. Churg-Strauss syndrome in childhood: A systematic literature review and clinical comparison with adult patients. Semin. Arthritis Rheum. 2009, 39, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Piram, M.; Maldini, C.; Mahr, A. Effect of race/ethnicity on risk, presentation and course of connective tissue diseases and primary systemic vasculitides. Curr. Opin. Rheumatol. 2012, 24, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, S.; Hellmich, B.; Arning, L.; Moosig, F.; Lamprecht, P.; Gross, W.L.; Epplen, J.T. Functionally relevant variations of the interleukin-10 gene associated with antineutrophil cytoplasmic antibody-negative Churg-Strauss syndrome, but not with Wegener’s granulomatosis. Arthritis Rheum. 2008, 58, 1839–1848. [Google Scholar] [CrossRef] [PubMed]
- Martorana, D.; Maritati, F.; Malerba, G.; Bonatti, F.; Alberici, F.; Oliva, E.; Sebastio, P.; Manenti, L.; Brugnano, R.; Catanoso, M.G.; et al. PTPN22 R620W polymorphism in the ANCA-associated vasculitides. Rheumatology 2012, 51, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, S.; Hoffjan, S.; Chan, A.; Rey, L.; Harper, L.; Fricke, H.; Holle, J.U.; Gross, W.L.; Epplen, J.T.; Lamprecht, P. Novel association of the CD226 (DNAM-1) Gly307Ser polymorphism in Wegener’s granulomatosis and confirmation for multiple sclerosis in German patients. Genes Immun. 2009, 10, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Wieczorek, S.; Holle, J.U.; Bremer, J.P.; Wibisono, D.; Moosig, F.; Fricke, H.; Assmann, G.; Harper, L.; Arning, L.; Gross, W.L.; et al. Contrasting association of a non-synonymous leptin receptor gene polymorphism with Wegener’s granulomatosis and Churg-Strauss syndrome. Rheumatology 2010, 49, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Hammanci, K.; Anil, H.; Kocak, A.; Dinleyici, E.C. Familial eosinophilic granilomatosis with polyangiitis in a mother and daughter. BMJ Case Rep. 2014, 2014. [Google Scholar] [CrossRef]
- Lane, S.E.; Watts, R.A.; Bentham, G.; Innes, N.J.; Scott, D.G. Are environmental factors important in primary systemic vasculitis? A casecontrol study. Arthritis Rheum. 2003, 48, 814–823. [Google Scholar] [CrossRef] [PubMed]
- Razek, A.A.; Castillo, M. Imaging appearance of granulomatous lesions of head and neck. Eur J Radiol 2010, 76, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Hubner, C.; Dietz, A.; Stremmel, W.; Stiehl, A.; Andrassy, H. Macrolide-induced Churg-Strauss syndrome in a patient with atopy. Lancet 1997, 350. [Google Scholar] [CrossRef]
- Lilly, C.M.; Churg, A.; Lazarovich, M.; Pauwels, R.; Hendeles, L.; Rossenwasser, L.J.; Ledford, D.; Wechsler, M.E. Asthma therapies and Churg-Strauss syndrome. J. Allergy Clin. Immunol. 2002, 109, S1–S20. [Google Scholar] [CrossRef] [PubMed]
- Stirling, R.G.; Chung, K.F. Leukotriene antagonists and Churg-Strauss syndrome: The smoking gun. Thorax 1999, 54, 865–866. [Google Scholar] [CrossRef] [PubMed]
- Josefson, D. Asthma drug linked with Churg-Strauss syndrome. BMJ 1997, 315, 330. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, M.E.; Wong, D.A.; Miller, M.K.; Lawrence-Miyasaki, L. Churg-Strauss síndrome in patients treated with omalizumab. Chest 2009, 136, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Hauser, T.; Mahr, A.; Metzler, C.; Coste, J.; Sommerstein, R.; Gross, W.L.; Guillevin, L.; Hellmich, B. The leucotriene receptor antagonist montelukast and the risk of Churg-Strauss syndrome: A casecrossover study. Thorax 2008, 63, 677–682. [Google Scholar] [CrossRef] [PubMed]
- Bibby, S.; Healy, B.; Steele, R.; Kumareswaran, K.; Nelson, H.; Beasley, R. Association between leukotriene receptor antagonist therapy and Churg-Strauss syndrome: An analysis of the FDA AERS database. Thorax 2010, 65, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Iglesis, E.; Camacho Lovillo, M.; Delgado Pellecín, I.; Lirola Cruz, M.J.; Falcon Neyra, M.D.; Salazar Quero, J.C.; Bernabeu-Wittel, J.; González Valencia, J.P.; Neth, O. Successful management of Churg-Strauss syndrome ursing omalizumab as adjuvant immunomodulatory therapy: First documented peadiatric case. Pediatr. Pulmonol. 2014, 49, E78–E81. [Google Scholar] [CrossRef] [PubMed]
- Graziani, A.; Quercia, O.; Girelli, F.; Martelli, A.; Mirici Cappa, F.; Stefanini, G.F. Omalizumab treatment in patient with severe asthma and eosinophilic granulomatosis with polyangiitis. A case report. Eur. Ann. Allergy Clin. Immunol. 2014, 46, 226–228. [Google Scholar] [PubMed]
- Vaglio, A.; Casazza, I.; Grasselli, C.; Corradi, D.; Sinico, R.A.; Buzio, C. Churg-Strauss syndrome. Kidney Int. 2009, 76, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Noth, I.; Strek, M.E.; Leff, A.R. Churg-Strauss syndrome. Lancet 2003, 361, 587–594. [Google Scholar] [CrossRef]
- Lie, J.T. Limited forms of Churg-Strauss syndrome. Pathol. Annu. 1993, 28, 199–220. [Google Scholar] [PubMed]
- Bacciu, A.; Bacciu, S.; Mercante, G.; Ingegnoli, F.; Grasselli, C.; Vaglio, A.; Pasanisi, E.; Vincenti, V.; Garini, G.; Ronda, N.; et al. Ear, nose and throat manifestations of Churg-Strauss syndrome. Acta Otolaryngol. 2006, 126, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Bacciu, A.; Buzio, C.; Giordano, D.; Pasanisi, E.; Vincenti, V.; Mercante, G.; Grasselli, C.; Bacciu, S. Nasal polyposis in Churg-Strauss syndrome. Laryngoscope 2008, 118, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Alobid, I.; Guilemany, J.M.; Mullol, J. Nasal manifestations of systemic illnesses. Curr. Allergy Asthma Rep. 2004, 4, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Alobid, I.; Mullol, J.; Cid, M.C. Rinitis of granulomatous and vasculitic diseases. Clin. Allergy Immunol. 2007, 19, 221–239. [Google Scholar] [PubMed]
- Bottero, P.; Bonini, M.; Vecchio, F.; Grittini, A.; Patruno, G.M.; Colombo, B.; Sinico, R.A. The common allergens in the Churg-Strauss syndrome. Allergy 2007, 62, 1288–1294. [Google Scholar] [CrossRef] [PubMed]
- Keogh, K.A.; Specks, U. Churg-Strauss syndrome. Semin. Respir. Crit. Care Med. 2006, 27, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Lee, K.S.; Chung, M.P.; Han, J.; Chong, S.; Chung, M.J.; Chin, A.Y.; Kim, H.Y. Pulmonary involvement in Churg-Strauss syndrome: An analysis of CT, clinical, and pathologic findings. Eur. Radiol. 2007, 17, 3157–3165. [Google Scholar] [CrossRef] [PubMed]
- Guillevin, L.; Cohen, P.; Gayraud, M.; Lhote, F.; Jarrousse, B.; Casassus, P. Churg-Strauss syndrome. Clinical study and long-term followup of 96 patients. Medicine 1999, 78, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Sinico, R.A.; di Toma, L.; Maggiore, U.; Bottero, P.; Radice, A.; Tosoni, C.; Grasselli, C.; Pavone, L.; Gregorini, G.; Monti, S.; et al. Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg-Strauss syndrome. Arthritis Rheum. 2005, 52, 2926–2935. [Google Scholar] [CrossRef] [PubMed]
- Guillevin, L.; Lhote, F.; Gayraud, M.; Cohen, P.; Jarrousse, B.; Lortholary, O.; Thibult, N.; Casassus, P. Prognostic factors in polyarteritis nodosa and Churg-Strauss syndrome. A prospective study in 342 patients. Medicine 1996, 75, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Neumann, T.; Manger, B.; Schmid, M.; Kroegel, C.; Hansch, A.; Kaiser, W.A.; Reinhardt, D.; Wolf, G.; Hein, G.; Mall, G.; et al. Cardiac involvement in Churg-Strauss syndrome: Impact of endomyocarditis. Medicine 2009, 88, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Dennert, R.M.; van Paassen, P.; Schalla, S.; Kuznetsova, T.; Alzand, B.S.; Staessen, J.A.; Velthuis, S.; Crijns, H.J.; Tervaert, J.W.C.; Heymans, S. Cardiac involvement in Churg-Strauss syndrome. Arthritis Rheum. 2010, 62, 627–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allenbach, Y.; Seror, R.; Pagnoux, C.; Teixeira, L.; Guilpain, P.; Guillevin, L. High frequency of venous thromboembolic events in Churg-Strauss syndrome, Wegener’s granulomatosis and microscopic polyangiitis but not polyarteritis nodosa: A systematic retrospective study on 1130 patients. Ann. Rheum. Dis. 2009, 68, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Vaglio, A.; Corradi, D.; Ronda, N.; Garini, G.; Buzio, C. Large bowel obstruction heralding Churg-Strauss syndrome. Am. J. Gastroenterol. 2004, 99, 562–563. [Google Scholar] [CrossRef] [PubMed]
- Sable-Fourtassou, R.; Cohen, P.; Mahr, A.; Pagnoux, C.; Mouthon, L.; Jayne, D.; Blockmans, D.; Cordier, J.F.; Delaval, P.; Puechal, X.; et al. Antineutrophil cytoplasmic antibodies and the Churg-Strauss syndrome. Ann. Intern. Med. 2005, 143, 632–638. [Google Scholar] [CrossRef] [PubMed]
- Sironen, R.K.; Seppa, A.; Kosma, V.M.; Kuopio, T. Churg-Strauss syndrome manifested by appendicitis, cholecystitis and superficial micronodular liver lesions–an unusual clinicopathological presentation. J. Clin. Pathol. 2010, 63, 848–850. [Google Scholar] [CrossRef] [PubMed]
- Keogh, K.A.; Specks, U. Churg-Strauss syndrome: Clinical presentation, antineutrophil cytoplasmic antibodies, and leukotriene receptor antagonists. Am. J. Med. 2003, 115, 284–290. [Google Scholar] [CrossRef]
- Cattaneo, L.; Chierici, E.; Pavone, L.; Grasselli, C.; Manganelli, P.; Buzio, C.; Pavesi, G. Peripheral neuropathy in Wegener’s granulomatosis, Churg-Strauss syndrome and microscopic polyangiitis. J. Neurol. Neurosurg. Psychiatry 2007, 78, 1119–1123. [Google Scholar] [CrossRef] [PubMed]
- Healy, B.; Bibby, S.; Steele, R.; Weatherall, M.; Nelson, H.; Beasley, R. Antineutrophil cytoplasmic autoantibodies and myeloperoxidase autoantibodies in clinical expression of Churg-Strauss syndrome. J. Allergy Clin. Immunol. 2013, 131. [Google Scholar] [CrossRef] [PubMed]
- Gendelman, S.; Zeft, A.; Spalding, S.J. Childhood-onset eosinophilic granulomatosis with polyangiitis (formerly Churg-Strauss syndrome): A contemporary single-center cohort. J. Rheumatol. 2013, 40, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Pagnoux, C.; Guilpain, P.; Guillevin, L. Churg-Strauss syndrome. Curr. Opin. Rheumatol. 2007, 19, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Vaglio, A.; Strehl, J.D.; Manger, B.; Maritati, F.; Alberici, F.; Beyer, C.; Rech, J.; Sinico, R.A.; Bonatti, F.; Battistelli, L.; et al. IgG4 immune response in Churg-Strauss syndrome. Ann. Rheum. Dis. 2012, 71, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Zwerina, J.; Axmann, R.; Manger, B.; Schett, G. The emergence of antineutrophil cytoplasmic antibodies may precede the clinical onset of Churg-Strauss syndrome. Arthritis Rheum. 2009, 60, 626–627. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, K.; Gross, W.L.; Moosig, F. Extended follow-up after stopping mepolizumab in relapsing/refractory Churg-Strauss Syndrome. Clin. Exp. Rheumatol. 2012, 30, S62–S65. [Google Scholar] [PubMed]
- Grayson, P.C.; Monach, P.A.; Pagnoux, C.; Cuthbertson, D.; Carette, S.; Hoffman, G.S.; Khalidi, N.A.; Koening, C.L.; Langford, C.A.; Maksimowicz-McKinnon, K.; et al. Value of commonly measured laboratory tests as biomarkers of disease activity and predictors of relapse in eosinophilic granulomatosis with polyangiitis. Rheumatology. 2015, 54, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Jeong, Y.J.; Kim, K.I.; Seo, I.J.; Lee, C.H.; Lee, K.N.; Kim, K.N.; Kim, J.S.; Kwon, W.J. Eosinophilic lung disease: A clinical, radiological and pathological view. Radiographic 2007, 27, 617–637. [Google Scholar] [CrossRef] [PubMed]
- Szczeklik, W.; Sokolowska, B.; Mastalerz, L.; Grzanka, P.; Górka, J.; Pacułt, K.; Miszalski-Jamka, T.; Soja, J.; Musiał, J. Pulmonary findings in Churg-Strauss syndrome in chest radiography and high resolution computed tomography at the time of initial diagnosis. Clin. Rheumatol. 2010, 29, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Furuiye, M.; Yoshimura, N.; Kobayashi, A.; Tamaoka, M.; Miyazaki, Y.; Ohtani, Y.; Miyake, S.; Inase, N.; Yoshizawa, Y. Churg-Strauss versus chronic eosinophilic pneumonia in high resolution computed tomography findings. J. Comput. Assist. Tomogr. 2010, 34, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Lund, V.J.; Mackay, I.S. Stating in rhinosinusitis. Rhinology 1993, 31, 183–184. [Google Scholar] [PubMed]
- Metson, R.; Gliklich, R.E.; Stankiewicz, J.A.; Kennedy, D.W.; Duncavange, J.A.; Hofman, S.R.; Ohnishi, T.; Terrell, J.E.; White, P.S. Comparison of sinus computer tomography staging system. Otolaryngol Head Neck Surg. 1997, 117, 372–379. [Google Scholar] [CrossRef]
- Fokkens, W.J.; Lund, V.J.; Mullol, J.; Bachert, C.; Alobid, I.; Baroody, F.; Cohen, N.; Cervin, A.; Douglas, R.; Gevaert, P.; et al. EPOS 2012: European position paper on rhinosinusitis and nasal polyps 2012. A summary for otorhinolaryngologists. Rhinology 2012, 50, 1–12. [Google Scholar] [PubMed]
- Szczeklik, W.; Sokolowska, B.M.; Zuk, J.; Mastalerz, L.; Szczeklik, A.; Musiał, J. The course of asthma in Churg Strauss syndrome. J. Asthma 2011, 48, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Lotvall, J.; Akdis, C.A.; Bacharier, L.B.; Bjermer, L.; Casale, T.B.; Custovic, A.; Lemanske, R.F.; Wardlaw, A.J.; Wenzel, S.E.; Greenberger, P.A. Endotypes asthma: A new approach to classification of disease entities asthma syndrome. J. Allergy Clin. Inmunol. 2011, 127, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Amelink, M.; de Groot, J.C.; de Nijs, S.B.; Lutter, R.; Zwinderman, A.H.; Sterk, P.J.; ten Brinke, A.; Bel, E.H. Severe adult-onset asthma: A distinct phenotype. J. Allergy Clin. Immunol. 2013, 132, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Szczeklik, W.; Grzanka, P.; Mastalerz, L.; Sokołowska, B.; Musial, J. Lung involvement in Churg-Strauss. syndrome as related to the activity of the disease. Allergy 2010, 65, 1484–1485. [Google Scholar] [CrossRef] [PubMed]
- Szczeklik, W.; Jakiela, B.; Adamek, D.; Musial, J. cutting-edge issues in the Churg Strauss syndrome. Clin. Rev. Allergy Immunol. 2013, 44, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Groh, M.; Pagnoux, C.; Baldini, C.; Bel, E.; Bottero, P.; Cottin, V.; Dalhoff, K.; Dunogué, B.; Gross, W.; Holle, J.; et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss) (EGPA) Consensus Task Force recommendations for evaluation and management. Eur. J. Intern. Med. 2015, 26, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Simon, H.U.; Rothenberg, M.E.; Bochner, B.S.; Weller, P.F.; Wardlaw, A.J.; Wechsler, M.E.; Rosenwasser, L.J.; Roufosse, F.; Gleich, G.J.; Klion, A.D. Refining the definition of hypereosinophilic syndrome. J. Allergy Clin. Immunol. 2010, 126, 45–49. [Google Scholar] [CrossRef] [PubMed]
- Gotlib, J. World Health Organization-defined eosinophilic disorders: 2012 update on diagnosis, risk stratification, and management. Am. J. Hematol. 2012, 87, 903–914. [Google Scholar] [CrossRef] [PubMed]
- Khoury, P.; Zagallo, P.; Talar-Williams, C.; Santos, C.S.; Dinerman, E.; Holland, N.C.; Klion, A.D. Serum biomarkers are similar in Churg-Strauss syndrome and hypereosinophilic syndrome. Allergy 2012, 67, 1149–1156. [Google Scholar] [CrossRef] [PubMed]
- Corradi, D.; Vaglio, A.; Maestri, R.; Legname, V.; Leonardi, G.; Bartoloni, G.; Buzio, C. Eosinophilic myocarditis in a patient with idiopathic hypereosinophilic syndrome: Insights into mechanisms of myocardial cell death. Hum. Pathol. 2004, 35, 1160–1163. [Google Scholar] [CrossRef] [PubMed]
- Polzer, K.; Karonitsch, T.; Neumann, T.; Eger, G.; Haberler, C.; Soleiman, A.; Hellmich, B.; Csernok, E.; Distler, J.; Manger, B.; et al. Eotaxin-3 is involved in Churg-Strauss síndrome a serum marker closely correlating with disease activity. Rheumatology 2008, 47, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Sinico, R.A.; Bottero, P. Churg-Strauss angiitis. Best Pract. Res. Clin. Rheumatol. 2009, 23, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Wechsler, M.E. Pulmonary eosinophilic syndromes. Immunol. Allergy Clin. N. Am 2007, 27, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.H.; Zen, Y.; Deshpande, V. IgG4-related disease. N. Engl. J. Med. 2012, 366, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Vaglio, A.; Zwerina, J. IgG4-related disease. N. Engl. J. Med. 2012, 366, 1646–1647. [Google Scholar] [PubMed]
- Fujita, A.; Sakai, O.; Chapman, M.N.; Sugimoto, H. IgG4-related disease of the head and neck: CT and MR imaging manifestations. RadioGraphics 2012, 32, 1945–1958. [Google Scholar] [CrossRef] [PubMed]
- Cohen, P.; Pagnoux, C.; Mahr, A.; Arene, J.P.; Mouthon, L.; le Guern, V.; André, M.H.; Gayraud, M.; Jayne, D.; Blöckmans, D.; et al. Churg-Strauss syndrome with poor-prognosis factors: A prospective multicenter trial comparing glucocorticoids and six or twelve cyclophosphamide pulses in forty-eight patients. Arthritis Rheum. 2007, 57, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Ribi, C.; Cohen, P.; Pagnoux, C.; Mahr, A.; Arene, J.P.; Lauque, D.; Puéchal, X.; Letellier, P.; Delaval, P.; Cordier, J.F.; et al. Treatment of Churg-Strauss syndrome without poor-prognosis factors: A multicenter, prospective, randomized, open-label study of seventy-two patients. Arthritis Rheum. 2008, 58, 586–594. [Google Scholar] [CrossRef] [PubMed]
- Metzler, C.; Hellmich, B.; Gause, A.; Gross, W.L.; de Groot, K. Churg-Strauss syndrome-successful induction of remission with methotrexate and unexpected high cardiac and pulmonary relapse ratio during maintenance treatment. Clin. Exp. Rheumatol. 2004, 22 (Suppl. S36), S52–S61. [Google Scholar] [PubMed]
- Danieli, M.G.; Cappelli, M.; Malcangi, G.; Logullo, F.; Salvi, A.; Danieli, G. Long term effectiveness of intravenous immunoglobulin in Churg-Strauss syndrome. Ann. Rheum. Dis. 2004, 63, 1649–1654. [Google Scholar] [CrossRef] [PubMed]
- Tatsis, E.; Schnabel, A.; Gross, W.L. Interferonalpha treatment of four patients with the Churg-Strauss syndrome. Ann. Intern. Med. 1998, 129, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Metzler, C.; Schnabel, A.; Gross, W.L.; Hellmich, B. A phase II study of interferon-alpha for the treatment of refractory Churg-Strauss syndrome. Clin. Exp. Rheumatol. 2008, 26 (Suppl. S49), S35–S40. [Google Scholar] [PubMed]
- Metzler, C.; Csernok, E.; Gross, W.L.; Hellmich, B. Interferon-alpha for maintenance of remission in Churg-Strauss syndrome: A long-term observational study. Clin. Exp. Rheumatol. 2010, 28 (Suppl. S57), 24–30. [Google Scholar] [PubMed]
- Corren, J. Anti-interleukin-5 antibody therapy in asthma and allergies. Curr. Opin. Allergy Clin. Immunol. 2011, 11, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Kahn, J.E.; Grandpeix-Guyodo, C.; Marroun, I.; Catherinot, E.; Mellot, F.; Roufosse, F.; Blétry, O. Sustained response to mepolizumab in refractory Churg-Strauss syndrome. J. Allergy Clin. Immunol. 2010, 125, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Marigowda, G.; Oren, E.; Israel, E.; Wechsler, M.E. Mepolizumab as a steroidsparing treatment option in patients with Churg-Strauss syndrome. J. Allergy Clin. Immunol. 2010, 125, 1336–1343. [Google Scholar] [CrossRef] [PubMed]
- Moosig, F.; Gross, W.L.; Herrmann, K.; Bremer, J.P.; Hellmich, B. Targeting interleukin-5 in refractory and relapsing Churg-Strauss syndrome. Ann. Intern. Med. 2011, 155, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Cartin-Ceba, R.; Keogh, K.A.; Specks, U.; Sethi, S.; Fervenza, F.C. Rituximab for the treatment of Churg-Strauss syndrome with renal involvement. Nephrol. Dial. Transplant. 2011, 26, 2865–2871. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.B.; Ferraro, A.J.; Chaudhry, A.N.; Brogan, P.; Salama, A.D.; Smith, K.G.; Savage, C.O.; Jayne, D.R. A multicenter survey of rituximab therapy for refractory antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2009, 60, 2156–2168. [Google Scholar] [CrossRef] [PubMed]
- Pepper, R.J.; Fabre, M.A.; Pavesio, C.; Gaskin, G.; Jones, R.B.; Jayne, D.; Pusey, C.D.; Salama, A.D. Rituximab is effective in the treatment of refractory Churg-Strauss syndrome and is associated with diminished T-cell interleukin-5 production. Rheumatology 2008, 47, 1104–1105. [Google Scholar] [CrossRef] [PubMed]
- Alobid, I.; Mullol, J. Role of medical therapy in the management of nasal polyps. Curr. Allergy Asthma Rep. 2012, 12, 144–153. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izquierdo-Domínguez, A.; Cordero Castillo, A.; Alobid, I.; Mullol, J. Churg-Strauss Syndrome or Eosinophilic Granulomatosis with Polyangiitis. Sinusitis 2016, 1, 24-43. https://doi.org/10.3390/sinusitis1010024
Izquierdo-Domínguez A, Cordero Castillo A, Alobid I, Mullol J. Churg-Strauss Syndrome or Eosinophilic Granulomatosis with Polyangiitis. Sinusitis. 2016; 1(1):24-43. https://doi.org/10.3390/sinusitis1010024
Chicago/Turabian StyleIzquierdo-Domínguez, Adriana, Arturo Cordero Castillo, Isam Alobid, and Joaquim Mullol. 2016. "Churg-Strauss Syndrome or Eosinophilic Granulomatosis with Polyangiitis" Sinusitis 1, no. 1: 24-43. https://doi.org/10.3390/sinusitis1010024
APA StyleIzquierdo-Domínguez, A., Cordero Castillo, A., Alobid, I., & Mullol, J. (2016). Churg-Strauss Syndrome or Eosinophilic Granulomatosis with Polyangiitis. Sinusitis, 1(1), 24-43. https://doi.org/10.3390/sinusitis1010024