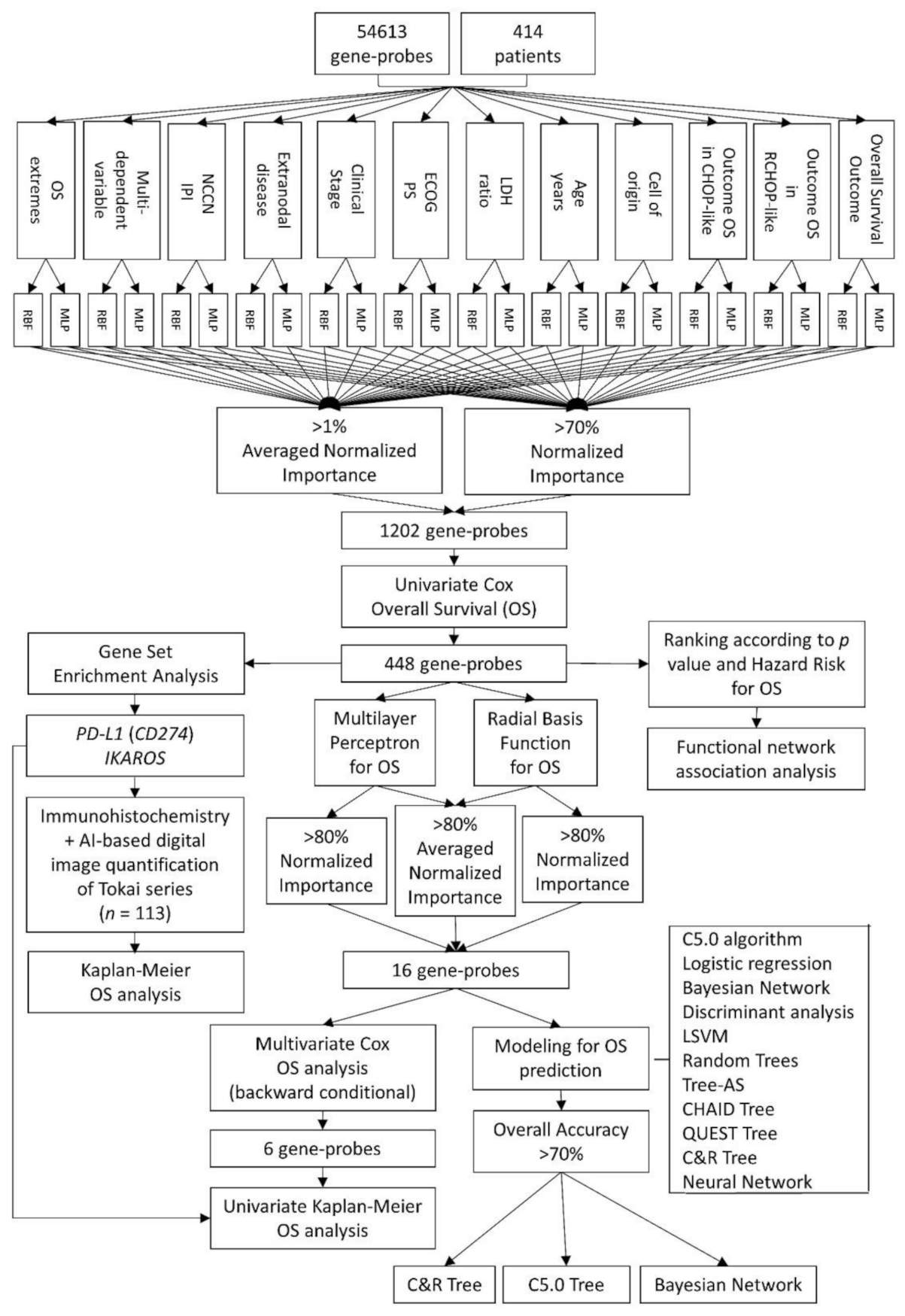
Figure 1.
Analysis algorithm. In this project, two types of artificial neural network analyses were performed: Multilayer Perceptron (MLP) and Radial Basis Function (RBF). The input data were the gene expression of 54,613 gene-probes from 414 patients with diffuse large B-cell lymphoma (DLBCL). The target variables were the outcome of the overall survival (dead versus alive), as well as several relevant clinicopathological characteristics, including the cell-of-origin classification and the International Prognostic Index (IPI). The gen-probes were ranked according to their normalized importance (NI). A cutoff of >70% of normalized importance and >1% of averaged normalized importance was applied. Cox regression analysis (univariate and multivariate) reduced the final list to the most relevant genes (n = 448). The gene-set enrichment analysis (GSEA) technique confirmed the association toward bad or good prognosis and PD-L1 and IKAROS were validated in an independent series of Tokai University. Additional data reduction was performed with Cox and Kaplan–Meier overall survival analyses, second round of artificial neural networks and predictive modeling in a multistep process up to a final 16 and 6 genes sets.
Figure 1.
Analysis algorithm. In this project, two types of artificial neural network analyses were performed: Multilayer Perceptron (MLP) and Radial Basis Function (RBF). The input data were the gene expression of 54,613 gene-probes from 414 patients with diffuse large B-cell lymphoma (DLBCL). The target variables were the outcome of the overall survival (dead versus alive), as well as several relevant clinicopathological characteristics, including the cell-of-origin classification and the International Prognostic Index (IPI). The gen-probes were ranked according to their normalized importance (NI). A cutoff of >70% of normalized importance and >1% of averaged normalized importance was applied. Cox regression analysis (univariate and multivariate) reduced the final list to the most relevant genes (n = 448). The gene-set enrichment analysis (GSEA) technique confirmed the association toward bad or good prognosis and PD-L1 and IKAROS were validated in an independent series of Tokai University. Additional data reduction was performed with Cox and Kaplan–Meier overall survival analyses, second round of artificial neural networks and predictive modeling in a multistep process up to a final 16 and 6 genes sets.

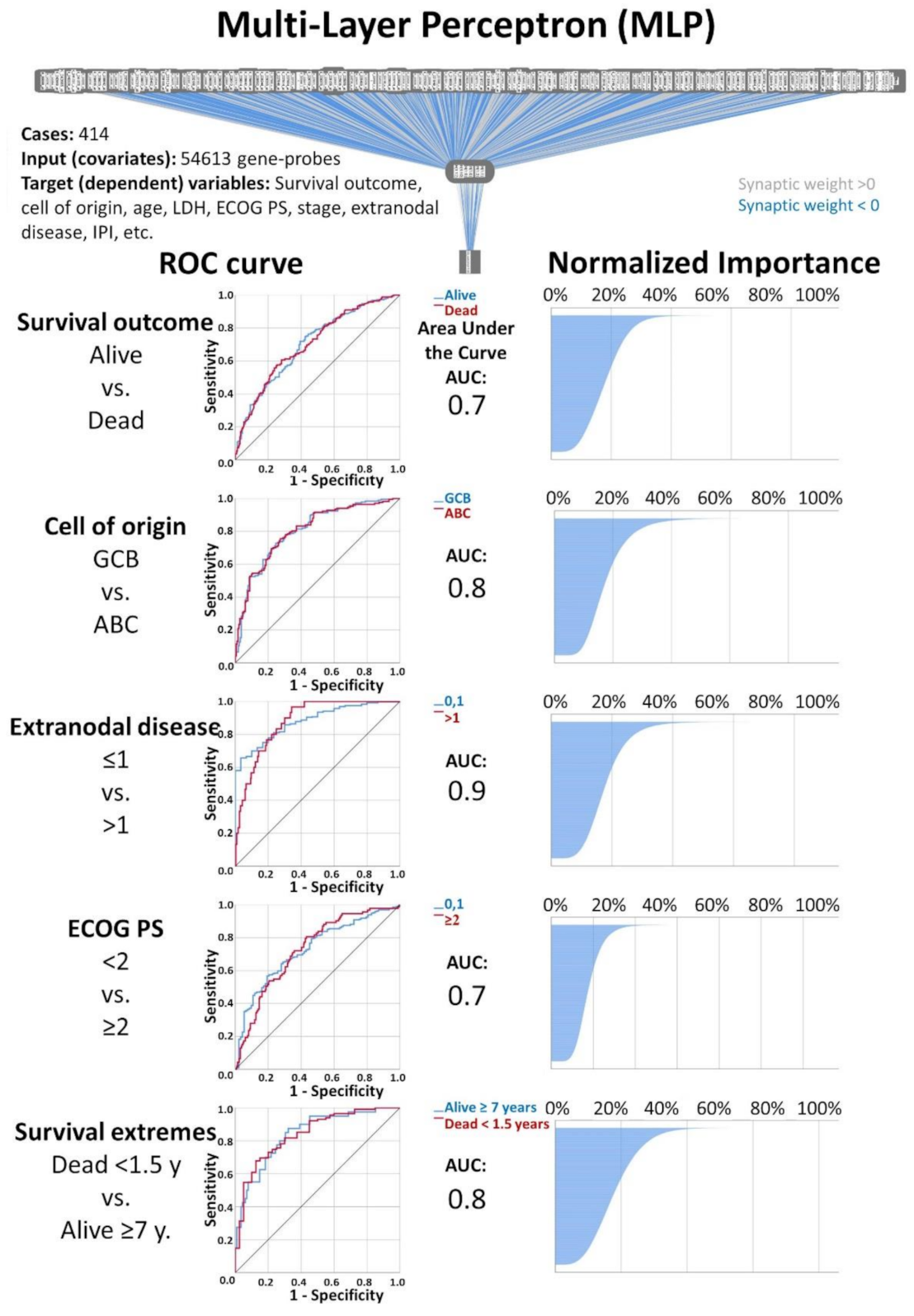
Figure 2.
Multilayer Perceptron (MLP) analysis: The artificial neural network analysis consisted of applying of Multilayer Perceptron (MLP) and Radial Basis Function (RBF) artificial neural networks on publicly available gene-expression data from DLBCL patients. For both MLP and RBF, the inputs (covariates) were the 54,613 gene-probes, and the target variables (dependent variables) were the overall survival outcome (dead vs. alive) and a series of clinicopathological variables, including the cell of origin molecular classification, age, LDH, ECOG Performance Status, clinical stage, extranodal disease and IPI. A total of 26 individual AI analyses were performed. The most relevant genes were selected according to their normalized importance, following a strategy as described in Material and Methods and in
Table 3. This figure shows part of the results of the MLP analysis. AUC, area under the curve.
Figure 2.
Multilayer Perceptron (MLP) analysis: The artificial neural network analysis consisted of applying of Multilayer Perceptron (MLP) and Radial Basis Function (RBF) artificial neural networks on publicly available gene-expression data from DLBCL patients. For both MLP and RBF, the inputs (covariates) were the 54,613 gene-probes, and the target variables (dependent variables) were the overall survival outcome (dead vs. alive) and a series of clinicopathological variables, including the cell of origin molecular classification, age, LDH, ECOG Performance Status, clinical stage, extranodal disease and IPI. A total of 26 individual AI analyses were performed. The most relevant genes were selected according to their normalized importance, following a strategy as described in Material and Methods and in
Table 3. This figure shows part of the results of the MLP analysis. AUC, area under the curve.
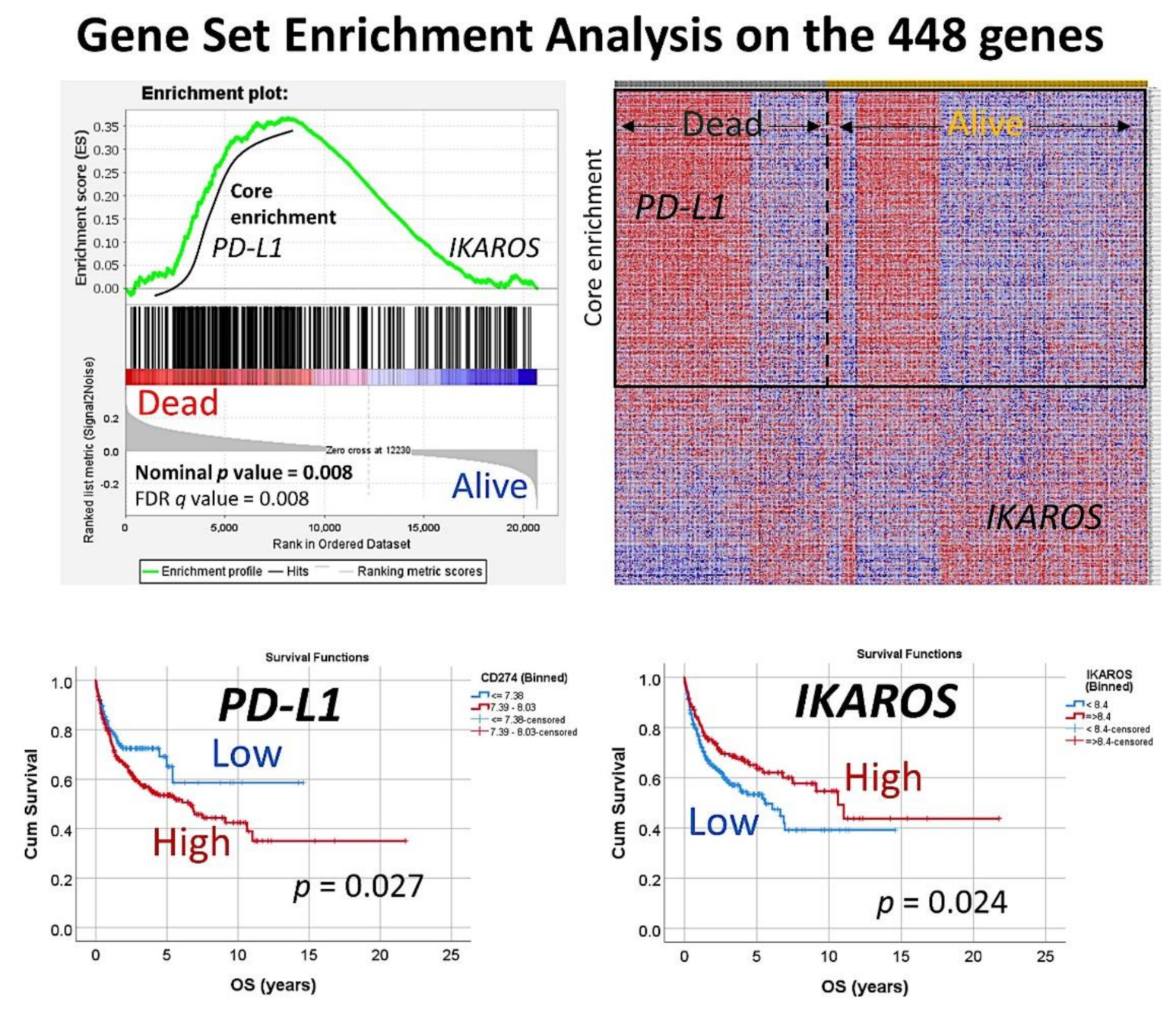
Figure 3.
Gene-set enrichment analysis on the set of 448 genes. The set of 448 genes was used in a GSEA analysis, to confirm the association of this set to the overall survival outcome of the patients (dead vs. alive phenotype). In the core enrichment associated to poor prognosis (dead), the PD-L1 (CD274) gene was identified. In the side of good prognosis (alive), the gene IKAROS was identified. Both markers that belong to the immune checkpoint pathway were further validated by immunohistochemistry in an independent series of DLBCL from Tokai University Hospital.
Figure 3.
Gene-set enrichment analysis on the set of 448 genes. The set of 448 genes was used in a GSEA analysis, to confirm the association of this set to the overall survival outcome of the patients (dead vs. alive phenotype). In the core enrichment associated to poor prognosis (dead), the PD-L1 (CD274) gene was identified. In the side of good prognosis (alive), the gene IKAROS was identified. Both markers that belong to the immune checkpoint pathway were further validated by immunohistochemistry in an independent series of DLBCL from Tokai University Hospital.
Figure 4.
Functional network association analysis on the set of 448 genes. In order to analyze the set of 448 genes according to the biological processes, molecular function, cellular component and pathways, a network analysis was made. The network was characterized by 390 nodes, 791 edges, an average node degree of 4.06, average local clustering coefficient of 0.376 and a protein-protein interaction (PPI) enrichment p-value of 1 × 10−16. In general, the set belonged to the Gene Ontology (GO) nuclei acid metabolic process (GO: 0090304, False Discovery Rate (FDR) = 0.00012). Of note, within the general network, five clusters could be identified.
Figure 4.
Functional network association analysis on the set of 448 genes. In order to analyze the set of 448 genes according to the biological processes, molecular function, cellular component and pathways, a network analysis was made. The network was characterized by 390 nodes, 791 edges, an average node degree of 4.06, average local clustering coefficient of 0.376 and a protein-protein interaction (PPI) enrichment p-value of 1 × 10−16. In general, the set belonged to the Gene Ontology (GO) nuclei acid metabolic process (GO: 0090304, False Discovery Rate (FDR) = 0.00012). Of note, within the general network, five clusters could be identified.
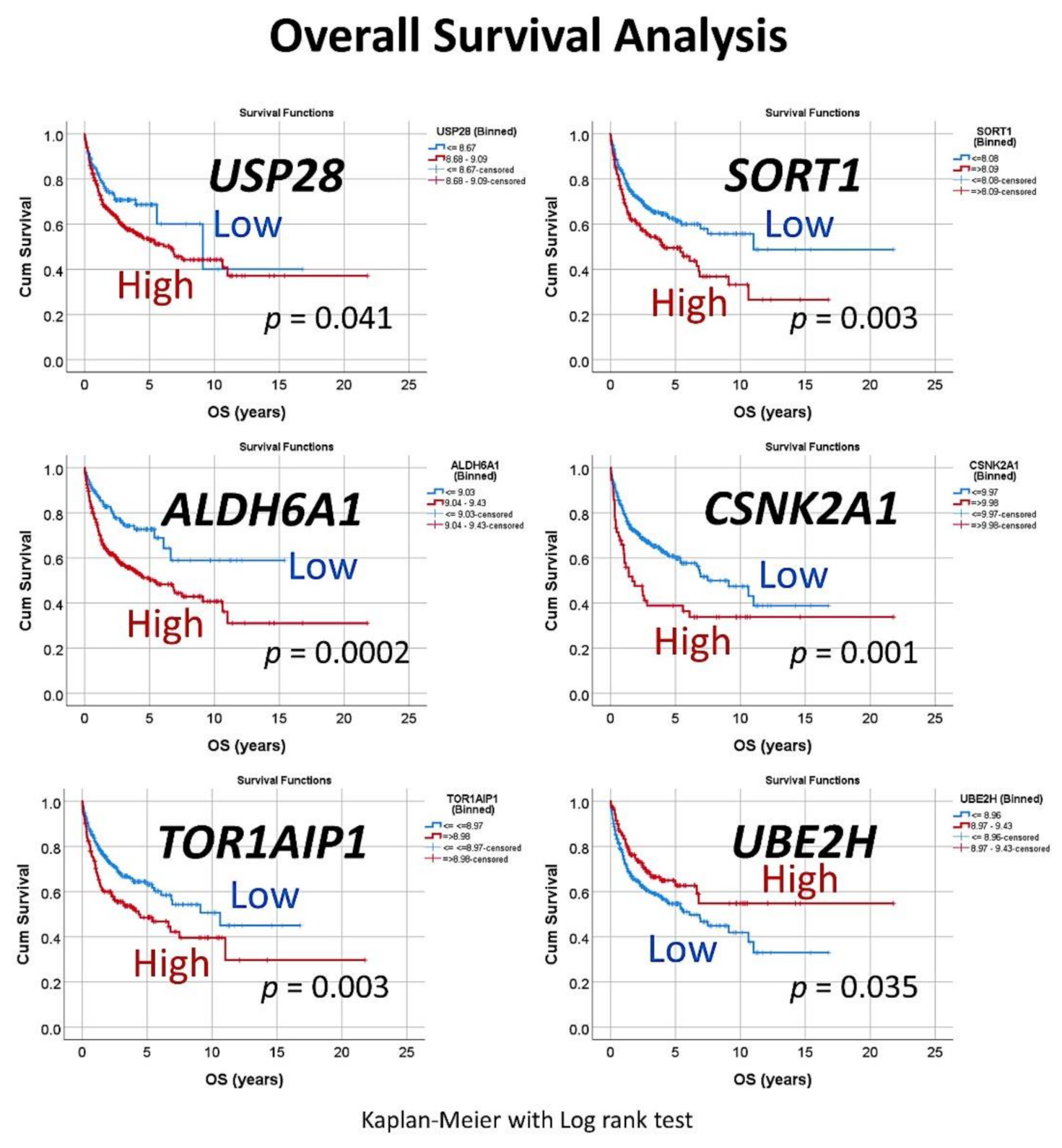
Figure 5.
Univariate overall survival analysis of the set of six genes. For each of the six genes, a cutoff was searched to stratify the patients into high and low expression. Then, the overall survival for each marker was analyzed, using the Kaplan–Meier with Log rank test.
Figure 5.
Univariate overall survival analysis of the set of six genes. For each of the six genes, a cutoff was searched to stratify the patients into high and low expression. Then, the overall survival for each marker was analyzed, using the Kaplan–Meier with Log rank test.
Figure 6.
Correlation with known pathogenic biomarkers of DLBCL. The clinical relevance for overall survival of known pathogenic biomarkers, including BCL2, MYC, CD163 and TNFAIP8, was tested in this series. After that, an unsupervised hierarchical clustering was performed with the set of six genes (USP28, SORT1, ALDH6A1, CSNK2A1, TOR1AIP1 and UBE2H), PD-L1 (CD204), IKAROS, BCL2, MYC, CD163 and TNFAIP8. The dendrogram for the rows showed how TNFAIP8, ALDH6A1, PD-L1 and USP2B clustered in the same group. In addition, CSNK2A1 and MYC were also close.
Figure 6.
Correlation with known pathogenic biomarkers of DLBCL. The clinical relevance for overall survival of known pathogenic biomarkers, including BCL2, MYC, CD163 and TNFAIP8, was tested in this series. After that, an unsupervised hierarchical clustering was performed with the set of six genes (USP28, SORT1, ALDH6A1, CSNK2A1, TOR1AIP1 and UBE2H), PD-L1 (CD204), IKAROS, BCL2, MYC, CD163 and TNFAIP8. The dendrogram for the rows showed how TNFAIP8, ALDH6A1, PD-L1 and USP2B clustered in the same group. In addition, CSNK2A1 and MYC were also close.
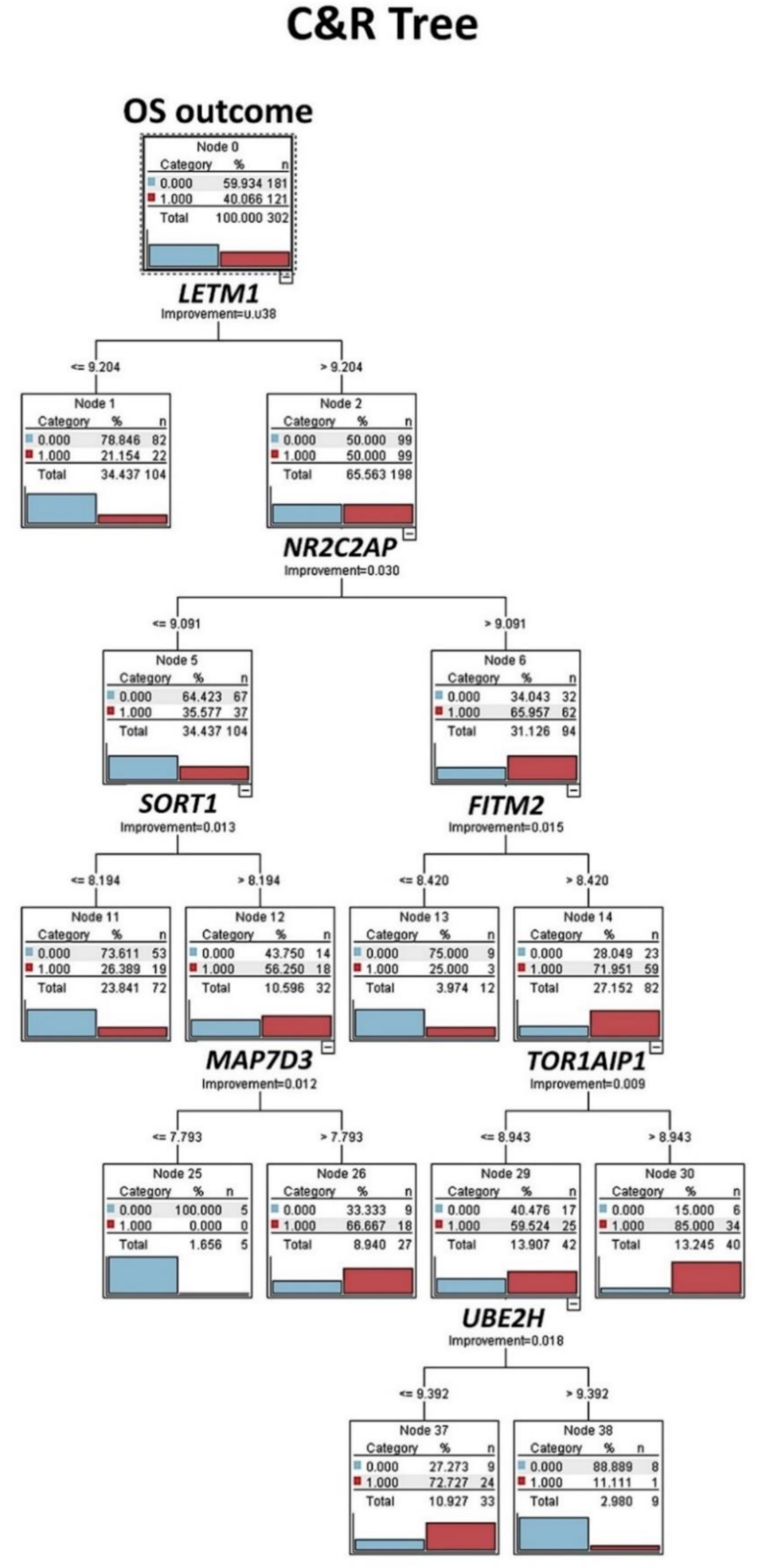
Figure 7.
Classification and regression tree (C&R tree): Prognostic modeling was performed, using the set of 16 genes, as shown in
Figure 1, and 12 different types of machine-learning analyses were tested. Then, the ones with >70% of overall accuracy were selected. This figure shows the result of the C&R tree. Decision list models identify subgroups or segments that show a higher or lower likelihood of a binary (yes or no) outcome relative to the overall sample. C&R tree node generates a decision tree that allows you to predict or classify future observations. The method uses recursive partitioning to split the training records into segments by minimizing the impurity at each step. Dead outcome, red color (number 1). Alive outcome, blue color (number 0).
Figure 7.
Classification and regression tree (C&R tree): Prognostic modeling was performed, using the set of 16 genes, as shown in
Figure 1, and 12 different types of machine-learning analyses were tested. Then, the ones with >70% of overall accuracy were selected. This figure shows the result of the C&R tree. Decision list models identify subgroups or segments that show a higher or lower likelihood of a binary (yes or no) outcome relative to the overall sample. C&R tree node generates a decision tree that allows you to predict or classify future observations. The method uses recursive partitioning to split the training records into segments by minimizing the impurity at each step. Dead outcome, red color (number 1). Alive outcome, blue color (number 0).
Figure 8.
C5 decision tree: The set of 16 genes was also tested, using C5 decision tree. This result had an overall accuracy above 70%. The C5.0 node builds either a decision tree or a rule set. The model works by splitting the sample based on the field that provides the maximum information gain at each level. The target field must be categorical (in our case, the overall survival outcome as dead vs. alive). Multiple splits into more than two subgroups are allowed. Dead outcome, red color (number 1). Alive outcome, blue color (number 0).
Figure 8.
C5 decision tree: The set of 16 genes was also tested, using C5 decision tree. This result had an overall accuracy above 70%. The C5.0 node builds either a decision tree or a rule set. The model works by splitting the sample based on the field that provides the maximum information gain at each level. The target field must be categorical (in our case, the overall survival outcome as dead vs. alive). Multiple splits into more than two subgroups are allowed. Dead outcome, red color (number 1). Alive outcome, blue color (number 0).
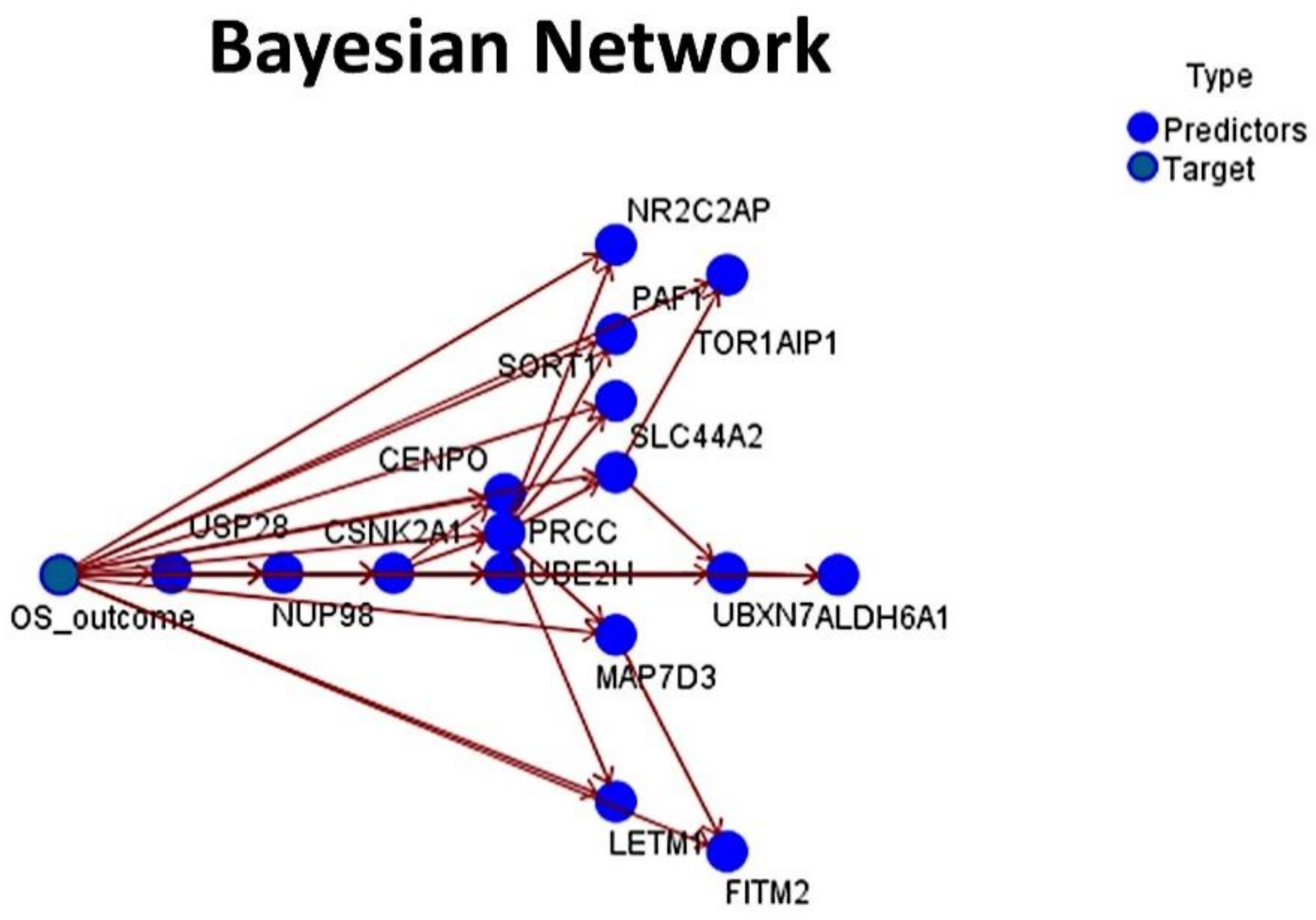
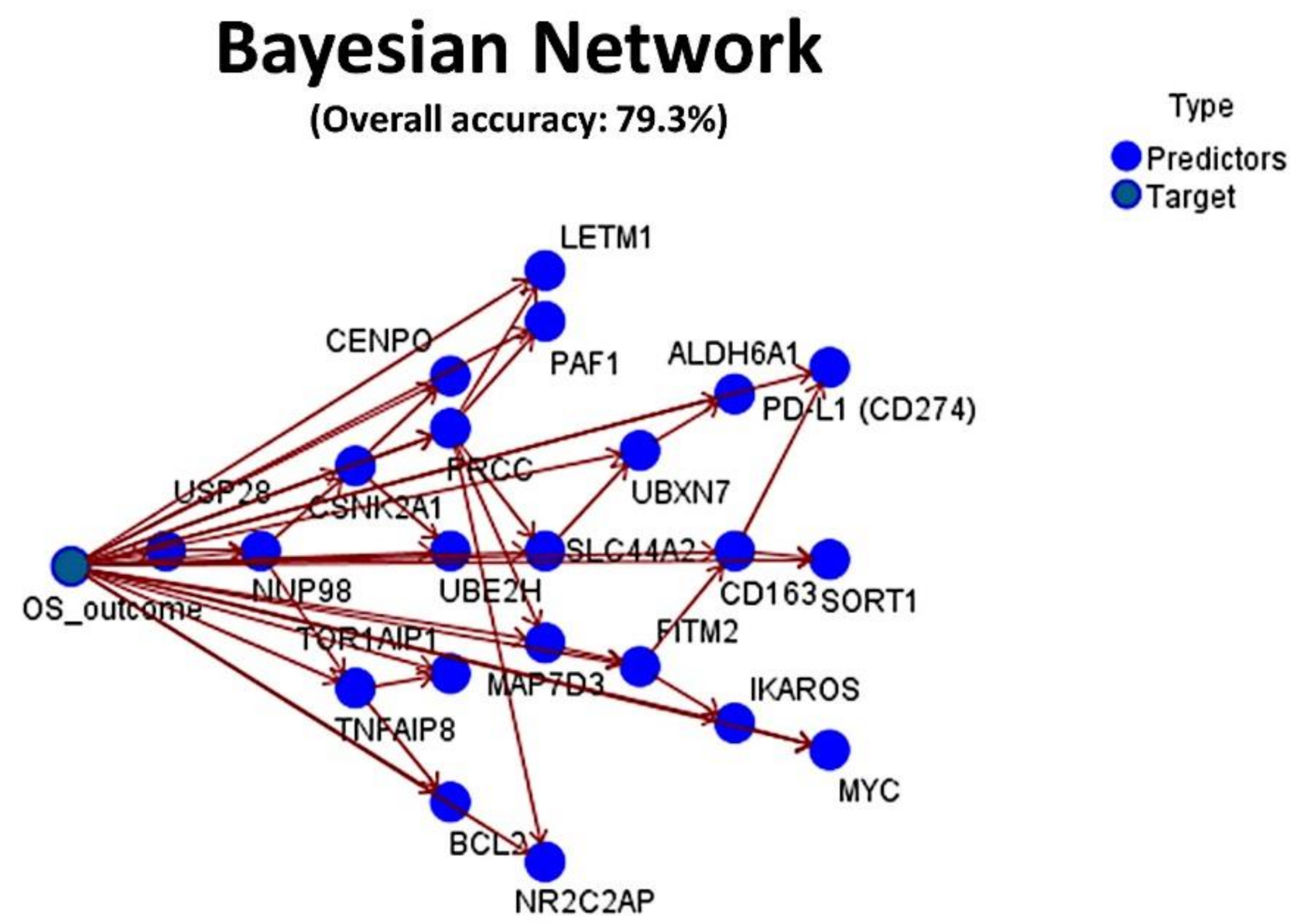
Figure 9.
Bayesian Network: The set of 16 genes was also tested, using a Bayesian Network. The result had an overall accuracy above 70%. A Bayesian Network is a graphical model that displays variables (nodes) in a dataset and the probabilistic, or conditional, independencies between them. Causal relationships between the several variables may be represented by a Bayesian Network; however, the links (arcs) between the nodes do not necessarily represent a direct cause and effect.
Figure 9.
Bayesian Network: The set of 16 genes was also tested, using a Bayesian Network. The result had an overall accuracy above 70%. A Bayesian Network is a graphical model that displays variables (nodes) in a dataset and the probabilistic, or conditional, independencies between them. Causal relationships between the several variables may be represented by a Bayesian Network; however, the links (arcs) between the nodes do not necessarily represent a direct cause and effect.
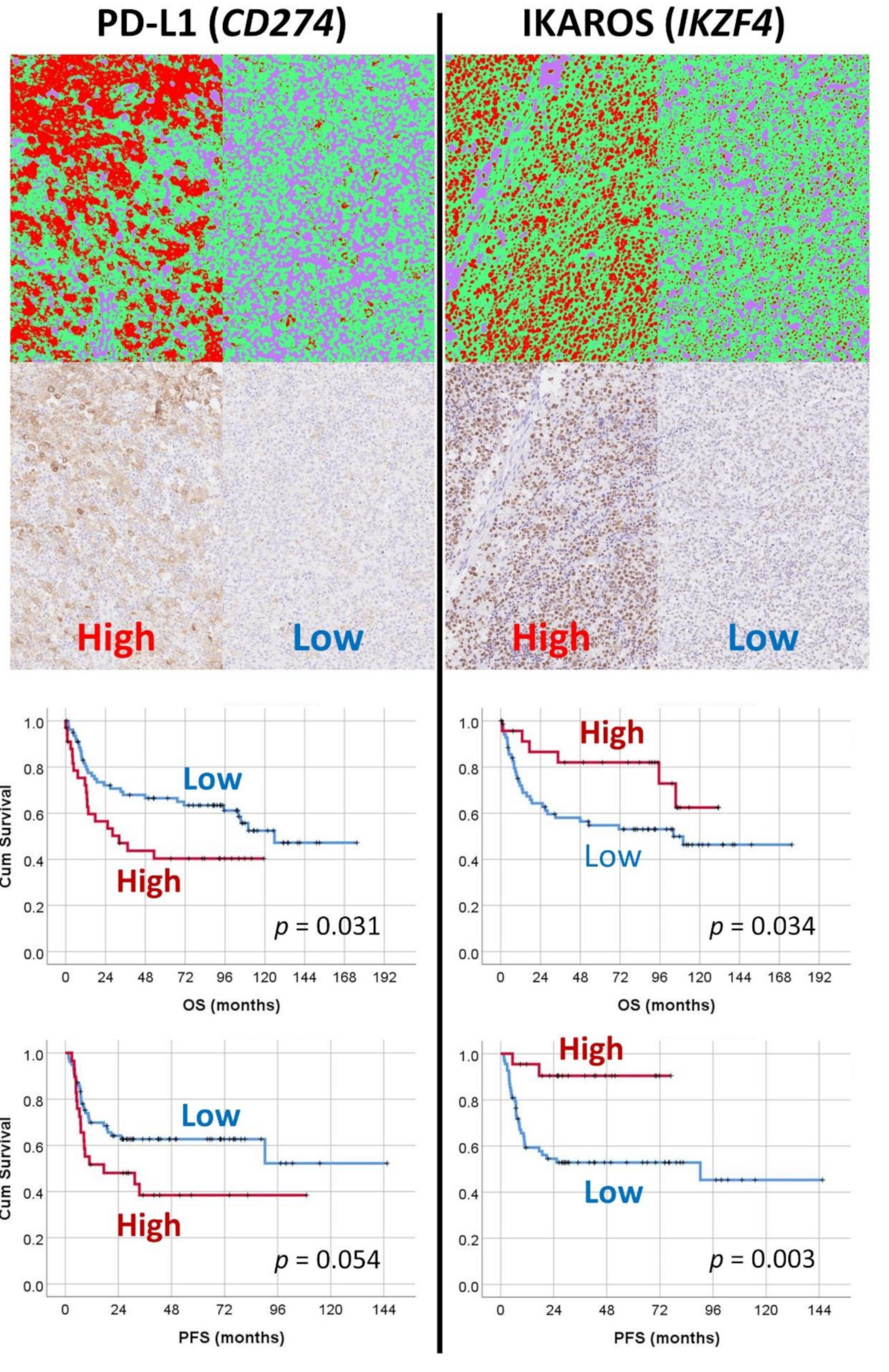
Figure 10.
Machine-learning-based digital-image analysis of immunohistochemical expression of PD-L1 and IKAROS and their correlation with the survival of the patients. The markers of PD-L1 and IKAROS were identified in the artificial neural network analysis of gene-expression data as bad prognosis and good prognosis markers, respectively. The immunohistochemical expression was tested in an independent DLBCL series from Tokai University. For the digital-image quantification, an AI-based segmentation method was used. Correlation with the survival of the patients confirmed the AI results.
Figure 10.
Machine-learning-based digital-image analysis of immunohistochemical expression of PD-L1 and IKAROS and their correlation with the survival of the patients. The markers of PD-L1 and IKAROS were identified in the artificial neural network analysis of gene-expression data as bad prognosis and good prognosis markers, respectively. The immunohistochemical expression was tested in an independent DLBCL series from Tokai University. For the digital-image quantification, an AI-based segmentation method was used. Correlation with the survival of the patients confirmed the AI results.
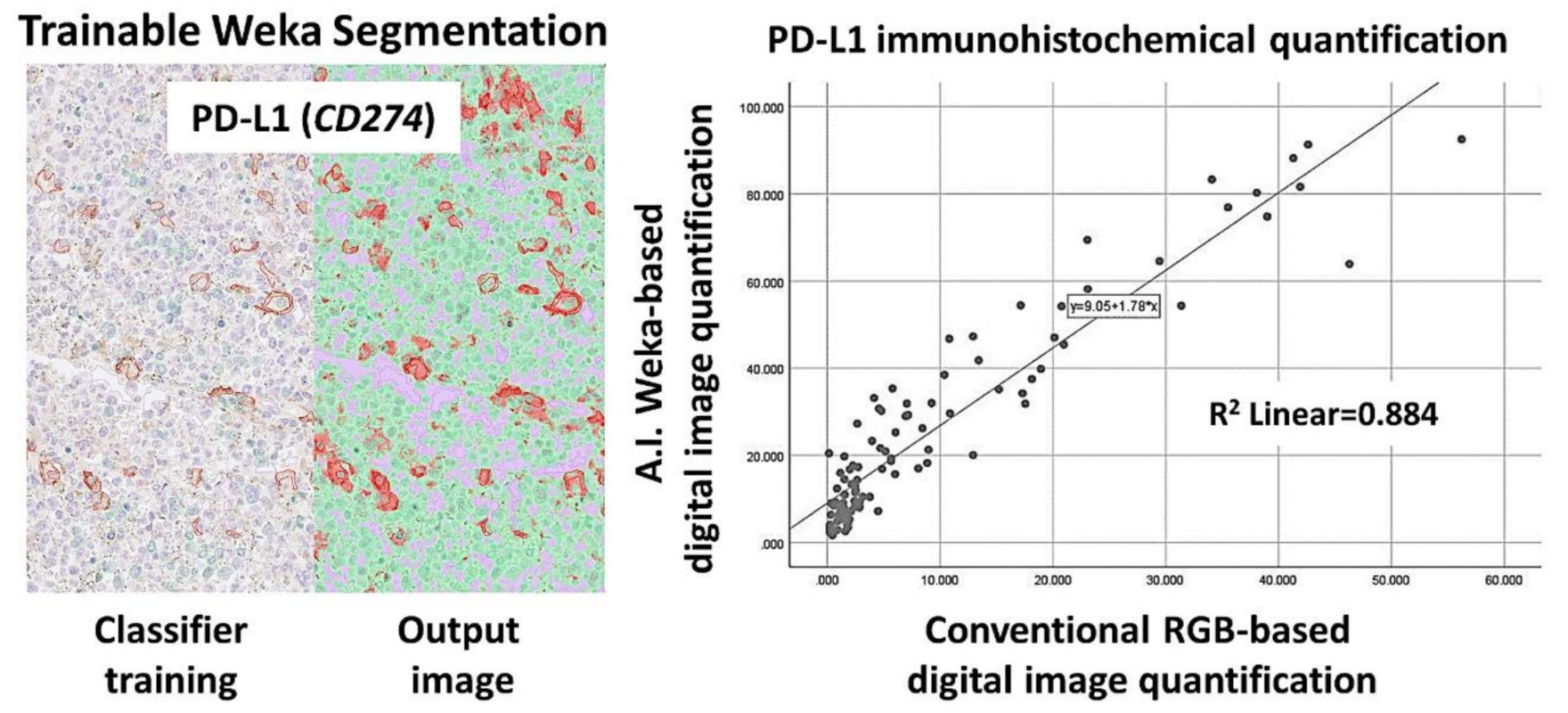
Figure 11.
PD-L1 (CD274) marker validation, using digital image. Digital image quantification of PD-L1. PD-L1 was analyzed, using a conventional RGB-based analysis, as well as with a machine-learning trainable segmentation method. Good correlation was found between both methods.
Figure 11.
PD-L1 (CD274) marker validation, using digital image. Digital image quantification of PD-L1. PD-L1 was analyzed, using a conventional RGB-based analysis, as well as with a machine-learning trainable segmentation method. Good correlation was found between both methods.
Figure 12.
Final integrated Bayesian Network. The set of 16 genes, PD-L1 (CD274) and IKAROS were merged with known biomarkers with prognostic relevance in diffuse large B-cell lymphoma (DLBCL), including BCL2, MYC, CD163 and TNFAIP8. The resulting machine-learning analysis had an overall accuracy for prediction of the overall survival of 79.3%.
Figure 12.
Final integrated Bayesian Network. The set of 16 genes, PD-L1 (CD274) and IKAROS were merged with known biomarkers with prognostic relevance in diffuse large B-cell lymphoma (DLBCL), including BCL2, MYC, CD163 and TNFAIP8. The resulting machine-learning analysis had an overall accuracy for prediction of the overall survival of 79.3%.
Figure 13.
Final integrated C5.0 decision tree. The set of 16 genes, PD-L1 (CD274) and IKAROS were merged with known biomarkers with prognostic relevance in diffuse large B-cell lymphoma (DLBCL), including BCL2, MYC, CD163 and TNFAIP8. The resulting machine-learning analysis had an overall accuracy for prediction of the overall survival of 82.1%. Dead outcome, red color (number 1). Alive outcome, blue color (number 0).
Figure 13.
Final integrated C5.0 decision tree. The set of 16 genes, PD-L1 (CD274) and IKAROS were merged with known biomarkers with prognostic relevance in diffuse large B-cell lymphoma (DLBCL), including BCL2, MYC, CD163 and TNFAIP8. The resulting machine-learning analysis had an overall accuracy for prediction of the overall survival of 82.1%. Dead outcome, red color (number 1). Alive outcome, blue color (number 0).
Table 1.
Clinicopathological characteristics of the discovery series (GSE10846).
Table 1.
Clinicopathological characteristics of the discovery series (GSE10846).
| Variable | no. | % | p-Value | Hazard Risk | 95.0% CI for HR |
|---|
| Lower | Upper |
|---|
| Sex Male | 224/414 | 54.6 | 0.9 | 1.021 | 0.744 | 1.402 |
| Age > 60 | 226/414 | 54.6 | 2 × 10−6 | 2.209 | 1.59 | 3.069 |
| LDH ratio > 1 | 182/351 | 51.9 | 5.1 × 10−8 | 2.723 | 1.899 | 3.905 |
| LDH ratio > 3 | 32/351 | 9.1 | 2.9 × 10−8 | 3.673 | 2.319 | 5.818 |
| ECOG Performance Status ≥ 2 | 93/389 | 23.9 | 3.1 × 10−10 | 2.835 | 2.049 | 3.921 |
| Clinical stage III or IV | 218/406 | 53.7 | 2.5 × 10−4 | 1.834 | 1.326 | 2.537 |
| Extranodal disease site > 1 | 30/383 | 7.8 | 0.014 | 1.927 | 1.144 | 3.246 |
| NCCN IPI | | | | | | |
| Low risk | 54/321 | 16.8 | 5.2 × 10−08 | - | - | - |
| Low-intermediate risk | 152/321 | 47.4 | 3.8 × 10−4 | 5.221 | 2.096 | 13.004 |
| High-intermediate risk | 98/321 | 30.5 | 4 × 10−6 | 8.74 | 3.493 | 21.871 |
| High risk | 17/321 | 5.3 | 6.9 × 10−8 | 17.761 | 6.244 | 50.521 |
| Cell-of-origin molecular subtype | | | | | | |
| Germinal center B-cell (GCB) | 183/414 | 44.2 | 2.8 × 10−8 | - | - | - |
| Activated B-cell (ABC) | 167/414 | 40.3 | 1.1 × 10−8 | 2.75 | 1.944 | 3.891 |
| Unclassified | 64/414 | 15.5 | 0.2 | 1.389 | 0.84 | 2.298 |
| Treatment | | | | | | |
| RCHOP-like | 233/414 | 56.3 | 7.8 × 10−5 | 0.52 | 0.376 | 0.719 |
| CHOP-like | 181/414 | 43.7 | - | - | - | - |
| Overall survival (outcome) | | | | | | |
| Dead | 165/414 | 39.9 | - | - | - | - |
| Alive | 249/414 | 60.1 | - | - | - | - |
| Overall survival | | | | | | |
| Dead < 1.5 years | 115/414 | 27.8 | 3.3 × 10−9 | 113.448 | 23.654 | 544.122 |
| Alive ≥ 7 years | 40/414 | 9.7 | - | - | - | - |
Table 2.
Clinicopathological characteristics of the validation series (Tokai cases).
Table 2.
Clinicopathological characteristics of the validation series (Tokai cases).
| Variable | no. | % | p-Value | Hazard Risk | 95.0% CI for HR |
|---|
| Lower | Upper |
|---|
| Sex Male | 63/113 | 55.8 | 0.9 | 1.045 | 0.6 | 1.821 |
| Age > 60 | 78/112 | 69.6 | 0.01 | 2.553 | 1.24 | 5.253 |
| Location | | | | | | |
| Nodal (+spleen) | 63/113 | 55.8 | 0.4 | - | - | - |
| Extranodal | | | | | | |
| Waldeyer’s ring | 11/113 | 9.7 | 0.2 | 0.486 | 0.147 | 1.613 |
| Gastrointestinal | 10/113 | 8.8 | 0.6 | 0.735 | 0.223 | 2.422 |
| Other extranodal | 29/113 | 25.7 | 0.4 | 1.326 | 0.725 | 2.427 |
| LDH High (>219) | 70/112 | 62.5 | 1.8 × 10−3 | 3.03 | 1.51 | 6.083 |
| Seric IL2RA High (>530) | 83/106 | 78.3 | 1.4 × 10−2 | 3.627 | 1.299 | 10.125 |
| ECOG Performance Status ≥ 2 | 15/90 | 16.7 | 6.2 × 10−4 | 3.466 | 1.701 | 7.062 |
| Clinical stage III or IV | 52/105 | 49.5 | 0.01 | 2.138 | 1.17 | 3.905 |
| Extranodal disease site >1 | 20/86 | 23.3 | 5.1 × 10−5 | 3.985 | 2.041 | 7.78 |
| B symptoms | 24/94 | 25.5 | 0.2 | 1.557 | 0.805 | 3.011 |
| International Prognostic Index (IPI) | | | | | | |
| Low risk (L) | 34/96 | 35.4 | 1.9−10-2 | - | - | - |
| Low-intermediate risk (LI) | 29/96 | 30.2 | 0.01 | 3.265 | 1.392 | 7.656 |
| High-intermediate risk (HI) | 20/96 | 20.8 | 0.02 | 2.99 | 1.193 | 7.495 |
| High risk (H) | 13/96 | 13.5 | 4.9 × 10−3 | 4.326 | 1.558 | 12.016 |
| Cell-of-origin subtype (Hans) | | | | | | |
| GCB | 37/110 | 33.6 | - | - | - | - |
| Non-GCB | 73/110 | 66.4 | 1.4 × 10−2 | 2.318 | 1.186 | 4.529 |
| Epstein–Barr virus, EBER+ | 16/111 | 14.4 | 2.5 × 10−2 | 2.291 | 1.11 | 4.729 |
| Treatment | | | | | | |
| RCHOP | 79/106 | 74.5 | 0.3 | - | - | - |
| RCHOP-like | 22/106 | 20.8 | 0.1 | 1.677 | 0.873 | 3.219 |
| Others | 5/106 | 4.7 | 0.5 | 1.701 | 0.406 | 7.134 |
| Response to treatment | | | | | | |
| CR | 72/101 | 63.7 | - | | | |
| PR+PD+SD+NC | 29/101 | 28.7 | 2.9 × 10−13 | 11.467 | 5.956 | 22.076 |
| Overall survival (outcome) | | | | | | |
| Dead | 51/113 | 45.1 | - | - | - | - |
| Alive | 62/113 | 54.9 | - | - | - | - |
Table 3.
Multilayer Perceptron (MLP) artificial neural network analysis for the prediction of DLBCL prognosis.
Table 3.
Multilayer Perceptron (MLP) artificial neural network analysis for the prediction of DLBCL prognosis.
| Multilayer Perception | Dependent Variable | Outcome Dead | Outcome Dead RCHOP-Like Only | Outcome Dead CHOP-Like Only | Cell-of-Origin Activated B-Cell-Like | Age > 60 | LDH Ratio ≥ 1 | LDH Ratio > 3 | ECOG ≥ 2 | Stage III/IV | Extranodal Sites > 1 | Sex Male | NCCN IPI–like HI+H | Dead < 1.5 vs. Alive => 7 y. | Multivariate |
|---|
| Case processing summary | Training | 283 | 161 | 131 | 252 | 279 | 253 | 239 | 264 | 295 | 260 | 268 | 222 | 104 | 187 |
| Training Percentage | 68.40 | 69.10 | 72.40 | 72.00 | 67.40 | 72.10 | 68.10 | 67.90 | 72.70 | 67.90 | 67.70 | 69.20 | 67.10 | 69.00 |
| Testing | 131 | 72 | 50 | 98 | 135 | 98 | 112 | 125 | 111 | 123 | 128 | 99 | 51 | 84 |
| Testing Percentage | 31.60 | 30.90 | 27.60 | 28.00 | 32.60 | 27.90 | 31.90 | 32.10 | 27.30 | 32.10 | 32.30 | 30.80 | 32.90 | 31.00 |
| Valid | 414 | 233 | 181 | 350 | 414 | 351 | 351 | 389 | 406 | 383 | 396 | 321 | 155 | 271 |
| Excluded | 6 | 0 | 0 | 0 | 6 | 69 | 69 | 31 | 14 | 37 | 24 | 99 | 265 | 149 |
| Total | 420 | 233 | 181 | 350 | 420 | 420 | 420 | 420 | 420 | 420 | 420 | 420 | 420 | 420 |
| Network information | Number of Units | 54,613 | 54,613 | 54,613 | 54,613 | 54,613 | 54,613 | 54,613 | 54,613 | 54,613 | 54,613 | 54,613 | 54,613 | 54,613 | 54,613 |
| Rescaling Method of Covariates | Standardized |
| Hidden layer | Number of Hidden Layers | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Number of Units in Hidden Layer | 6 | 6 | 9 | 15 | 5 | 9 | 15 | 9 | 8 | 8 | 8 | 8 | 10 | 7 |
| Activation Function | Hyperbolic tangent |
| Output layer | Dependent variable | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 8 |
| Number of Units | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 16 |
| Activation Function | Softmax |
| Error Function | Cross-entropy |
| Model summary training | Cross-Entropy Error | 174.16 | 85.80 | 79.90 | 140.50 | 178.40 | 163.90 | 74.50 | 132.80 | 193.90 | 47.50 | 186.20 | 136.50 | 46.49 | 840.80 |
| Percent of Incorrect Predictions | 33.90 | 24.80 | 33.60 | 27.80 | 38.40 | 36.80 | 10.50 | 24.60 | 39.30 | 6.50 | 45.40 | 33.80 | 19.20 | 30.20 |
| Stopping Rule Used * | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Time in Minutes | 9.12 | 4.92 | 4.60 | 7.10 | 8.20 | 8.02 | 6.80 | 7.92 | 9.35 | 7.63 | 9.33 | 7.32 | 2.98 | 7.83 |
| Model summary testing | Cross-Entropy Error | 77.39 | 36.10 | 25.30 | 49.80 | 87.30 | 63.00 | 22.70 | 61.50 | 69.60 | 27.60 | 80.60 | 59.10 | 18.80 | 397.90 |
| Percent of Incorrect Predictions | 32.10 | 26.40 | 26.00 | 25.50 | 39.30 | 35.70 | 6.30 | 23.20 | 34.20 | 8.90 | 35.20 | 30.30 | 19.60 | 32.90 |
| Classification | Training Overall Percent | 66.10 | 75.20 | 66.40 | 72.20 | 61.60 | 63.20 | 89.50 | 75.40 | 60.70 | 93.50 | 54.50 | 66.20 | 80.80 | 69.80 |
| Testing Overall Percent | 67.90 | 73.60 | 74.00 | 74.50 | 60.70 | 64.30 | 93.80 | 76.80 | 65.80 | 91.10 | 64.80 | 69.70 | 80.40 | 67.10 |
| Area under the curve | Alive | 0.70 | 0.69 | 0.76 | 0.80 | 0.68 | 0.67 | 0.75 | 0.73 | 0.66 | 0.88 | 0.60 | 0.68 | 0.84 | 0.66 |
| Dead | 0.70 | 0.69 | 0.76 | 0.80 | 0.68 | 0.67 | 0.75 | 0.73 | 0.66 | 0.88 | 0.60 | 0.68 | 0.84 | 0.66 |
Table 4.
Radial Basis Function (RBF) artificial neural network analysis for the prediction of DLBCL prognosis.
Table 4.
Radial Basis Function (RBF) artificial neural network analysis for the prediction of DLBCL prognosis.
| Radial Basis Function | Dependent Variable | Outcome Dead | Outcome Dead RCHOP-Like Only | Outcome Dead CHOP-Like Only | Cell-of-Origin Activated B-Cell-Like | Age > 60 | LDH Ratio ≥ 1 | LDH Ratio > 3 | ECOG ≥ 2 | Stage III/IV | Extranodal Sites > 1 | Sex Male | NCCN IPI–Like HI+H | Dead < 1.5 vs. Alive => 7 years | Multivariate |
|---|
| Case processing summary | Training | 283 | 161 | 131 | 239 | 301 | 239 | 240 | 264 | 295 | 261 | 269 | 222 | 104 | 187 |
| Training Percentage | 68.30 | 69.10 | 72.40 | 68.30 | 72.70 | 68.10 | 68.40 | 67.90 | 72.70 | 68.10 | 67.90 | 69.20 | 67.10 | 69.00 |
| Testing | 131 | 72 | 50 | 111 | 113 | 112 | 111 | 125 | 111 | 122 | 127 | 99 | 51 | 84 |
| Testing Percentage | 31.60 | 30.90 | 27.60 | 31.70 | 27.30 | 31.90 | 31.60 | 32.10 | 27.30 | 31.90 | 32.10 | 30.80 | 32.90 | 31.00 |
| Valid | 414 | 233 | 181 | 350 | 414 | 351 | 351 | 389 | 406 | 383 | 396 | 321 | 155 | 271 |
| Excluded | 6 | 0 | 0 | 70 | 6 | 69 | 69 | 31 | 14 | 37 | 24 | 99 | 265 | 149 |
| Total | 420 | 233 | 181 | 420 | 420 | 420 | 420 | 420 | 420 | 420 | 420 | 420 | 420 | 420 |
| Network information | Number of Units | 54,613 | 54,613 | 54,613 | 54,613 | 54,613 | 54,613 | 54,613 | 54,613 | 54,613 | 54,613 | 54,613 | 54,613 | 54,613 | 54,613 |
| Rescaling Method of Covariates | Standardized |
| Hidden layer | Number of Hidden Layers | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Number of Units in Hidden Layer | 10 | 2 | 3 | 10 | 2 | 10 | 9 | 3 | 2 | 10 | 2 | 8 | 8 | 8 |
| Activation Function | Softmax |
| Output layer | Dependent variable | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 8 |
| Number of Units | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 2 | 16 |
| Activation Function | Identity |
| Error Function | Sum of Squares |
| Model summary training | Cross Entropy Error | 57.50 | 31.20 | 31.30 | 47.60 | 74.20 | 55.90 | 23.70 | 48.40 | 73.30 | 15.50 | 66.60 | 49.40 | 16.60 | 293.60 |
| Percent of Incorrect Predictions | 33.60 | 26.70 | 41.20 | 27.60 | 46.20 | 39.70 | 11.30 | 24.20 | 46.10 | 7.70 | 46.10 | 34.70 | 23.10 | 31.10 |
| Stopping Rule Used * | - |
| Time in Minutes | 171.08 | 32.70 | 21.07 | 127.07 | 221.92 | 127.37 | 117.73 | 145.20 | 145.75 | 141.83 | 171.65 | 107.20 | 13.33 | 57.70 |
| Model summary testing | Cross Entropy Error | 27.80 | 12.90 | 12.50 | 24.90 | 28.00 | 27.00 | 5.30 | 22.30 | 27.70 | 8.60 | 31.20 | 22.60 | 9.30 | 138.30 |
| Percent of Incorrect Predictions | 32.10 | 23.60 | 44.00 | 36.00 | 43.30 | 41.10 | 4.50 | 23.20 | 46.80 | 8.20 | 48.80 | 35.40 | 29.40 | 33.60 |
| Classification | Training Overall Percent | 66.40 | 73.30 | 58.80 | 72.40 | 53.80 | 60.30 | 88.80 | 75.80 | 53.90 | 92.30 | 53.90 | 65.30 | 76.90 | 68.90 |
| Testing Overall Percent | 67.90 | 76.40 | 56.00 | 64.00 | 56.60 | 58.90 | 95.00 | 76.80 | 53.20 | 91.80 | 51.20 | 64.60 | 70.60 | 66.40 |
| Area under the curve | Alive | 0.71 | 0.58 | 0.54 | 0.75 | 0.54 | 0.62 | 0.59 | 0.52 | 0.49 | 0.83 | 0.52 | 0.58 | 0.75 | 0.64 |
| Dead | 0.71 | 0.58 | 0.54 | 0.75 | 0.54 | 0.62 | 0.59 | 0.52 | 0.49 | 0.83 | 0.52 | 0.58 | 0.75 | 0.64 |
Table 5.
Comparison of Performance between MLP and RBF artificial neural networks for prediction of DLBCL prognosis.
Table 5.
Comparison of Performance between MLP and RBF artificial neural networks for prediction of DLBCL prognosis.
| Artificial Neural Network Parameters | MLP | RBF | p-Value |
|---|
| (n = 14) | (n = 14) |
|---|
| Training set number | 228.4 ± 59.7 | 228.3 ± 60.9 | 0.995 |
| Training set percentage | 69.4% ± 2.0 | 69.2% ± 1.9 | 0.864 |
| Testing set number | 101.2 ± 28.2 | 101.4 ± 26.9 | 0.989 |
| Testing set percentage | 30.6% ± 2.0 | 30.7% ± 1.9 | 0.872 |
| Valid number of cases | 329.6 ± 86.7 | 329.6 ± 86.7 | 1 |
| Number of gene-probes (units) | 54,613 | 54,613 | 1 |
| Rescaling method for covariates | Standardized | Standardized | N/A |
| Hidden layer(s) | | | |
| Number of hidden layers | 1 | 1 | N/A |
| Number of units in hidden layer | 8.8 ± 2.9 | 6.2 ± 3.6 | 0.048 |
| Activation function | Hyperbolic tangent | Softmax | N/A |
| Output layer | | | |
| Dependent (target) variable | 1 (8 for multivariate) | 1 (8 for multivariate) | 1 |
| Number of units | 2 (16 for multivariate) | 2 (16 for multivariate) | 1 |
| Activation function | Softmax | Identity | N/A |
| Error function | Cross-entropy | Sum of Squares | N/A |
| Model summary | | | |
| Training | | | |
| Cross-entropy error | 177.2 ± 197.7 | 63.2 ± 69.1 | 0.058 |
| Percent of incorrect predictions | 28.9% ± 11.0 | 31.4% ± 12.3 | 0.559 |
| Training time (min) | 7.2 ± 1.8 | 114.4 ± 61.9 | 7.5 × 10−7 |
| Testing | | | |
| Cross-entropy error | 76.9 ± 95.2 | 28.5 ± 32.8 | 0.091 |
| Percent of incorrect predictions | 26.8% ± 9.8 | 32.1% ± 13.5 | 0.245 |
| Classification | | | |
| Training sample, overall percent correct | 71.1% ± 11.0 | 68.6% ± 12.3 | 0.583 |
| Testing sample, overall percent correct | 73.2% ± 9.8 | 67.8% ± 13.4 | 0.239 |
| Area under the curve (ROC) | 0.7 ± 0.1 | 0.6 ± 0.1 | 0.007 |
Table 6.
Top 10 genes according to the p-value (set of 448 gene-probes).
Table 6.
Top 10 genes according to the p-value (set of 448 gene-probes).
| Gene | Probe | B | SE | Wald | df | p-Value | Hazard Risk | Lower | Upper |
|---|
| ALG3 | 207396_s_at | 0.74 | 0.134 | 30.257 | 1 | 3.8 × 10−8 | 2.095 | 1.61 | 2.727 |
| UCK2 | 209825_s_at | 0.589 | 0.11 | 28.454 | 1 | 9.6 × 10−8 | 1.803 | 1.452 | 2.238 |
| ZMYND19 | 227477_at | 0.687 | 0.13 | 27.96 | 1 | 1.2 × 10−7 | 1.988 | 1.541 | 2.564 |
| ELFN1-AS1 | 231443_at | 0.551 | 0.107 | 26.369 | 1 | 2.8 × 10−7 | 1.735 | 1.406 | 2.141 |
| PHTF2 | 1554780_a_at | -0.55 | 0.108 | 25.968 | 1 | 3.5 × 10−7 | 0.577 | 0.467 | 0.713 |
| EXOSC7 | 212627_s_at | 0.546 | 0.11 | 24.453 | 1 | 7.6 × 10−7 | 1.726 | 1.39 | 2.144 |
| BCAT2 | 203576_at | 0.594 | 0.121 | 23.946 | 1 | 9.9 × 10−7 | 1.81 | 1.427 | 2.296 |
| TBRG4 | 220789_s_at | 0.645 | 0.133 | 23.646 | 1 | 1 × 10−6 | 1.906 | 1.47 | 2.471 |
| THOC1 | 204064_at | 0.906 | 0.186 | 23.676 | 1 | 1 × 10−6 | 2.476 | 1.718 | 3.566 |
| KIF13B | 202962_at | 0.413 | 0.086 | 23.247 | 1 | 1 × 10−6 | 1.511 | 1.278 | 1.787 |
Table 7.
Top 10 bad prognostic genes according to the Hazard Risk (set of 448 gene-probes).
Table 7.
Top 10 bad prognostic genes according to the Hazard Risk (set of 448 gene-probes).
| Gene | Probe | B | SE | Wald | df | p-Value | Hazard Risk | Lower | Upper |
|---|
| THOC1 | 204064_at | 0.906 | 0.186 | 23.676 | 1 | 1 × 10−6 | 2.476 | 1.718 | 3.566 |
| TMX2 | 201175_at | 0.878 | 0.184 | 22.698 | 1 | 2 × 10−6 | 2.407 | 1.677 | 3.455 |
| HNRNPC | 214737_x_at | 0.748 | 0.202 | 13.762 | 1 | 2 × 10−4 | 2.113 | 1.423 | 3.138 |
| ALG3 | 207396_s_at | 0.74 | 0.134 | 30.257 | 1 | 3.8 × 10−8 | 2.095 | 1.61 | 2.727 |
| NELFA | 203112_s_at | 0.719 | 0.153 | 22.04 | 1 | 3 × 10−6 | 2.052 | 1.52 | 2.77 |
| PPP6R2 | 202791_s_at | 0.695 | 0.158 | 19.292 | 1 | 1.1 × 10−6 | 2.003 | 1.469 | 2.73 |
| ZMYND19 | 227477_at | 0.687 | 0.13 | 27.96 | 1 | 1.2 × 10−7 | 1.988 | 1.541 | 2.564 |
| TBRG4 | 220789_s_at | 0.645 | 0.133 | 23.646 | 1 | 1 × 10−6 | 1.906 | 1.47 | 2.471 |
| GLO1 | 200681_at | 0.643 | 0.157 | 16.795 | 1 | 4 × 10−5 | 1.903 | 1.399 | 2.588 |
| BORCS8 | 1553978_at | 0.62 | 0.163 | 14.513 | 1 | 1 × 10−5 | 1.859 | 1.351 | 2.558 |
Table 8.
Top 10 good prognostic genes according to the Hazard Risk (set of 448 gene-probes).
Table 8.
Top 10 good prognostic genes according to the Hazard Risk (set of 448 gene-probes).
| Gene | Probe | B | SE | Wald | df | p-Value | Hazard Risk | Lower | Upper |
|---|
| TTC3 | 208663_s_at | −0.124 | 0.034 | 13.115 | 1 | 0.0002 | 0.884 | 0.826 | 0.945 |
| YTHDC1 | 214814_at | −0.134 | 0.043 | 9.514 | 1 | 0.002 | 0.875 | 0.803 | 0.952 |
| B3GALNT1 | 223374_s_at | −0.146 | 0.058 | 6.326 | 1 | 0.012 | 0.864 | 0.771 | 0.968 |
| ZNF277 | 1555193_a_at | −0.152 | 0.074 | 4.246 | 1 | 0.039 | 0.859 | 0.744 | 0.993 |
| RAB39B | 238695_s_at | −0.154 | 0.063 | 5.87 | 1 | 0.015 | 0.857 | 0.757 | 0.971 |
| ITPR1 | 211323_s_at | −0.156 | 0.065 | 5.671 | 1 | 0.017 | 0.856 | 0.753 | 0.973 |
| CLIC5 | 213317_at | −0.157 | 0.066 | 5.745 | 1 | 0.017 | 0.854 | 0.751 | 0.972 |
| SEL1L | 202062_s_at | −0.16 | 0.077 | 4.335 | 1 | 0.037 | 0.852 | 0.732 | 0.991 |
| N/A | 242693_at | −0.167 | 0.069 | 5.962 | 1 | 0.015 | 0.846 | 0.74 | 0.968 |
| MFSD6 | 219858_s_at | −0.171 | 0.064 | 7.251 | 1 | 0.0071 | 0.843 | 0.744 | 0.955 |
Table 9.
Set of 16 prognostic genes.
Table 9.
Set of 16 prognostic genes.
| Gene | Probe | B | SE | Wald | df | p-Value | Hazard Risk | Lower | Upper |
|---|
| PAF1 | 202093_s_at | 0.237 | 0.118 | 4.01 | 1 | 0.045 | 1.267 | 1.005 | 1.597 |
| USP28 | 1552678_a_at | 0.422 | 0.133 | 10.083 | 1 | 0.0015 | 1.526 | 1.175 | 1.98 |
| SORT1 | 212807_s_at | 0.177 | 0.081 | 4.758 | 1 | 0.03 | 1.194 | 1.018 | 1.401 |
| MAP7D3 | 219626_at | 0.376 | 0.135 | 7.79 | 1 | 0.005 | 1.456 | 1.118 | 1.896 |
| FITM2 | 226805_at | 0.302 | 0.148 | 4.162 | 1 | 0.04 | 1.352 | 1.012 | 1.807 |
| CENPO | 226118_at | 0.324 | 0.108 | 8.946 | 1 | 0.003 | 1.383 | 1.118 | 1.71 |
| PRCC | 208938_at | 0.229 | 0.117 | 3.856 | 1 | 0.05 | 1.258 | 1 | 1.581 |
| ALDH6A1 | 221588_x_at | 0.515 | 0.158 | 10.57 | 1 | 0.0012 | 1.673 | 1.227 | 2.282 |
| CSNK2A1 | 212075_s_at | 0.418 | 0.134 | 9.715 | 1 | 0.0018 | 1.52 | 1.168 | 1.977 |
| TOR1AIP1 | 212409_s_at | 0.384 | 0.162 | 5.607 | 1 | 0.018 | 1.468 | 1.068 | 2.017 |
| NUP98 | 203194_s_at | 0.339 | 0.131 | 6.718 | 1 | 0.009 | 1.404 | 1.086 | 1.814 |
| UBE2H | 221962_s_at | -0.415 | 0.121 | 11.699 | 1 | 0.0006 | 0.66 | 0.521 | 0.838 |
| UBXN7 | 217100_s_at | 0.269 | 0.108 | 6.187 | 1 | 0.013 | 1.309 | 1.059 | 1.618 |
| SLC44A2 | 224609_at | 0.251 | 0.107 | 5.503 | 1 | 0.019 | 1.286 | 1.042 | 1.586 |
| NR2C2AP | 226839_at | 0.355 | 0.149 | 5.687 | 1 | 0.017 | 1.427 | 1.065 | 1.911 |
| LETM1 | 222006_at | 0.282 | 0.137 | 4.275 | 1 | 0.038 | 1.326 | 1.015 | 1.733 |
Table 10.
Set of six prognostic genes.
Table 10.
Set of six prognostic genes.
| Gene | Probe | B | SE | Wald | df | p-Value | Hazard Risk | Lower | Upper |
|---|
| USP28 | 1552678_a_at | 0.44309 | 0.155462 | 8.123335 | 1 | 0.004 | 1.558 | 1.148 | 2.112 |
| SORT1 | 212807_s_at | 0.196301 | 0.082305 | 5.688408 | 1 | 0.017 | 1.217 | 1.036 | 1.430 |
| ALDH6A1 | 221588_x_at | 0.403888 | 0.167332 | 5.825904 | 1 | 0.016 | 1.498 | 1.079 | 2.079 |
| CSNK2A1 | 212075_s_at | 0.304248 | 0.152804 | 3.964474 | 1 | 0.047 | 1.356 | 1.005 | 1.829 |
| TOR1AIP1 | 212409_s_at | 0.313668 | 0.169528 | 3.423401 | 1 | 0.06 | 1.368 | 0.982 | 1.908 |
| UBE2H | 221962_s_at | −0.63261 | 0.113921 | 30.83679 | 1 | 2.8 × 10−8 | 0.531 | 0.425 | 0.664 |
Table 11.
Biological function of the set of 16 prognostic genes.
Table 11.
Biological function of the set of 16 prognostic genes.
| Gene | Function |
|---|
| PAF1 | Positive regulation of cell cycle G1/S phase transition |
| USP28 | DNA damage response checkpoint and MYC proto-oncogene stability |
| SORT1 | Endocytosis |
| MAP7D3 | Microtubule cytoskeleton organization |
| FITM2 | Cytoskeleton organization and lipid and energy homeostasis |
| CENPO | Mitotic progression and chromosome segregation |
| PRCC | Regulation of cell cycle progression |
| ALDH6A1 | Pyrimidine metabolism, RNA binding |
| CSNK2A1 | Cell cycle, apoptosis process |
| TOR1AIP1 | Regulation of nuclear membrane integrity, protein localization to nucleus |
| NUP98 | Role in the nuclear pore complex (NPC) assembly and/or maintenance |
| UBE2H | ATP binding, ubiquitin-protein transferase activity |
| UBXN7 | Ubiquitin binding |
| SLC44A2 | Positive regulation of I-kappaB kinase/NF-kappaB signaling |
| NR2C2AP | Transcription initiation from RNA polymerase II promoter |
| LETM1 | Regulation of concentration of calcium ion. |
Table 12.
Correlation between PD-L1 and the clinicopathological features of the patients in the validation series (Tokai cases).
Table 12.
Correlation between PD-L1 and the clinicopathological features of the patients in the validation series (Tokai cases).
| Predictors for High PD-L1 | p-Value | Odds Ratio | 95% CI for OR |
|---|
| Lower | Upper |
|---|
| Sex Male | 0.211 | 1.699 | 0.741 | 3.898 |
| Age > 60 | 0.994 | 1.004 | 0.415 | 2.429 |
| Location | | | | |
| Nodal (+spleen) (Reference) | - | - | - | - |
| Extranodal | | | | |
| Waldeyer’s ring | 0.999 | 0 | 0 | - |
| Gastrointestinal | 0.756 | 1.242 | 0.317 | 4.875 |
| Other extranodal | 0.487 | 0.71 | 0.27 | 1.864 |
| LDH High (>219) | 0.115 | 2.037 | 0.841 | 4.933 |
| Seric IL2RA high (>530) | 0.016 | 12.453 | 1.598 | 97.065 |
| ECOG Performance Status ≥ 2 | 0.207 | 2.111 | 0.661 | 6.741 |
| Clinical stage III or IV | 0.006 | 3.585 | 1.452 | 8.851 |
| Extranodal disease site > 1 | 0.741 | 0.825 | 0.263 | 2.588 |
| B symptoms | 0.004 | 4.333 | 1.618 | 11.606 |
| IPI HI+H | 0.041 | 2.579 | 1.037 | 6.411 |
| Non-GCB Subtype (Hans’s algorithm) | 0.014 | 3.757 | 1.307 | 10.794 |
| Epstein–Barr virus, EBER+ | 0.005 | 4.931 | 1.620 | 15.005 |
| High RGS1 protein expression | 0.015 | 3.003 | 1.241 | 7.264 |
| Absence of clinical response to treatment | 0.078 | 2.284 | 0.912 | 5.717 |
Table 13.
Correlation between IKAROS and the clinicopathological features of the patients in the validation series (Tokai cases).
Table 13.
Correlation between IKAROS and the clinicopathological features of the patients in the validation series (Tokai cases).
| Predictors for High IKAROS | p-Value | Odds Ratio | 95% CI for OR |
|---|
| Lower | Upper |
|---|
| Sex Male | 0.216 | 0.549 | 0.213 | 1.418 |
| Age > 60 | 0.768 | 1.173 | 0.405 | 3.4 |
| Location | | | | |
| Nodal (+spleen) (Reference) | - | - | - | - |
| Extranodal | | | | |
| Waldeyer’s ring | 0.317 | 0.33 | 0.038 | 2.887 |
| Gastrointestinal | 0.869 | 1.133 | 0.256 | 5.004 |
| Other extranodal | 0.483 | 0.661 | 0.208 | 2.101 |
| LDH high (>219) | 0.407 | 0.669 | 0.259 | 1.728 |
| Seric IL2RA high (>530) | 0.189 | 0.481 | 0.162 | 1.433 |
| ECOG Performance Status ≥ 2 | 0.632 | 0.711 | 0.176 | 2.873 |
| Clinical stage III or IV | 0.955 | 0.972 | 0.368 | 2.566 |
| Extranodal disease site >1 | 0.802 | 0.86 | 0.264 | 2.796 |
| B symptoms | 0.635 | 0.739 | 0.213 | 2.566 |
| IPI HI+H | 0.731 | 1.206 | 0.414 | 3.512 |
| GCB subtype (Hans’s algorithm) | 0.008 | 3.756 | 1.405 | 10.04 |
| Epstein–Barr virus, EBER+ | 0.276 | 0.418 | 0.087 | 2.008 |
| High RGS1 protein expression | 0.112 | 0.459 | 0.176 | 1.199 |
| Clinical response to treatment | 0.031 | 9.767 | 1.226 | 77.796 |


 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}