Functional and Safety Characterization of Weissella paramesenteroides Strains Isolated from Dairy Products through Whole-Genome Sequencing and Comparative Genomics




Abstract
1. Introduction
2. Materials and Methods
2.1. Microbial Strains and Culture Conditions
2.2. Whole Genome Sequencing, Assembly, and Quality Control
2.3. In Silico Typing and Characterization
2.4. Phylogenetic Analysis and Comparative Genomics
2.5. Statistical Analysis
3. Results and Discussion
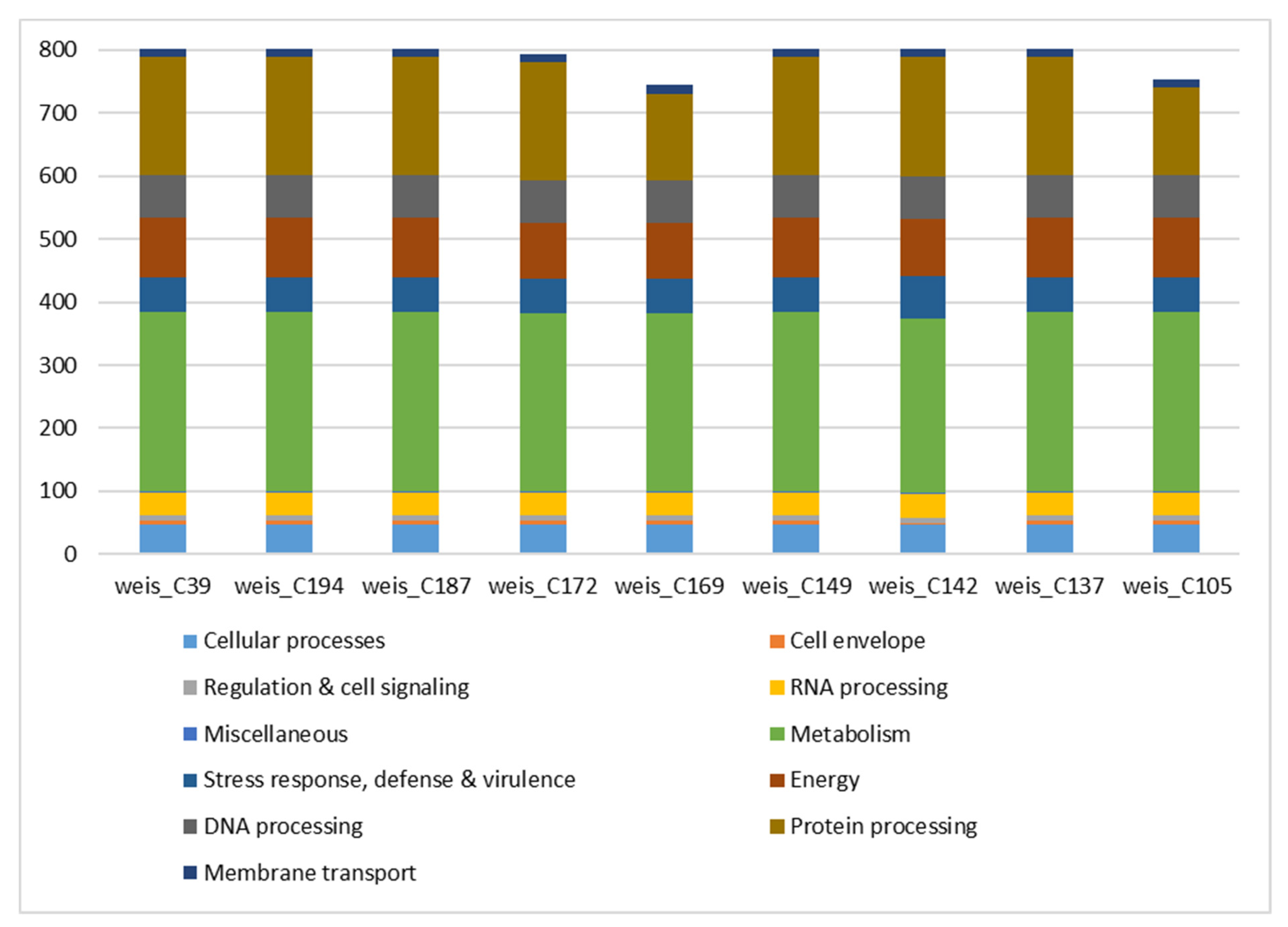
3.1. Species Identification, Assembly Statistics, and Subsystem Analysis
3.2. Presence of Resistance and Virulence Genes
3.3. Other Genomic Features
3.3.1. Bacteriocins, Prophages, and CRISPR-Cas
3.3.2. Plasmids and Other MGEs
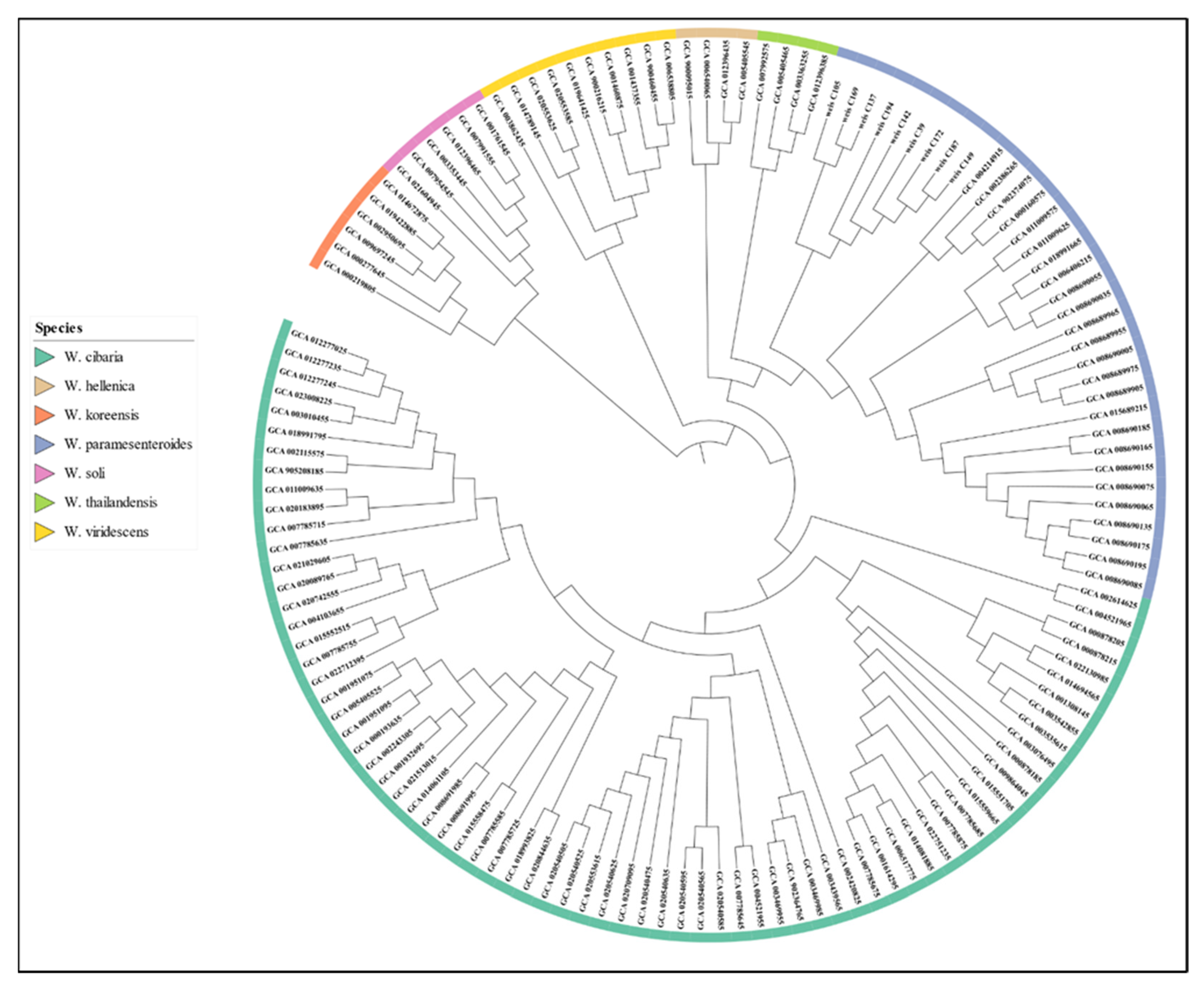
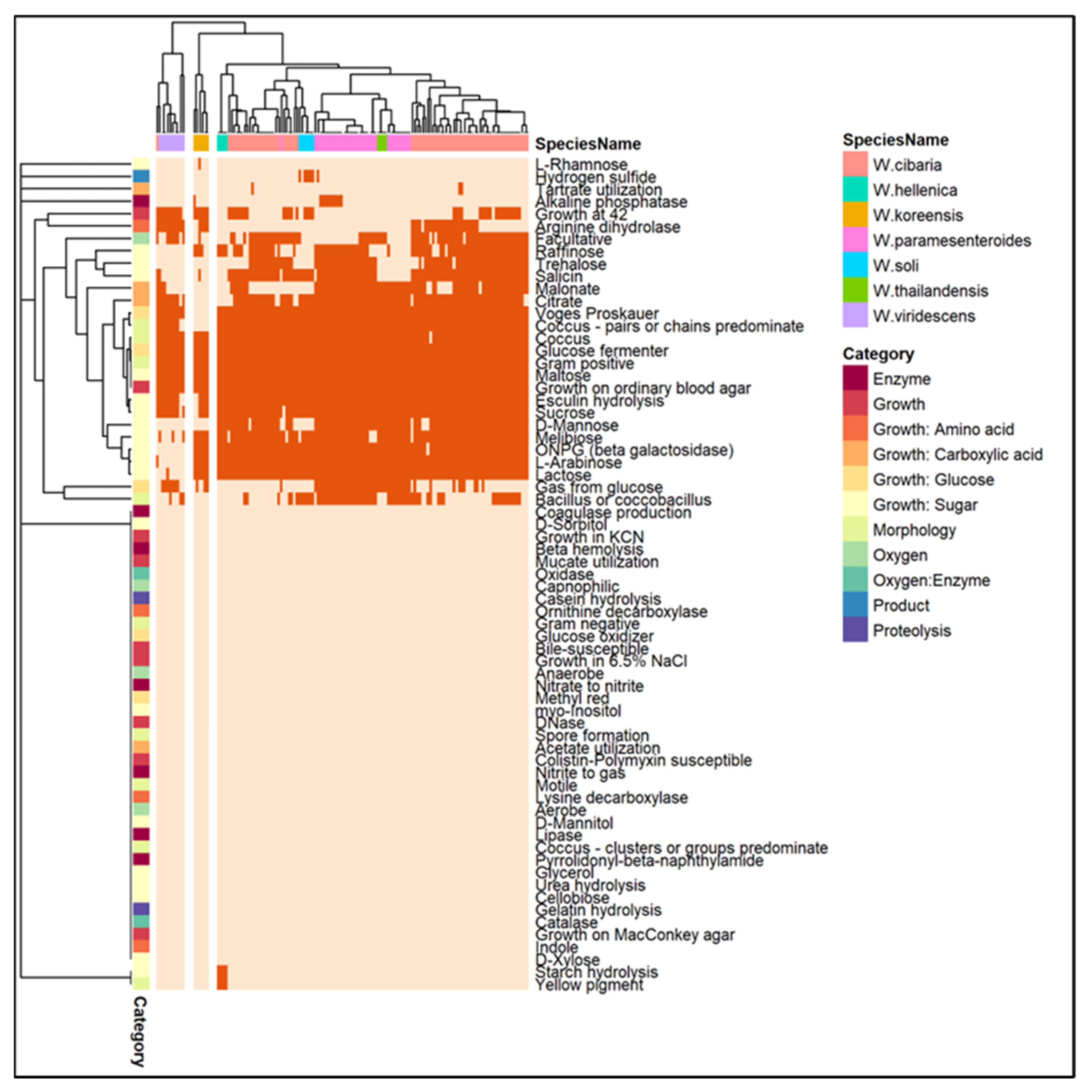
3.4. Phylogenetic Analysis and Comparative Genomics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lonvaud-Funel, A. Leuconostocaceae Family. In Encyclopedia of Food Microbiology; Elsevier: Amsterdam, The Netherlands, 2014; Volume 2, pp. 455–465. ISBN 9780123847331. [Google Scholar]
- Teixeira, C.G.; da Silva, R.R.; Fusieger, A.; Martins, E.; de Freitas, R.; de Carvalho, A.F. The Weissella genus in the food industry: A review. Res. Soc. Dev. 2021, 10, e8310514557. [Google Scholar] [CrossRef]
- Fessard, A.; Remize, F. Why Are Weissella Spp. Not Used as Commercial Starter Cultures for Food Fermentation? Fermentation 2017, 3, 38. [Google Scholar] [CrossRef]
- Abriouel, H.; Lerma, L.L.; Casado Muñoz, M. del C.; Montoro, B.P.; Kabisch, J.; Pichner, R.; Cho, G.S.; Neve, H.; Fusco, V.; Franz, C.M.A.P.; et al. The Controversial Nature of the Weissella Genus: Technological and Functional Aspects versus Whole Genome Analysis-Based Pathogenic Potential for Their Application in Food and Health. Front. Microbiol. 2015, 6, 1–14. [Google Scholar] [CrossRef]
- Tarrah, A.; Pakroo, S.; Lemos Junior, W.J.F.; Guerra, A.F.; Corich, V.; Giacomini, A. Complete Genome Sequence and Carbohydrates-Active EnZymes (CAZymes) Analysis of Lactobacillus paracasei DTA72, a Potential Probiotic Strain with Strong Capability to Use Inulin. Curr. Microbiol. 2020, 77, 2867–2875. [Google Scholar] [CrossRef]
- Bintsis, T. Lactic Acid Bacteria as Starter Cultures: An Update in Their Metabolism and Genetics. AIMS Microbiol. 2018, 4, 665–684. [Google Scholar] [CrossRef] [PubMed]
- Tenea, G.N.; Hurtado, P. Next-Generation Sequencing for Whole-Genome Characterization of Weissella cibaria UTNGt21O Strain Originated From Wild Solanum quitoense Lam. Fruits: An Atlas of Metabolites With Biotechnological Significance. Front. Microbiol. 2021, 12, 1240. [Google Scholar] [CrossRef] [PubMed]
- Surachat, K.; Kantachote, D.; Wonglapsuwan, M.; Chukamnerd, A.; Deachamag, P.; Mittraparp-arthorn, P.; Jeenkeawpiam, K. Complete Genome Sequence of Weissella cibaria NH9449 and Comprehensive Comparative-Genomic Analysis: Genomic Diversity and Versatility Trait Revealed. Front. Microbiol. 2022, 13, 1–15. [Google Scholar] [CrossRef]
- Graham, K.; Stack, H.; Rea, R. Safety, Beneficial and Technological Properties of Enterococci for Use in Functional Food Applications—A Review. Crit. Rev. Food Sci. Nutr. 2020, 60, 3836–3861. [Google Scholar] [CrossRef]
- Collineau, L.; Boerlin, P.; Carson, C.A.; Chapman, B.; Fazil, A.; Hetman, B.; McEwen, S.A.; Jane Parmley, E.; Reid-Smith, R.J.; Taboada, E.N.; et al. Integrating Whole-Genome Sequencing Data into Quantitative Risk Assessment of Foodborne Antimicrobial Resistance: A Review of Opportunities and Challenges. Front. Microbiol. 2019, 10, 1–18. [Google Scholar] [CrossRef]
- Tsigkrimani, M.; Bakogianni, M.; Paramithiotis, S.; Bosnea, L.; Pappa, E.; Drosinos, E.H.; Skandamis, P.N.; Mataragas, M. Microbial Ecology of Artisanal Feta and Kefalograviera Cheeses, Part I: Bacterial Community and Its Functional Characteristics with Focus on Lactic Acid Bacteria as Determined by Culture-Dependent Methods and Phenotype Microarrays. Microorganisms 2022, 10, 161. [Google Scholar] [CrossRef]
- Tsigkrimani, M.; Panagiotarea, K.; Paramithiotis, S.; Bosnea, L.; Pappa, E.; Drosinos, E.H.; Skandamis, P.N.; Mataragas, M. Microbial Ecology of Sheep Milk, Artisanal Feta, and Kefalograviera Cheeses. Part II: Technological, Safety, and Probiotic Attributes of Lactic Acid Bacteria Isolates. Foods 2022, 11, 459. [Google Scholar] [CrossRef] [PubMed]
- Syrokou, M.K.; Themeli, C.; Paramithiotis, S.; Mataragas, M.; Bosnea, L.; Argyri, A.A.; Chorianopoulos, N.G.; Skandamis, P.N.; Drosinos, E.H. Microbial Ecology of Greek Wheat Sourdoughs, Identified by a Culture-Dependent and a Culture-Independent Approach. Foods 2020, 9, 1603. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data 2019. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 27 May 2022).
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Completing Bacterial Genome Assemblies with Multiplex MinION Sequencing. Microb. Genomics 2017, 3, e000132. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.J.; Wattam, A.R.; Aziz, R.K.; Brettin, T.; Butler, R.; Butler, R.M.; Chlenski, P.; Conrad, N.; Dickerman, A.; Dietrich, E.M.; et al. The PATRIC Bioinformatics Resource Center: Expanding Data and Analysis Capabilities. Nucleic Acids Res. 2020, 48, D606–D612. [Google Scholar] [CrossRef]
- Bosi, E.; Donati, B.; Galardini, M.; Brunetti, S.; Sagot, M.F.; Lió, P.; Crescenzi, P.; Fani, R.; Fondi, M. MeDuSa: A Multi-Draft Based Scaffolder. Bioinformatics 2015, 31, 2443–2451. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043. [Google Scholar] [CrossRef]
- Lu, J.; Salzberg, S.L. SkewIT: The Skew Index Test for Large-Scale GC Skew Analysis of Bacterial Genomes. PLOS Comput. Biol. 2020, 16, e1008439. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved Metagenomic Analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. TYGS Is an Automated High-Throughput Platform for State-of-the-Art Genome-Based Taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.; Ouk Kim, Y.; Park, S.-C.; Chun, J. OrthoANI: An Improved Algorithm and Software for Calculating Average Nucleotide Identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Kiryutin, B.; Koonin, E.V.; Krylov, D.M.; Mazumder, R.; Smirnov, S.; Nikolskaya, A.N.; et al. The COG Database: An Updated Vesion Includes Eukaryotes. BMC Bioinform. 2003, 4, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an Update of CRISRFinder, Includes a Portable Version, Enhanced Performance and Integrates Search for Cas Proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef]
- Arndt, D.; Marcu, A.; Liang, Y.; Wishart, D.S. PHAST, PHASTER and PHASTEST: Tools for Finding Prophage in Bacterial Genomes. Brief. Bioinform. 2019, 20, 1560–1567. [Google Scholar] [CrossRef]
- Seemann, T. Abricate, Github 2020. Available online: https://github.com/tseemann/abricate (accessed on 27 May 2022).
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of Acquired Antimicrobial Resistance Genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and Refined Dataset for Big Data Analysis—10 Years On. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef]
- Johansson, M.H.K.; Bortolaia, V.; Tansirichaiya, S.; Aarestrup, F.M.; Roberts, A.P.; Petersen, T.N. Detection of Mobile Genetic Elements Associated with Antibiotic Resistance in Salmonella enterica Using a Newly Developed Web Tool: MobileElementFinder. J. Antimicrob. Chemother. 2021, 76, 101–109. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; Garciá-Fernández, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. In Silico Detection and Typing of Plasmids Using Plasmidfinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef]
- Cosentino, S.; Voldby Larsen, M.; Møller Aarestrup, F.; Lund, O. PathogenFinder—Distinguishing Friend from Foe Using Bacterial Whole Genome Sequence Data. PLoS ONE 2013, 8, e77302. [Google Scholar] [CrossRef]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.G.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid Large-Scale Prokaryote Pan Genome Analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Croucher, N.J.; Page, A.J.; Connor, T.R.; Delaney, A.J.; Keane, J.A.; Bentley, S.D.; Parkhill, J.; Harris, S.R. Rapid Phylogenetic Analysis of Large Samples of Recombinant Bacterial Whole Genome Sequences Using Gubbins. Nucleic Acids Res. 2015, 43, e15. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum Evolution Trees with Profiles Instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Yohe, T.; Huang, L.; Entwistle, S.; Wu, P.; Yang, Z.; Busk, P.K.; Xu, Y.; Yin, Y. DbCAN2: A Meta Server for Automated Carbohydrate-Active Enzyme Annotation. Nucleic Acids Res. 2018, 46, W95–W101. [Google Scholar] [CrossRef]
- Weimann, A.; Mooren, K.; Frank, J.; Pope, P.B.; Bremges, A.; McHardy, A.C. From Genomes to Phenotypes: Traitar, the Microbial Trait Analyzer. mSystems 2016, 1, e00101-16. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. EggNOG 5.0: A Hierarchical, Functionally and Phylogenetically Annotated Orthology Resource Based on 5090 Organisms and 2502 Viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. ClusterProfiler 4.0: A Universal Enrichment Tool for Interpreting Omics Data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Reijnders, M.J.M.F.; Waterhouse, R.M. Summary Visualizations of Gene Ontology Terms With GO-Figure! Front. Bioinforma. 2021, 1, 638255. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy Platform for Accessible, Reproducible and Collaborative Biomedical Analyses: 2018 Update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef] [PubMed]
- Brynildsrud, O.; Bohlin, J.; Scheffer, L.; Eldholm, V. Rapid Scoring of Genes in Microbial Pan-Genome-Wide Association Studies with Scoary. Genome Biol. 2016, 17, 238. [Google Scholar] [CrossRef] [PubMed]
- Overbeek, R.; Begley, T.; Butler, R.M.; Choudhuri, J.V.; Chuang, H.Y.; Cohoon, M.; de Crécy-Lagard, V.; Diaz, N.; Disz, T.; Edwards, R.; et al. The Subsystems Approach to Genome Annotation and Its Use in the Project to Annotate 1000 Genomes. Nucleic Acids Res. 2005, 33, 5691–5702. [Google Scholar] [CrossRef]
- Silva, C.C.G.; Silva, S.P.M.; Ribeiro, S.C. Application of Bacteriocins and Protective Cultures in Dairy Food Preservation. Front. Microbiol. 2018, 9, 594. [Google Scholar] [CrossRef]
- Henning, C.; Gautam, D.; Muriana, P. Identification of Multiple Bacteriocins in Enterococcus spp. Using an Enterococcus-Specific Bacteriocin PCR Array. Microorganisms 2015, 3, 1. [Google Scholar] [CrossRef]
- He, Q.; Hou, Q.; Wang, Y.; Li, J.; Li, W.; Kwok, L.-Y.; Sun, Z.; Zhang, H.; Zhong, Z. Comparative Genomic Analysis of Enterococcus faecalis: Insights into Their Environmental Adaptations. BMC Genom. 2018, 19, 527. [Google Scholar] [CrossRef]
- Ghattargi, V.C.; Gaikwad, M.A.; Meti, B.S.; Nimonkar, Y.S.; Dixit, K.; Prakash, O.; Shouche, Y.S.; Pawar, S.P.; Dhotre, D.P. Comparative Genome Analysis Reveals Key Genetic Factors Associated with Probiotic Property in Enterococcus faecium Strains. BMC Genom. 2018, 19, 652. [Google Scholar] [CrossRef]
- Harmer, C.J.; Hall, R.M. An Analysis of the IS6/IS26 Family of Insertion Sequences: Is It a Single Family? Microb. Genom. 2019, 5, e000291. [Google Scholar] [CrossRef]
- Haandrikman, A.J.; van Leeuwen, C.; Kok, J.; Vos, P.; de Vos, W.M.; Venema, G. Insertion Elements on Lactococcal Proteinase Plasmids. Appl. Environ. Microbiol. 1990, 56, 1890–1896. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liang, Q.; Lu, B.; Shen, H.; Liu, S.; Shi, Y.; Leptihn, S.; Li, H.; Wei, J.; Liu, C.; et al. Whole-Genome Analysis of Probiotic Product Isolates Reveals the Presence of Genes Related to Antimicrobial Resistance, Virulence Factors, and Toxic Metabolites, Posing Potential Health Risks. BMC Genom. 2021, 22, 210. [Google Scholar] [CrossRef] [PubMed]
- Carattoli, A.; Bertini, A.; Villa, L.; Falbo, V.; Hopkins, K.L.; Threlfall, E.J. Identification of Plasmids by PCR-Based Replicon Typing. J. Microbiol. Methods 2005, 63, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Harris, H.M.B.; McCann, A.; Guo, C.; Argimón, S.; Zhang, W.; Yang, X.; Jeffery, I.B.; Cooney, J.C.; Kagawa, T.F.; et al. Expanding the Biotechnology Potential of Lactobacilli through Comparative Genomics of 213 Strains and Associated Genera. Nat. Commun. 2015, 6, 8322. [Google Scholar] [CrossRef]
- Akesson, M.; Dufour, M.; Sloan, G.L.; Simmonds, R.S. Targeting of Streptococci by Zoocin A. FEMS Microbiol. Lett. 2007, 270, 155–161. [Google Scholar] [CrossRef]
- Kemperman, R.; Kuipers, A.; Karsens, H.; Nauta, A.; Kuipers, O.; Kok, J. Identification and Characterization of Two Novel Clostridial Bacteriocins, Circularin A and Closticin 574. Appl. Environ. Microbiol. 2003, 69, 1589–1597. [Google Scholar] [CrossRef]
- Zhang, X.; Vrijenhoek, J.E.P.; Bonten, M.J.M.; Willems, R.J.L.; van Schaik, W. A Genetic Element Present on Megaplasmids Allows Enterococcus faecium to Use Raffinose as Carbon Source. Environ. Microbiol. 2011, 13, 518–528. [Google Scholar] [CrossRef]
- Chilambi, G.S.; Nordstrom, H.R.; Evans, D.R.; Ferrolino, J.A.; Hayden, R.T.; Marón, G.M.; Vo, A.N.; Gilmore, M.S.; Wolf, J.; Rosch, J.W.; et al. Evolution of Vancomycin-Resistant Enterococcus faecium during Colonization and Infection in Immunocompromised Pediatric Patients. Proc. Natl. Acad. Sci. USA 2020, 117, 11703–11714. [Google Scholar] [CrossRef]
- Kiousi, D.E.; Efstathiou, C.; Tegopoulos, K.; Mantzourani, I.; Alexopoulos, A.; Plessas, S.; Kolovos, P.; Koffa, M.; Galanis, A. Genomic Insight Into Lacticaseibacillus paracasei SP5, Reveals Genes and Gene Clusters of Probiotic Interest and Biotechnological Potential. Front. Microbiol. 2022, 13, 2038. [Google Scholar] [CrossRef]
- Pal, V.K.; Bandyopadhyay, P.; Singh, A. Hydrogen Sulfide in Physiology and Pathogenesis of Bacteria and Viruses. IUBMB Life 2018, 70, 393–410. [Google Scholar] [CrossRef]
- Ferreira, A.S. Insights into the Role of Extracellular Polysaccharides in Burkholderia Adaptation to Different Environments. Front. Cell. Infect. Microbiol. 2011, 1, 16. [Google Scholar] [CrossRef] [PubMed]
- Elshaghabee, F.M.; Ghadimi, D.; Habermann, D.; de Vrese, M.; Bockelmann, W.; Kaatsch, H.-J.; Heller, K.J.; Schrezenmeir, J. Effect of Oral Administration of Weissella confusa on Fecal and Plasma Ethanol Concentrations, Lipids and Glucose Metabolism in Wistar Rats Fed High Fructose and Fat Diet. Hepatic Med. Evid. Res. 2020, 12, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Ram, S.; Mitra, M.; Shah, F.; Tirkey, S.R.; Mishra, S. Bacteria as an Alternate Biofactory for Carotenoid Production: A Review of Its Applications, Opportunities and Challenges. J. Funct. Foods 2020, 67, 103867. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain ID | Genus & Species | Genome Size (Mb) | GC Content (%) | No of Scaffolds | N50 (Mb) | No of CDSs |
|---|---|---|---|---|---|---|
| weis_C39 | W. paramesenteroides | 1.95 | 37.98 | 25 | 1.37 | 1965 |
| weis_C194 | W. paramesenteroides | 1.95 | 37.99 | 26 | 1.92 | 1969 |
| weis_C187 | W. paramesenteroides | 1.95 | 37.98 | 24 | 1.92 | 1968 |
| weis_C172 | W. paramesenteroides | 1.91 | 38 | 21 | 1.87 | 1915 |
| weis_C169 | W. paramesenteroides | 1.91 | 38 | 26 | 1.50 | 1920 |
| weis_C149 | W. paramesenteroides | 1.95 | 37.98 | 25 | 1.13 | 1964 |
| weis_C142 | W. paramesenteroides | 1.75 | 38.37 | 7 | 1.75 | 1690 |
| weis_C137 | W. paramesenteroides | 1.95 | 37.98 | 25 | 1.34 | 1969 |
| weis_C105 | W. paramesenteroides | 1.95 | 37.98 | 27 | 1.92 | 1975 |
| Strain ID | weis_C39 | weis_C105 | weis_C137 | weis_C142 | weis_C149 | weis_C169 | weis_C172 | weis_C187 | weis_C194 |
|---|---|---|---|---|---|---|---|---|---|
| weis_C39 | 99.99 | 99.99 | 99.91 | 99.99 | 99.99 | 99.99 | 99.99 | 99.99 | |
| weis_C105 | 99.99 | 99.91 | 99.99 | 99.98 | 99.99 | 99.99 | 99.97 | ||
| weis_C137 | 99.89 | 99.99 | 99.99 | 99.98 | 99.98 | 99.99 | |||
| weis_C142 | 99.89 | 99.90 | 99.89 | 99.90 | 99.89 | ||||
| weis_C149 | 99.98 | 99.99 | 99.99 | 99.98 | |||||
| weis_C169 | 99.99 | 99.98 | 99.99 | ||||||
| weis_C172 | 99.99 | 99.99 | |||||||
| weis_C187 | 99.98 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Apostolakos, I.; Paramithiotis, S.; Mataragas, M. Functional and Safety Characterization of Weissella paramesenteroides Strains Isolated from Dairy Products through Whole-Genome Sequencing and Comparative Genomics. Dairy 2022, 3, 799-813. https://doi.org/10.3390/dairy3040055
Apostolakos I, Paramithiotis S, Mataragas M. Functional and Safety Characterization of Weissella paramesenteroides Strains Isolated from Dairy Products through Whole-Genome Sequencing and Comparative Genomics. Dairy. 2022; 3(4):799-813. https://doi.org/10.3390/dairy3040055
Chicago/Turabian StyleApostolakos, Ilias, Spiros Paramithiotis, and Marios Mataragas. 2022. "Functional and Safety Characterization of Weissella paramesenteroides Strains Isolated from Dairy Products through Whole-Genome Sequencing and Comparative Genomics" Dairy 3, no. 4: 799-813. https://doi.org/10.3390/dairy3040055
APA StyleApostolakos, I., Paramithiotis, S., & Mataragas, M. (2022). Functional and Safety Characterization of Weissella paramesenteroides Strains Isolated from Dairy Products through Whole-Genome Sequencing and Comparative Genomics. Dairy, 3(4), 799-813. https://doi.org/10.3390/dairy3040055