
Targeted Drug Delivery of Anticancer Agents Using C5N2 Substrate: Insights from Density Functional Theory



Abstract
1. Introduction
2. Computational Methodology
3. Results and Discussion
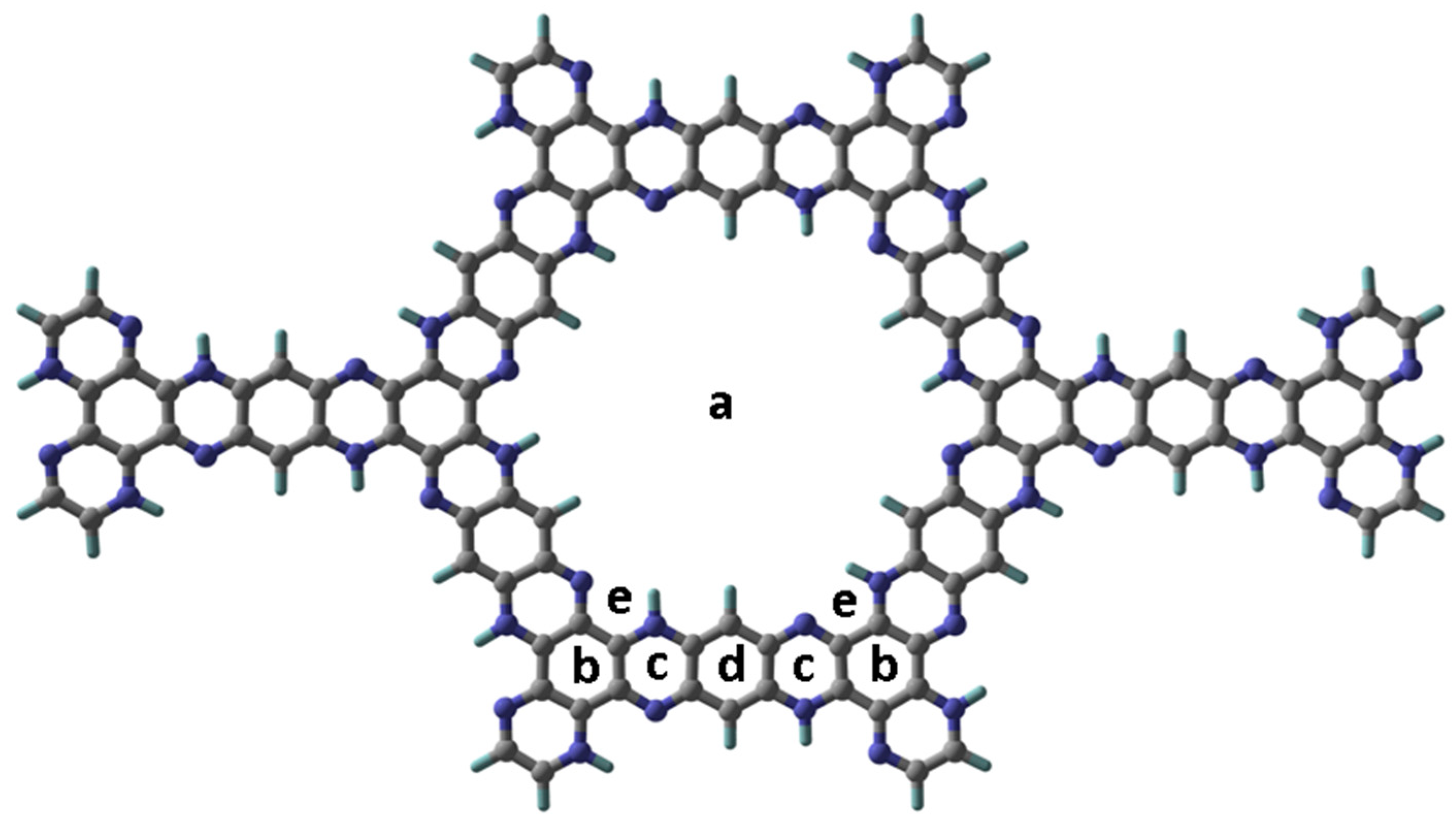
3.1. Topological Analysis
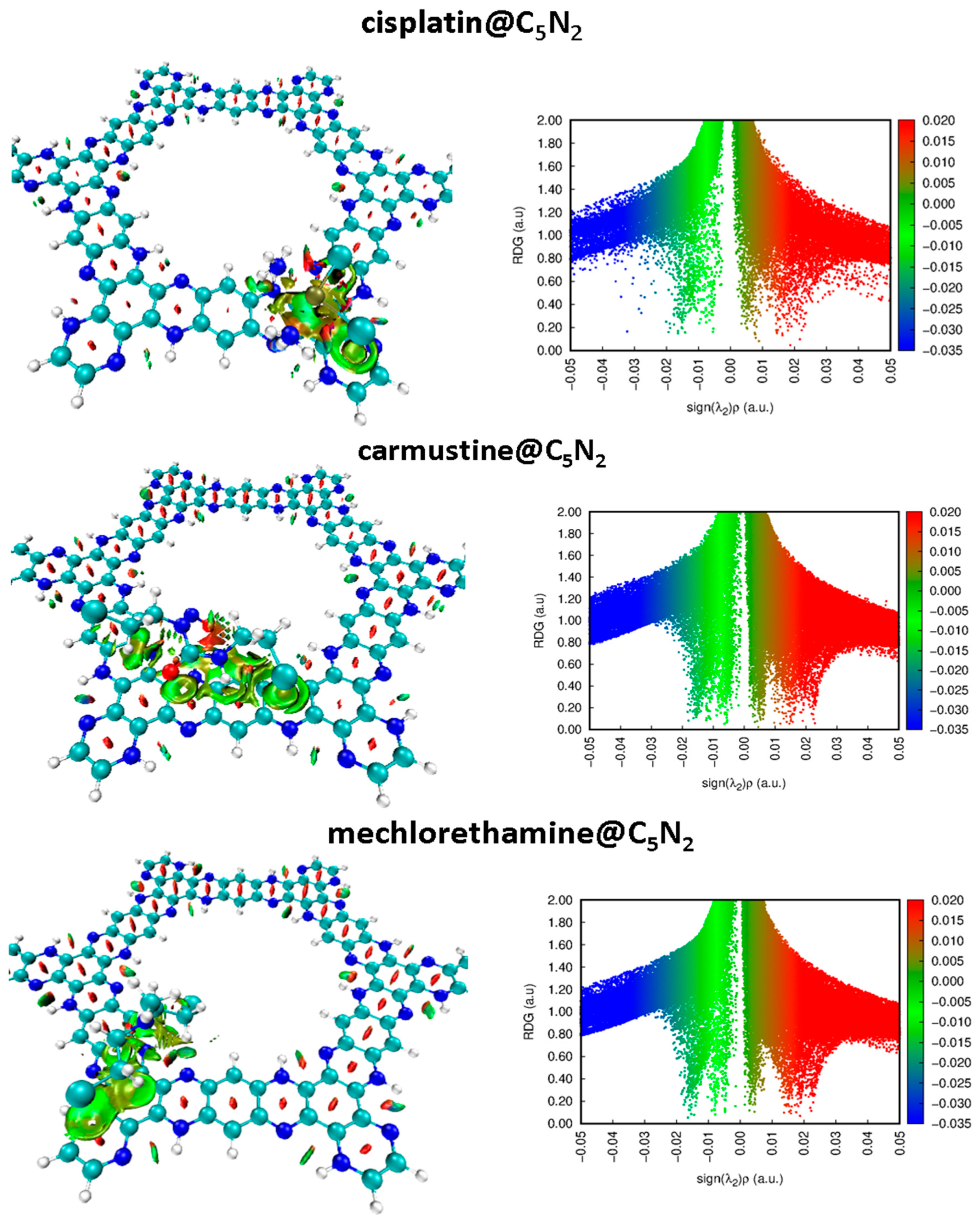
3.1.1. Non-Covalent Interaction (NCI) Analysis
3.1.2. Quantum Theory of Atoms in Molecules (QTAIM) Analysis
3.1.3. Electron Localization Function (ELF) Analysis
3.2. Analysis of Electronic Properties
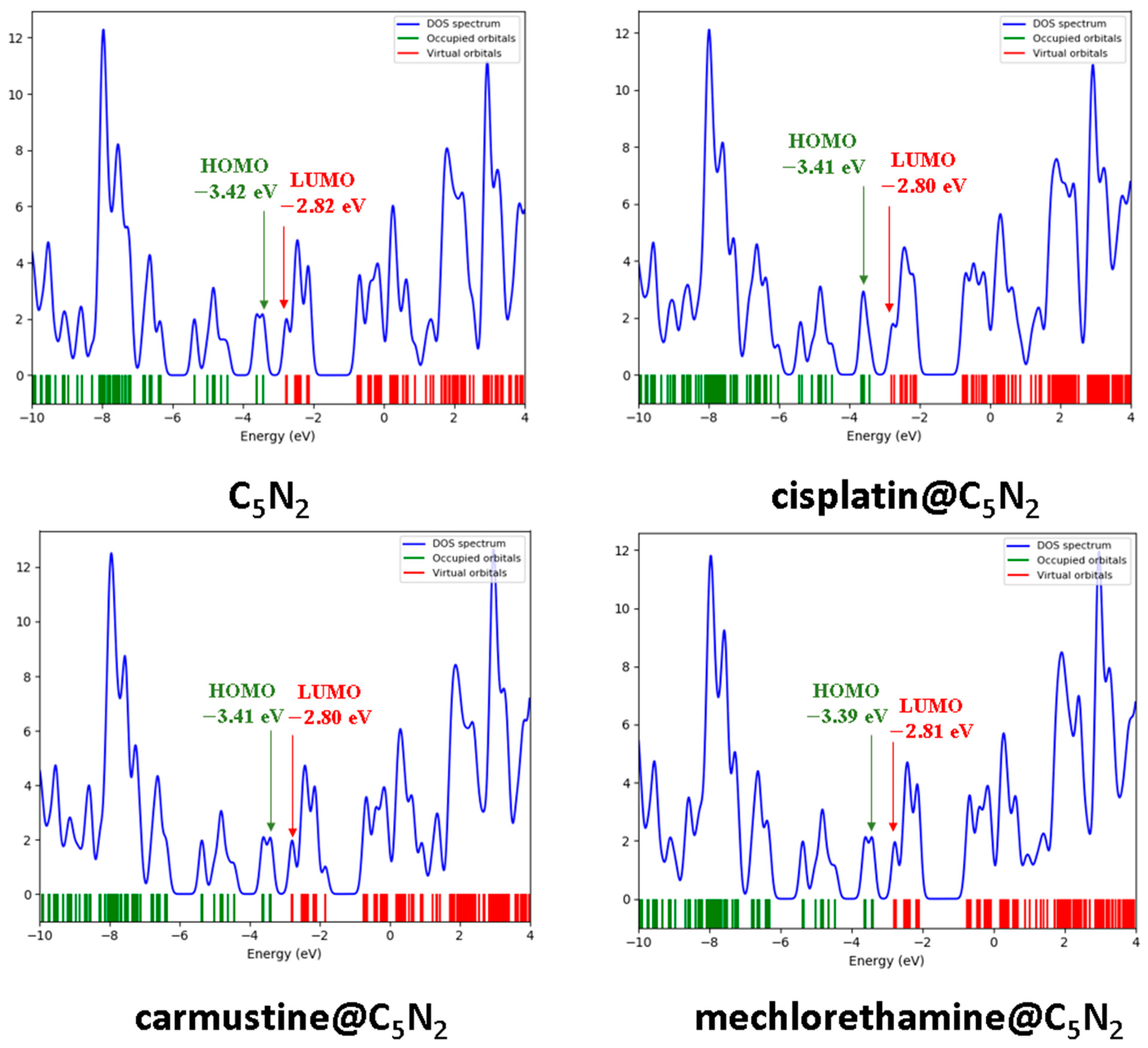
3.2.1. Frontier Molecular Orbital (FMOs) Analysis and Chemical Reactivity Descriptors
3.2.2. Density of States (DOS) Analysis
3.2.3. NBO and EDD Analyses
4. Recovery Time
5. Solvent Effect
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA A Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2023. CA A Cancer J. Clin. 2023, 73, 233–254. [Google Scholar] [CrossRef]
- Cao, S.; Wei, Y.; Huang, J.; Yue, Y.; Deng, A.; Zeng, H.; Wei, W. A bibliometric worldview of breast-conserving surgery for breast cancer from 2013 to 2023. Front. Oncol. 2024, 14, 1405351. [Google Scholar] [CrossRef]
- Srinivasan, D.; Subbarayan, R.; Srivastava, N.; Radhakrishnan, A.; Adtani, P.N.; Chauhan, A.; Krishnamoorthy, L. A comprehensive overview of radiation therapy impacts of various cancer treatments and pivotal role in the immune system. Cell Biochem. Funct. 2024, 42, e4103. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Jasrotia, S.; Kumar, A. Effects of chemotherapy on the immune system: Implications for cancer treatment and patient outcomes. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2024, 397, 2551–2566. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhou, H.; Tan, L.; Siu, K.T.H.; Guan, X.-Y. Exploring treatment options in cancer: Tumor treatment strategies. Signal Transduct. Target. Ther. 2024, 9, 175. [Google Scholar] [CrossRef] [PubMed]
- Kar, A.; Agarwal, D.G.; Agarwal, D.S. A Review On Nanostructure Drug Carriers for Treatment and Management of Neuroendocrine Cancer. Int. J. Pharm. Sci. 2023, 14, 1–9. [Google Scholar] [CrossRef]
- Anand, U.; Dey, A.; Chandel, A.K.S.; Sanyal, R.; Mishra, A.; Pandey, D.K.; De Falco, V.; Upadhyay, A.; Kandimalla, R.; Chaudhary, A. Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics. Genes Dis. 2023, 10, 1367–1401. [Google Scholar] [CrossRef]
- Romani, A.M. Cisplatin in cancer treatment. Biochem. Pharmacol. 2022, 206, 115323. [Google Scholar] [CrossRef]
- Strojan, P.; Vermorken, J.B.; Beitler, J.J.; Saba, N.F.; Haigent, M., Jr.; Bossi, P.; Worden, F.P.; Langendijk, J.A.; Eisbruch, A.; Mendenhall, W.M. Cumulative cisplatin dose in concurrent chemoradiotherapy for head and neck cancer: A systematic review. Head Neck 2016, 38, E2151–E2158. [Google Scholar] [CrossRef]
- Song, M.; Cui, M.; Liu, K. Therapeutic strategies to overcome cisplatin resistance in ovarian cancer. Eur. J. Med. Chem. 2022, 232, 114205. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zheng, Z.; Chen, W.; Li, D.; Zhang, H.; Zhu, Y.; Mo, Q.; Zhao, X.; Fan, Q.; Deng, F. Regulation of cisplatin resistance in bladder cancer by epigenetic mechanisms. Drug Resist. Updates 2023, 68, 100938. [Google Scholar] [CrossRef] [PubMed]
- Sala, L.; Perecko, T.; Mestek, O.; Pinkas, D.; Homola, T.; Kocisek, J. Cisplatin-cross-linked DNA origami nanostructures for drug delivery applications. ACS Appl. Nano Mater. 2022, 5, 13267–13275. [Google Scholar] [CrossRef]
- Lemjabbar-Alaoui, H.; Hassan, O.U.; Yang, Y.-W.; Buchanan, P. Lung cancer: Biology and treatment options. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2015, 1856, 189–210. [Google Scholar] [CrossRef] [PubMed]
- Shirbin, S.J.; Ladewig, K.; Fu, Q.; Klimak, M.; Zhang, X.; Duan, W.; Qiao, G.G. Cisplatin-induced formation of biocompatible and biodegradable polypeptide-based vesicles for targeted anticancer drug delivery. Biomacromolecules 2015, 16, 2463–2474. [Google Scholar] [CrossRef]
- Wagstaff, A.J.; Brown, S.D.; Holden, M.R.; Craig, G.E.; Plumb, J.A.; Brown, R.E.; Schreiter, N.; Chrzanowski, W.; Wheate, N.J. Cisplatin drug delivery using gold-coated iron oxide nanoparticles for enhanced tumour targeting with external magnetic fields. Inorganica Chim. Acta 2012, 393, 328–333. [Google Scholar] [CrossRef]
- Perveen, M.; Nazir, S.; Arshad, A.W.; Khan, M.I.; Shamim, M.; Ayub, K.; Khan, M.A.; Iqbal, J. Therapeutic potential of graphitic carbon nitride as a drug delivery system for cisplatin (anticancer drug): A DFT approach. Biophys. Chem. 2020, 267, 106461. [Google Scholar] [CrossRef]
- Weiss, R.B.; Issell, B.F. The nitrosoureas: Carmustine (BCNU) and lomustine (CCNU). Cancer Treat. Rev. 1982, 9, 313–330. [Google Scholar] [CrossRef] [PubMed]
- Khanizadeh, A.; Ghaemi, A.; Pourmadadi, M.; Javadi, S.; Rahdar, A.; Yazdian, F.; Ghazy, E.; Pandey, S. Advancing cancer therapy: Unveiling the cutting-edge potential of carmustine nano carriers for targeted treatment. J. Drug Deliv. Sci. Technol. 2024, 99, 105943. [Google Scholar] [CrossRef]
- Ahmad, S.; Khan, I.; Pandit, J.; Emad, N.A.; Bano, S.; Dar, K.I.; Rizvi, M.M.A.; Ansari, M.D.; Aqil, M.; Sultana, Y. Brain targeted delivery of carmustine using chitosan coated nanoparticles via nasal route for glioblastoma treatment. Int. J. Biol. Macromol. 2022, 221, 435–445. [Google Scholar] [CrossRef]
- Chen, S.; Qiu, Q.; Wang, D.; She, D.; Yin, B.; Chai, M.; He, H.; Heo, D.N.; Wang, J. Long acting carmustine loaded natural extracellular matrix hydrogel for inhibition of glioblastoma recurrence after tumor resection. Front. Chem. Sci. Eng. 2022, 16, 536–545. [Google Scholar] [CrossRef]
- Bayat, M.; Taherpour, A.A.; Elahi, S.M.; Fellowes, T. Separation of anticancer medicines carmustine, lomustine, semustine and melphalan by PAMAM dendrimer: A theoretical study. J. Iran. Chem. Soc. 2018, 15, 1223–1234. [Google Scholar] [CrossRef]
- Kamel, M.; Mohammadi, M.; Mohammadifard, K.; Mahmood, E.A.; Heravi, M.R.P.; JM, A.H.; Hossaini, Z. Comprehensive theoretical prediction of the stability and electronic properties of hydroxyurea and carmustine drugs on pristine and Chitosan-functionalized graphitic carbon nitride in vacuum and aqueous environment. Vacuum 2023, 207, 111565. [Google Scholar] [CrossRef]
- Rani, V.; Venkatesan, J.; Prabhu, A. Carmustine-Loaded Liposomal Delivery Effectively Targets Malignant Glioma Cells and Seizes Endothelial Sprouting In vitro. J. Clust. Sci. 2024, 35, 1211–1221. [Google Scholar] [CrossRef]
- Mortazavifar, A.; Raissi, H.; Akbari, A. DFT and MD investigations on the functionalized boron nitride nanotube as an effective drug delivery carrier for Carmustine anticancer drug. J. Mol. Liq. 2019, 276, 577–587. [Google Scholar] [CrossRef]
- Solomon, J.; Jacobs, E.M.; Bateman, J.R.; Lukes, R.J.; Weiner, J.M.; Donohue, D.M. Chemotherapy of lymphoma with mechlorethamine and vinblastine. Arch. Intern. 1973, 131, 407–417. [Google Scholar] [CrossRef]
- Jacobs, E.M.; Peters, F.C.; Luce, J.K.; Zippin, C.; Wood, D.A. Mechlorethamine HCl and cyclophosphamide in the treatment of Hodgkin’s disease and the lymphomas. JAMA 1968, 203, 392–398. [Google Scholar] [CrossRef]
- Vonderheid, E.C.; Tan, E.T.; Kantor, A.F.; Shrager, L.; Micaily, B.; Van Scott, E.J. Long-term efficacy, curative potential, and carcinogenicity of topical mechlorethamine chemotherapy in cutaneous T cell lymphoma. J. Am. Acad. Dermatol. 1989, 20, 416–428. [Google Scholar] [CrossRef] [PubMed]
- Mekkey, S.M.; Al-dolaimy, F.; Hussein, U.A.-R.; Younis, S.M.D.; Kadhim, A.J.; Kareem, M.W.A.; Abed, N.K.; Asiri, M.; Alkhayyat, S.; Alsalamy, A.H. Investigation of Drug Delivery of Mechlorethamine (Anticancer Drug) by Si76, C76, Al38N38 Nanocages. Silicon 2024, 16, 585–592. [Google Scholar] [CrossRef]
- de Vries Schultink, A.; Suleiman, A.; Schellens, J.; Beijnen, J.; Huitema, A. Pharmacodynamic modeling of adverse effects of anti-cancer drug treatment. Eur. J. Clin. Pharmacol. 2016, 72, 645–653. [Google Scholar] [CrossRef]
- Zhong, Q.; Zee, K.; Rasmussen, K.; McKinley, B.J.; Linger, R.M.; Ray, S.D. Side effects of anti-cancer medications. In Side Effects of Drugs Annual; Elsevier: Amsterdam, The Netherlands, 2022; Volume 44, pp. 431–445. [Google Scholar]
- Torino, F.; Barnabei, A.; Paragliola, R.M.; Marchetti, P.; Salvatori, R.; Corsello, S.M. Endocrine side-effects of anti-cancer drugs: mAbs and pituitary dysfunction: Clinical evidence and pathogenic hypotheses. Eur. J. Endocrinol. 2013, 169, R153–R164. [Google Scholar] [CrossRef] [PubMed]
- Kashkooli, F.M.; Soltani, M.; Souri, M. Controlled anti-cancer drug release through advanced nano-drug delivery systems: Static and dynamic targeting strategies. J. Control. Release. 2020, 327, 316–349. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.-Y.; Zhang, A.-Q.; Cheng, S.-X.; Rong, L.; Zhang, X.-Z. Drug self-delivery systems for cancer therapy. Biomaterials 2017, 112, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Rashidi, N.; Davidson, M.; Apostolopoulos, V.; Kelley, M.R.; Nurgali, K. Targeted combination nano-drug delivery system to enhance anti-cancer efficacy and reduce side effects. Cancer Res. 2024, 84, 478. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Q.; Fan, G.; Chu, X. Theoretical study of boron nitride nanotubes as drug delivery vehicles of some anticancer drugs. Theor. Chem. Acc. 2018, 137, 104. [Google Scholar] [CrossRef]
- Li, Y.; Xiao, K.; Luo, J.; Lee, J.; Pan, S.; Lam, K.S. A novel size-tunable nanocarrier system for targeted anticancer drug delivery. J. Control. Release 2010, 144, 314–323. [Google Scholar] [CrossRef]
- Zhang, H.; Fan, T.; Chen, W.; Li, Y.; Wang, B. Recent advances of two-dimensional materials in smart drug delivery nano-systems. Bioact. Mater. 2020, 5, 1071–1086. [Google Scholar] [CrossRef]
- Molaei, M.J. Two-dimensional (2D) materials beyond graphene in cancer drug delivery, photothermal and photodynamic therapy, recent advances and challenges ahead: A review. J. Drug Deliv. Sci. Technol. 2021, 61, 101830. [Google Scholar] [CrossRef]
- Gangrade, A.; Mandal, B.B. Drug delivery of anticancer drugs from injectable 3D porous silk scaffold for prevention of gastric cancer growth and recurrence. ACS Biomater. Sci. Eng. 2020, 6, 6195–6206. [Google Scholar] [CrossRef]
- Shi, K.; Aviles-Espinosa, R.; Rendon-Morales, E.; Woodbine, L.; Maniruzzaman, M.; Nokhodchi, A. Novel 3D printed device with integrated macroscale magnetic field triggerable anti-cancer drug delivery system. Colloids Surf. B Biointerfaces 2020, 192, 111068. [Google Scholar] [CrossRef]
- Weng, Q.; Wang, B.; Wang, X.; Hanagata, N.; Li, X.; Liu, D.; Wang, X.; Jiang, X.; Bando, Y.; Golberg, D. Highly water-soluble, porous, and biocompatible boron nitrides for anticancer drug delivery. ACS Nano 2014, 8, 6123–6130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Yan, W.; Ji, Y. A novel manganese dioxide-based drug delivery strategy via in situ coating γ-polyglutamic acid/cisplatin for intelligent anticancer therapy. J. Mater. Chem. B 2023, 11, 667–674. [Google Scholar] [CrossRef]
- Chen, H.; Liu, T.; Su, Z.; Shang, L.; Wei, G. 2D transition metal dichalcogenide nanosheets for photo/thermo-based tumor imaging and therapy. Nanoscale Horiz. 2018, 3, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Chandrakala, V.; Aruna, V.; Angajala, G. Review on metal nanoparticles as nanocarriers: Current challenges and perspectives in drug delivery systems. Emergent Mater. 2022, 5, 1593–1615. [Google Scholar] [CrossRef]
- Wu, M.; Yang, J.; Ye, T.; Wang, B.; Tang, Y.; Ying, X. Efficient Drug Delivery of Ti3C2T x MXenes for Synergistic Treatment of Human Hypopharyngeal Squamous Cell Carcinoma. ACS Appl. Mater. Interfaces 2023, 15, 29939–29947. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Luo, X. Theoretical studies of MoS2 and phosphorene drug delivery for antituberculosis drugs. J. Phys. Chem. C 2020, 124, 8279–8287. [Google Scholar] [CrossRef]
- Wang, G.; Li, R.; Parseh, B.; Du, G. Prospects and challenges of anticancer agents’ delivery via chitosan-based drug carriers to combat breast cancer: A review. Carbohydr. Polym. 2021, 268, 118192. [Google Scholar] [CrossRef]
- Vuong, B.X.; Hajali, N.; Asadi, A.; Baqer, A.A.; Hachim, S.K.; Canli, G. Drug delivery assessment of an iron-doped fullerene cage towards thiotepa anticancer drug. Inorg. Chem. Commun. 2022, 141, 109558. [Google Scholar] [CrossRef]
- Gong, P.; Du, J.; Wang, D.; Cao, B.; Tian, M.; Wang, Y.; Sun, L.; Ji, S.; Liu, Z. Fluorinated graphene as an anticancer nanocarrier: An experimental and DFT study. J. Mater. Chem. B 2018, 6, 2769–2777. [Google Scholar] [CrossRef]
- Guven, A.; Villares, G.J.; Hilsenbeck, S.G.; Lewis, A.; Landua, J.D.; Dobrolecki, L.E.; Wilson, L.J.; Lewis, M.T. Carbon nanotube capsules enhance the in vivo efficacy of cisplatin. Acta Biomater. 2017, 58, 466–478. [Google Scholar] [CrossRef]
- Guo, X.-L.; Kang, X.-X.; Wang, Y.-Q.; Zhang, X.-J.; Li, C.-J.; Liu, Y.; Du, L.-B. Co-delivery of cisplatin and doxorubicin by covalently conjugating with polyamidoamine dendrimer for enhanced synergistic cancer therapy. Acta Biomater. 2019, 84, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Farooq, M.A.; Aquib, M.; Farooq, A.; Haleem Khan, D.; Joelle Maviah, M.B.; Sied Filli, M.; Kesse, S.; Boakye-Yiadom, K.O.; Mavlyanova, R.; Parveen, A. Recent progress in nanotechnology-based novel drug delivery systems in designing of cisplatin for cancer therapy: An overview. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1674–1692. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, S.; Wu, Y.; Chen, F. Emerging Roles of Graphitic Carbon Nitride-based Materials in Biomedical Applications. ACS Biomater. Sci. Eng. 2024, 10, 4645–4661. [Google Scholar] [CrossRef] [PubMed]
- Ning, X.-F.; Zhu, Y.-Q.; Sun, H.; Yang, Y.; Liu, M.-X. The Latest Applications of Carbon-Nitride-Based Materials for Combination Treatment of Cancer. ACS Appl. Mater. Interfaces 2024, 16, 64410–64423. [Google Scholar] [CrossRef]
- Zaboli, A.; Raissi, H.; Farzad, F.; Hashemzadeh, H. Assessment of adsorption behavior of 5-fluorouracil and pyrazinamide on carbon nitride and folic acid-conjugated carbon nitride nanosheets for targeting drug delivery. J. Mol. Liq. 2020, 301, 112435. [Google Scholar] [CrossRef]
- Ahsan, F.; Yar, M.; Gulzar, A.; Ayub, K. Therapeutic potential of C2N as targeted drug delivery system for fluorouracil and nitrosourea to treat cancer: A theoretical study. J. Nanostruct. Chem. 2023, 13, 89–102. [Google Scholar] [CrossRef]
- Pourmadadi, M.; Rahmani, E.; Eshaghi, M.M.; Shamsabadipour, A.; Ghotekar, S.; Rahdar, A.; Ferreira, L.F.R. Graphitic carbon nitride (g-C3N4) synthesis methods, surface functionalization, and drug delivery applications: A review. J. Drug Deliv. Sci. Technol. 2023, 79, 104001. [Google Scholar] [CrossRef]
- Wang, X.; Liu, S.; Wang, J.; Liu, Y.; Guan, S.; Zhang, T. Spherical g-C3N4@ PDA nanocarrier for synergistic chemo-photothermal tumor therapy. J. Photochem. Photobiol. A Chem. 2024, 454, 115736. [Google Scholar] [CrossRef]
- Asghar, S.; Roudgar-Amoli, M.; Alizadeh, A.; Shariatinia, Z. Water purification through adsorption of organic pollutant onto novel and effective phosphorus-containing g-C3N4/FeMo0. 5O3 nanocomposites. Water Air Soil Pollut. 2023, 234, 43. [Google Scholar] [CrossRef]
- Singh, J.A.; Overbury, S.H.; Dudney, N.J.; Li, M.; Veith, G.M. Gold nanoparticles supported on carbon nitride: Influence of surface hydroxyls on low temperature carbon monoxide oxidation. ACS Catal. 2012, 2, 1138–1146. [Google Scholar] [CrossRef]
- Ou, H.; Ning, S.; Zhu, P.; Chen, S.; Han, A.; Kang, Q.; Hu, Z.; Ye, J.; Wang, D.; Li, Y. Carbon nitride photocatalysts with integrated oxidation and reduction atomic active centers for improved CO2 conversion. Angew. Chem. Int. Ed. 2022, 61, e202206579. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Wang, A.; Cheng, R.; Tian, X.; Jing, S.; Tsiakaras, P. Efficient photocatalytic H2O2 production ability of a novel graphitic carbon nitride/carbon composites under visible light. Small 2023, 19, 2303813. [Google Scholar] [CrossRef]
- Peng, G.; Wu, J.; Wang, M.; Niklas, J.; Zhou, H.; Liu, C. Nitrogen-defective polymeric carbon nitride nanolayer enabled efficient electrocatalytic nitrogen reduction with high faradaic efficiency. Nano Lett. 2020, 20, 2879–2885. [Google Scholar] [CrossRef]
- Tan, L.; Nie, C.; Ao, Z.; Sun, H.; An, T.; Wang, S. Novel two-dimensional crystalline carbon nitrides beyond gC 3 N 4: Structure and applications. J. Meter. Chem. A 2021, 9, 17–33. [Google Scholar] [CrossRef]
- Shamim, M.; Perveen, M.; Nazir, S.; Hussnain, M.; Mehmood, R.; Khan, M.I.; Iqbal, J. DFT study of therapeutic potential of graphitic carbon nitride (g-C3N4) as a new drug delivery system for carboplatin to treat cancer. J. Mol. Liq. 2021, 331, 115607. [Google Scholar] [CrossRef]
- Perveen, M.; Aslam, F.; Nazir, S.; Khan, M.I.; Zahra, G.; Iqbal, J. DFT study of therapeutic potential of graphitic carbon nitride as a carrier for controlled release of melphalan: An anticancer drug. J. Mol. Model. 2022, 28, 359. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, X.; He, H.; Dai, L.; Hu, J.; Si, C. Graphitic carbon nitride enters the scene: A promising versatile tool for biomedical applications. Langmuir 2024, 40, 15389–15406. [Google Scholar] [CrossRef]
- Bian, C.; Wang, Y.; Yi, Y.; Shao, S.; Sun, P.; Xiao, Y.; Wang, W.; Dong, X. Enhanced photocatalytic activity of S-doped graphitic carbon nitride hollow microspheres: Synergistic effect, high-concentration antibiotic elimination and antibacterial behavior. J. Colloid Interface Sci. 2023, 643, 256–266. [Google Scholar] [CrossRef]
- Dong, J.; Zhao, Y.; Wang, K.; Chen, H.; Liu, L.; Sun, B.; Yang, M.; Sun, L.; Wang, Y.; Yu, X. Fabrication of graphitic carbon nitride quantum dots and their application for simultaneous fluorescence imaging and pH-responsive drug release. ChemistrySelect 2018, 3, 12696–12703. [Google Scholar] [CrossRef]
- Asif, K.; Perveen, M.; Khera, R.A.; Nazir, S.; Ayub, A.R.; Asif, T.; Shabbir, M.; Iqbal, J. Computational and theoretical study of graphitic carbon nitride (g-C3N4) as a drug delivery carrier for lonidamine drug to treat cancer. Comput. Theor. Chem. 2021, 1206, 113459. [Google Scholar] [CrossRef]
- Li, X. Aluminium nitride as an efficient catalyst in the synthesis of some chromeno [4, 3-b] chromenes and potential nanocarrier for delivery of flutamide. Chem. Pap. 2024, 78, 1157–1166. [Google Scholar] [CrossRef]
- Chen, W.; Liu, J.; Wang, Y.; Jiang, C.; Yu, B.; Sun, Z.; Lu, L. A C5N2 nanoparticle based direct nucleus delivery platform for synergistic cancer therapy. Angew. Chem. 2019, 131, 6356–6360. [Google Scholar] [CrossRef]
- Karimi, M.; Solati, N.; Amiri, M.; Mirshekari, H.; Mohamed, E.; Taheri, M.; Hashemkhani, M.; Saeidi, A.; Estiar, M.A.; Kiani, P. Carbon nanotubes part I: Preparation of a novel and versatile drug-delivery vehicle. Expert Opin. Drug Deliv. 2015, 12, 1071–1087. [Google Scholar] [CrossRef]
- Watermann, A.; Brieger, J. Mesoporous silica nanoparticles as drug delivery vehicles in cancer. Nanomaterials 2017, 7, 189. [Google Scholar] [CrossRef]
- Sun, C.-Y.; Qin, C.; Wang, X.-L.; Su, Z.-M. Metal-organic frameworks as potential drug delivery systems. Expert Opin. Drug Deliv. 2013, 10, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Cui, L.; Losic, D. Graphene and graphene oxide as new nanocarriers for drug delivery applications. Acta Biomater. 2013, 9, 9243–9257. [Google Scholar] [CrossRef] [PubMed]
- Garriga, R.; Herrero-Continente, T.; Palos, M.; Cebolla, V.L.; Osada, J.; Muñoz, E.; Rodríguez-Yoldi, M.J. Toxicity of carbon nanomaterials and their potential application as drug delivery systems: In vitro studies in Caco-2 and MCF-7 cell lines. Nanomater. 2020, 10, 1617. [Google Scholar] [CrossRef]
- Sayers, E.W.; Beck, J.; Bolton, E.E.; Brister, J.R.; Chan, J.; Connor, R.; Feldgarden, M.; Fine, A.M.; Funk, K.; Hoffman, J. Database resources of the National Center for Biotechnology Information in 2025. Nucleic Acids Res. 2024, 53, D20. [Google Scholar] [CrossRef] [PubMed]
- Qi, S.; Jin, Z.; Hou, Y.; Li, C.; Wang, X.; Zhou, Q. Converting an Almost Noncytotoxic Ru (II) Complex with Photolabile Ligands into a Highly Efficient PACT Agent. Part. Part. Syst. Charact. 2021, 38, 2100193. [Google Scholar] [CrossRef]
- Chen, W.; Sun, Z.; Jiang, C.; Sun, W.; Yu, B.; Wang, W.; Lu, L. An All-in-One Organic Semiconductor for Targeted Photoxidation Catalysis in Hypoxic Tumor. Angew. Chem. 2021, 133, 16777–16784. [Google Scholar] [CrossRef]
- Rosenkranz, A.; Ulasov, A.; Slastnikova, T.; Khramtsov, Y.V.; Sobolev, A. Use of intracellular transport processes for targeted drug delivery into a specified cellular compartment. Biochemistry (Moscow) 2014, 79, 928–946. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Liu, J.; Han, G.; Zhang, W.; Long, Y.; Deng, Y.; Wang, B.; Weng, Q. Design of red-emitting 1D zinc coordination polymer for targeted drug delivery to nucleus. Chem. Eng. J. 2023, 470, 144177. [Google Scholar] [CrossRef]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView, Version. 6.0; Semichem Inc.: Shawnee Mission, KS, USA, 2016. [Google Scholar]
- Yaghoubi, A.; Ramazani, A. Using Gaussian and GaussView software for effective teaching of chemistry by drawing molecules. Res. Chem. Educ. 2024, 6, 69–90. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian, Version 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Marsman, M.; Paier, J.; Stroppa, A.; Kresse, G. Hybrid functionals applied to extended systems. J. Phys. Condens. Matter 2008, 20, 064201. [Google Scholar] [CrossRef]
- Adamo, C.; Cossi, M.; Scalmani, G.; Barone, V. Accurate static polarizabilities by density functional theory: Assessment of the PBE0 model. Chem. Phys. Lett. 1999, 307, 265–271. [Google Scholar] [CrossRef]
- Melissen, S.; Le Bahers, T.; Steinmann, S.N.; Sautet, P. Relationship between Carbon Nitride Structure and Exciton Binding Energies: A DFT Perspective. J. Physs. Chem. C 2015, 119, 25188–25196. [Google Scholar] [CrossRef]
- Tuma, C.; Boese, A.D.; Handy, N.C. Predicting the binding energies of H-bonded complexes: A comparative DFT study. Phys. Chem. Chem. Phys. 1999, 1, 3939–3947. [Google Scholar] [CrossRef]
- Lin, T.-J.; Chiu, C.-c. Influence of nonmetal dopants on charge separation of graphitic carbon nitride by time-dependent density functional theory. Phys. Chem. Chem. Phys. 2020, 22, 647–657. [Google Scholar] [CrossRef]
- Veccham, S.P.; Head-Gordon, M. Density functionals for hydrogen storage: Defining the H2Bind275 test set with ab initio benchmarks and assessment of 55 functionals. J. Chem. Theory Comput. 2020, 16, 4963–4982. [Google Scholar] [CrossRef]
- Galimov, E.R.; Kostjukov, V.V. Computational analysis of photoisomerization of unsubstituted spirooxazine by TD-DFT: Solvent effect and functional choice. Theor. Chem. Acc. 2023, 143, 2. [Google Scholar] [CrossRef]
- Xu, H.; Tu, X.; Fan, G.; Wang, Q.; Wang, X.; Chu, X. Adsorption properties study of boron nitride fullerene for the application as smart drug delivery agent of anti-cancer drug hydroxyurea by density functional theory. J. Mol. Liq. 2020, 318, 114315. [Google Scholar] [CrossRef]
- Barman, R.; Bej, R.; Dey, P.; Ghosh, S. Cisplatin-conjugated polyurethane capsule for dual drug delivery to a cancer cell. ACS Appl. Mater. Interfaces 2023, 15, 25193–25200. [Google Scholar] [CrossRef]
- Chu, Y.-C.; Lin, T.-J.; Lin, Y.-R.; Chiu, W.-L.; Nguyen, B.-S.; Hu, C. Influence of P,S,O-Doping on g-C3N4 for hydrogel formation and photocatalysis: An experimental and theoretical study. Carbon 2020, 169, 338–348. [Google Scholar] [CrossRef]
- Ibarra-Rodríguez, M.; Sánchez, M. Adsorption of metformin on graphitic carbon nitride functionalized with metals of group 1–3 (Li, Na, K, Be, Mg, Ca, B, Al, and Ga), DFT calculations. Comput. Theor. Chem. 2022, 1207, 113532. [Google Scholar] [CrossRef]
- Ibarra-Rodríguez, M.; Sánchez, M. Graphitic carbon nitride functionalized with four boron atoms for adsorption and separation of CO2/CH4: DFT calculations. Adsorption 2020, 26, 597–605. [Google Scholar] [CrossRef]
- Ibarra-Rodríguez, M.; Sánchez, M. Adsorption of H2, N2, CO, H2S, NH3, SO2 and CH4 on Li-functionalized graphitic carbon nitride investigated by density functional theory. Bull. Mater. Sci. 2020, 43, 144. [Google Scholar] [CrossRef]
- Gorai, D.K.; Kundu, T.K. Platinum-silicon doped graphitic carbon nitride: A first principle calculation. Phys. B Condens. Matter 2022, 627, 413547. [Google Scholar] [CrossRef]
- Pauly, M.; White, E.; Deegbey, M.; Fosu, E.A.; Keller, L.; McGuigan, S.; Dianat, G.; Gabilondo, E.; Wong, J.C.; Murphey, C.G.E.; et al. Coordination of copper within a crystalline carbon nitride and its catalytic reduction of CO2. Dalton Trans. 2024, 53, 6779–6790. [Google Scholar] [CrossRef]
- Ali, B.; Siddique, S.A.; Ahmed Siddique, M.B.; Ullah, S.; Ali, M.A.; Rauf, A.; Kamran, M.A.; Arshad, M. Insight on the structural, electronic and optical properties of Zn, Ga-doped/dual-doped graphitic carbon nitride for visible-light applications. J. Mol. Graph. Model. 2023, 125, 108603. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Liu, W.-W.; Niu, S.-T.; Xu, Z.-Q.; Zou, R.; Cui, C.-Y.; Lei, Y.-X.; Zhang, X.-B.; Ran, F. Highly-dispersed nickel on 2D graphitic carbon nitrides (g-C3N4) for facilitating reaction kinetics of lithium-sulfur batteries. Appl. Surf. Sci. 2023, 609, 155327. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Legault, C. CYLview, Version 1.0 b; Université de Sherbrooke: Sherbrooke, QC, Canada, 2009. [Google Scholar]
- O’Boyle, N.M.; Tenderholt, A.L.; Langner, K.M. cclib: A library for package-independent computational chemistry algorithms. J Comput. Chem. 2008, 29, 839–845. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.-P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Vargas, R.; Garza, J.; Martínez, A.; Ibarra, I.A. Computational tools to study non-covalent interactions and confinement effects in chemical systems. Chem. Commun. 2024, 60, 3008–3018. [Google Scholar] [CrossRef]
- Hajji, M.; Abad, N.; Habib, M.A.; Elmgirhi, S.M.H.; Guerfel, T. Computational chemistry methods for modelling non-covalent interactions and chemical reactivity—An overview. J. Indian Chem. Soc. 2021, 98, 100208. [Google Scholar] [CrossRef]
- Popelier, P.L.A. The QTAIM Perspective of Chemical Bonding. In The Chemical Bond; John Wiley & Sons: Hoboken, NJ, USA, 2014; pp. 271–308. [Google Scholar]
- Kumar, P.S.V.; Raghavendra, V.; Subramanian, V. Bader’s Theory of Atoms in Molecules (AIM) and its Applications to Chemical Bonding. J. Chem. Sci. 2016, 128, 1527–1536. [Google Scholar] [CrossRef]
- Foroutan-Nejad, C.; Shahbazian, S.; Marek, R. Toward a Consistent Interpretation of the QTAIM: Tortuous Link between Chemical Bonds, Interactions, and Bond/Line Paths. Chem. A Eur. J. 2014, 20, 10140–10152. [Google Scholar] [CrossRef] [PubMed]
- Wick, C.R.; Clark, T. On bond-critical points in QTAIM and weak interactions. J. Mol. Model. 2018, 24, 142. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F. Comment on: Revisiting the variational nature of the quantum theory of atoms in molecules. Chem. Phys. Lett. 2006, 426, 226–228. [Google Scholar] [CrossRef]
- Phillips, J.C. Generalized Koopmans’ Theorem. Phys. Rev. 1961, 123, 420–424. [Google Scholar] [CrossRef]
- Hackett, J.C. Chemical Reactivity Theory: A Density Functional View. J. Am. Chem. Soc. 2010, 132, 7558. [Google Scholar] [CrossRef]
- Parr, R.G.; Szentpály, L.v.; Liu, S. Electrophilicity index. J. Am. Chem. Soc. 1999, 121, 1922–1924. [Google Scholar] [CrossRef]
- Bendjeddou, A.; Abbaz, T.; Gouasmia, A.; Villemin, D. Quantum chemical studies on molecular structure and reactivity descriptors of some P-nitrophenyl tetrathiafulvalenes by density functional theory (DFT). Acta Chim. Pharm. Indica 2016, 6, 32–44. [Google Scholar]
- de Miranda, D.B.; Quintal, S.; Ferreira, G.B. Electronic analysis of n-propyl xanthate complexes with group 12 metals: A theoretical–experimental study. J. Mol. Model. 2024, 30, 163. [Google Scholar] [CrossRef]
- Shilpa, D.; Sadasivam, K.; Thirumoorthy, M. Topology analysis of six phytochemicals through ELF and LOL basins–A DFT study. Indian J. Chem. (IJC) 2023, 62, 1171–1177. [Google Scholar]
- Jumabaev, A.; Koyambo-Konzapa, S.-J.; Hushvaktov, H.; Absanov, A.; Khudaykulov, B.; Holikulov, U.; Ernazarov, Z.; Issaoui, N.; Al-Dossary, O.M.; Nsangou, M. Intermolecular interactions in water and ethanol solution of ethyl acetate: Raman, DFT, MEP, FMO, AIM, NCI-RDG, ELF, and LOL analyses. J. Mol. Model. 2024, 30, 349. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, F. Natural bond orbital analysis: A critical overview of relationships to alternative bonding perspectives. J. Comput. Chem. 2012, 33, 2363–2379. [Google Scholar] [CrossRef] [PubMed]
- Koch, D.; Pavanello, M.; Shao, X.; Ihara, M.; Ayers, P.W.; Matta, C.F.; Jenkins, S.; Manzhos, S. The Analysis of Electron Densities: From Basics to Emergent Applications. Chem. Rev. 2024, 124, 12661–12737. [Google Scholar] [CrossRef]
- Leppert, J.; Urbinati, C.R.; Häfner, S.; Ohlenschläger, O.; Swanson, M.S.; Görlach, M.; Ramachandran, R. Identification of NH...N hydrogen bonds by magic angle spinning solid state NMR in a double-stranded RNA associated with myotonic dystrophy. Nucleic Acids Res. 2004, 32, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Pakiari, A.; Eskandari, K. The chemical nature of very strong hydrogen bonds in some categories of compounds. J. Mol. Struct. THEOCHEM 2006, 759, 51–60. [Google Scholar] [CrossRef]
- Grabowski, S.J. Hydrogen Bond—Definitions, Criteria of Existence and Various Types. In Understanding Hydrogen Bonds; The Royal Society of Chemistry: London, UK, 2020. [Google Scholar]
- Schiemenz, G.P. The sum of van der Waals radii–A pitfall in the search for bonding. Z. Naturforschung B 2007, 62, 235–243. [Google Scholar] [CrossRef]
- Kaviani, S.; Shahab, S.; Sheikhi, M.; Potkin, V.; Zhou, H. A DFT study of Se-decorated B12N12 nanocluster as a possible drug delivery system for ciclopirox. Comput. Theor. Chem. 2021, 1201, 113246. [Google Scholar] [CrossRef]
- Nayini, M.M.R.; Sayadian, H.; Razavipour, N.; Rezazade, M. Chemical-sensing of Amphetamine drug by inorganic AlN nano-cage: A DFT/TDDFT study. Inorg. Chem. Commun. 2020, 121, 108237. [Google Scholar] [CrossRef]
- Wang, L.; Liu, X.; Ji, L.; Luo, Q.; Duan, Y.; An, J.; Chen, X.; Zhang, Y.; Ren, J.; Wang, D. Resin with short-range π-π stacking aggregates for an efficient photocatalyst. Chem. Eng. J. 2022, 433, 134502. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Kalhor, S.; Fattahi, A. Combinatorial MD/QM studies to develop novel ionic liquid-based anticancer drug delivery systems with aminium derived from carbohydrates as cationic components. Sci. Rep. 2024, 14, 28980. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef] [PubMed]
- Oğuz, I.C.; Vassetti, D.; Labat, F. Assessing the performances of different continuum solvation models for the calculation of hydration energies of molecules, polymers and surfaces: A comparison between the SMD, VASPsol and FDPB models. Theor. Chem. Acc. 2021, 140, 99. [Google Scholar] [CrossRef]
- Gwee, E.S.; Seeger, Z.L.; Appadoo, D.R.; Wood, B.R.; Izgorodina, E.I. Influence of DFT Functionals and Solvation Models on the Prediction of Far-Infrared Spectra of Pt-Based Anticancer Drugs: Why Do Different Complexes Require Different Levels of Theory? ACS Omega 2019, 4, 5254–5269. [Google Scholar] [CrossRef]
- Zafar, A.; Reynisson, J. Hydration free energy as a molecular descriptor in drug design: A feasibility study. Mol. Inform. 2016, 35, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Bibi, S.; Ur-Rehman, S.; Khalid, L.; Bhatti, I.A.; Bhatti, H.N.; Iqbal, J.; Bai, F.Q.; Zhang, H.-X. Investigation of the adsorption properties of gemcitabine anticancer drug with metal-doped boron nitride fullerenes as a drug-delivery carrier: A DFT study. RSC Adv. 2022, 12, 2873–2887. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Gong, Y.; Zeng, Z.; Chen, S.; Ye, J.; Wang, Z.; Dionysiou, D.D. Dissolved organic matter promotes photocatalytic degradation of refractory organic pollutants in water by forming hydrogen bonding with photocatalyst. Water Res. 2023, 242, 120297. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Chen, Y.; Lu, Z.; Ma, L.; Tao, Q.; Li, Z.; Kong, L.; Liu, L.; Yang, X.; Ding, S. Monolithic three-dimensional tier-by-tier integration via van der Waals lamination. Nature 2024, 630, 340–345. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, K.; Davis, C.; Sherlock, S.; Cao, Q.; Chen, X.; Dai, H. Drug delivery with carbon nanotubes for in vivo cancer treatment. Cancer Res. 2008, 68, 6652–6660. [Google Scholar] [CrossRef]
- Saikia, N. Functionalized carbon nanomaterials in drug delivery: Emergent perspectives from application. In Novel Nanomaterials—Synthesis and Applications; IntechOpen: London, UK, 2018; pp. 231–255. [Google Scholar]
- Lee, Y.; Kwon, D.-G.; Kim, G.; Kwon, Y.-K. Ab initio study of aspirin adsorption on single-walled carbon and carbon nitride nanotubes. Phys. Chem. Chem. Phys. 2017, 19, 8076–8081. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug@C5N2 Complex | C5N2−Drug | Bond Lengths (Å) | Interaction Energy (kcal mol−1) |
|---|---|---|---|
| cisplatin@C5N2 | C2−H1 N4−H3 C6−Pt5 C8−Cl7 C10−Cl9 | 2.36 1.92 3.39 3.45 3.40 | −27.60 |
| carmustine@C5N2 | C13−H1 C14−H2 C12−O3 H11−O4 H10−O4 C9−H5 C8−Cl6 N7−Cl6 | 2.91 2.60 3.15 2.37 2.74 2.73 3.47 3.44 | −19.69 |
| mechlorethamine@C5N2 | C6−Cl1 C7−H2 C8−H3 N9−H4 N10−H5 | 3.35 2.69 2.99 2.59 2.24 | −17.73 |
| Drugs@C5N2 | C5N2−Drug | ρ (a.u.) | ∇2ρ (a.u.) | G (a.u.) | V (a.u.) | H (a.u.) | −V/G |
|---|---|---|---|---|---|---|---|
| cisplatin@C5N2 | C2−H1 N4−H3 C6−Pt5 C8−Cl7 C10−Cl9 | 0.0137 0.0329 0.0123 0.0075 0.0082 | 0.037 0.087 0.034 0.022 0.025 | 0.008 0.021 0.007 0.004 0.005 | −0.008 −0.021 −0.007 −0.004 −0.004 | 0.0004 0.0002 0.0007 0.0007 0.0008 | 0.95 0.98 0.90 0.83 0.85 |
| carmustine@C5N2 | C13−H1 C14−H2 C12−O3 H11−O4 H10−O4 C9−H5 C8−Cl6 N7−Cl6 | 0.0058 0.0030 0.0078 0.0100 0.0050 0.0070 0.0063 0.0064 | 0.018 0.016 0.026 0.041 0.025 0.023 0.022 0.021 | 0.003 0.004 0.005 0.009 0.004 0.004 0.004 0.004 | −0.002 −0.003 −0.004 −0.008 −0.003 −0.003 −0.003 −0.004 | 0.0008 0.0006 0.0008 0.0009 0.0012 0.0009 0.0008 0.0005 | 0.75 0.79 0.85 0.89 0.73 0.79 0.80 0.87 |
| mechlorethamine@C5N2 | C6−Cl1 C7−H2 C8−H3 N9−H4 N10−H5 | 0.0081 0.0082 0.0048 0.0091 0.0170 | 0.026 0.022 0.015 0.028 0.045 | 0.005 0.005 0.003 0.006 0.011 | −0.004 −0.004 −0.002 −0.005 −0.011 | 0.0009 0.0005 0.0007 0.0005 0.0001 | 0.84 0.90 0.77 0.90 0.01 |
| Complexes | EHOMO (eV) | ELUMO (eV) | Egap (eV) | μ (eV) | ω (eV) | η (eV) | S (eV) | NBO (e−) |
|---|---|---|---|---|---|---|---|---|
| C5N2 | −3.42 | −2.82 | 0.60 | −3.12 | 16.23 | 0.30 | 1.67 | − |
| cisplatin@C5N2 | −3.41 | −2.80 | 0.57 | −3.09 | 17.68 | 0.27 | 1.65 | −0.039 |
| carmustine@C5N2 | −3.41 | −2.80 | 0.57 | −3.09 | 17.68 | 0.27 | 1.65 | −0.031 |
| mechlorethamine@C5N2 | −3.39 | −2.81 | 0.58 | −3.10 | 16.51 | 0.29 | 1.72 | 0.479 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehdi Zaidi, S.H.; Ajmal, M.; Hashmi, M.A.; Lakhani, A. Targeted Drug Delivery of Anticancer Agents Using C5N2 Substrate: Insights from Density Functional Theory. Chemistry 2025, 7, 98. https://doi.org/10.3390/chemistry7030098
Mehdi Zaidi SH, Ajmal M, Hashmi MA, Lakhani A. Targeted Drug Delivery of Anticancer Agents Using C5N2 Substrate: Insights from Density Functional Theory. Chemistry. 2025; 7(3):98. https://doi.org/10.3390/chemistry7030098
Chicago/Turabian StyleMehdi Zaidi, Syeda Huda, Muhammad Ajmal, Muhammad Ali Hashmi, and Ahmed Lakhani. 2025. "Targeted Drug Delivery of Anticancer Agents Using C5N2 Substrate: Insights from Density Functional Theory" Chemistry 7, no. 3: 98. https://doi.org/10.3390/chemistry7030098
APA StyleMehdi Zaidi, S. H., Ajmal, M., Hashmi, M. A., & Lakhani, A. (2025). Targeted Drug Delivery of Anticancer Agents Using C5N2 Substrate: Insights from Density Functional Theory. Chemistry, 7(3), 98. https://doi.org/10.3390/chemistry7030098