Abstract
3,3-Dialkoxy-2-oxindoles are prevalent in natural products and exhibit unique biological activities. Among them, acyclic alkoxy analogues show instability in acidic conditions, making access to acyclic isatin ketals highly challenging. Conventional methods for the synthesis of 3,3-dialkoxy-2-oxindoles usually require strongly acidic and harsh reaction conditions, resulting in a low overall efficiency. Herein, we report on an acid- and metal-free protocol for the synthesis of 3,3-dialkoxy-2-oxindoles from isatins through an iodine-catalyzed ketalization. This photochemical protocol does not require the use of any specific reagents such as metal catalysts. Furthermore, the total synthesis of an unprecedented 2-oxindole alkaloid bearing 3,3-dimethoxy moiety is achieved.
1. Introduction
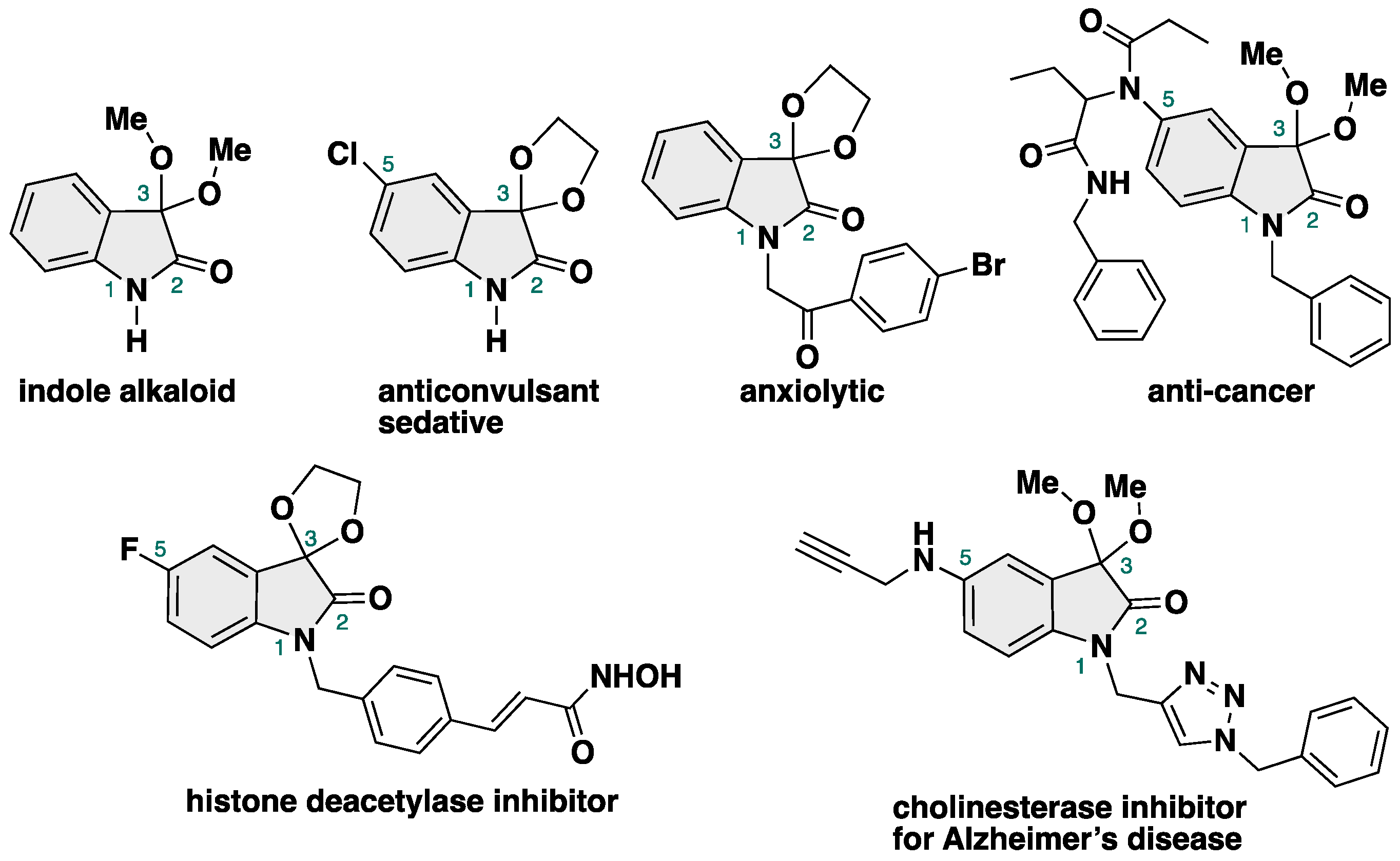
3,3-Dimethoxy-2-oxindole is an 2-oxindole-type alkaloid isolated from a native orchid of Taiwan Phaius mishmensis in 2016 by Chuang and co-workers (Figure 1) [1].
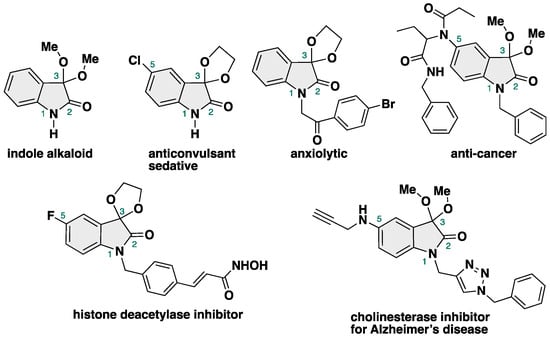
Figure 1.
Indole alkaloids and biologically active compounds bearing 3,3-dialkoxy-2-oxindole skeletons.
It has an unprecedented 3,3-dialkoxy moiety at the C3 position of isatin, which is known as a ketal in organic chemistry, a protective group of the ketone moiety [2]. Furthermore, 3,3-dialkoxy-2-oxindoles possess anticonvulsant [3], sedative/hypnotic [4], anxiolytic [5], anti-cancer [6], histone-diacetylase-inhibitory [7], and cholinesterase-inhibitory activities, meaning that they have the potential for use as anti-Alzheimer’s disease therapeutics [8]. Due to their potent biological activities [9], 3,3-dialkoxy-2-oxindoles are attractive targets for both organic [10,11,12,13,14,15] and medicinal [16,17,18,19] chemistry. However, in contrast with the rich chemistry of ketalization and the acetalization of carbonyl compounds [20,21,22,23,24,25,26,27], methods for the synthesis of 3,3-dialkoxy-2-oxindoles are less developed due to their intrinsic obstacles in the production of NH-free 3,3-dialkoxy-2-oxindoles, such as the equilibrium between ketones and ketals and the acidic properties of NH-isatin, which result in the partial decomposition of the products by isatin itself.
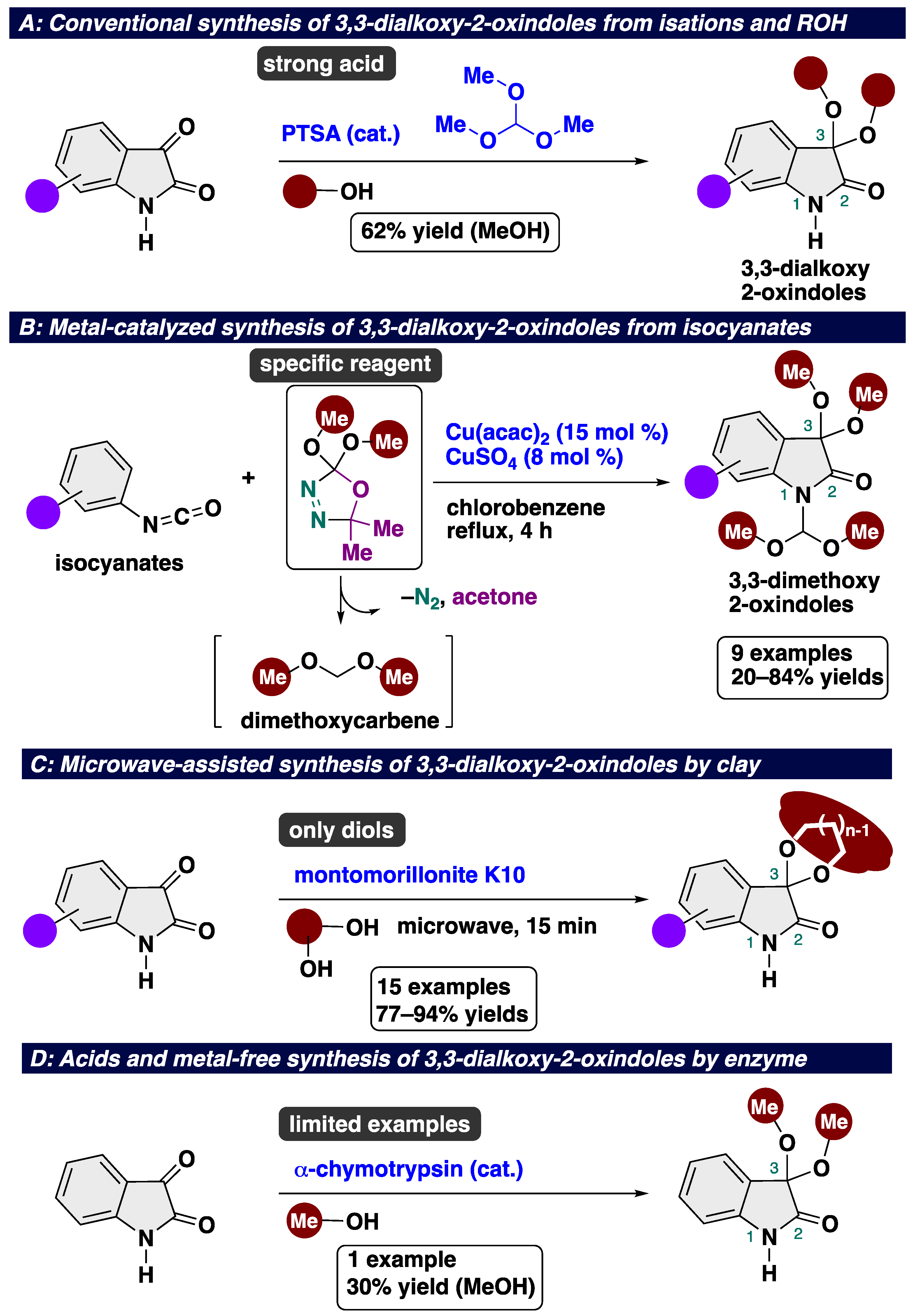
Conventional methods for accessing 3,3-dialkoxy-2-oxindoles include the ketalization of isatins with alcohol in the presence of strong acids and/or orthoesters (Scheme 1A) [28]. Almost all previous reports on the synthesis of 3,3-dialkoxy-2-oxindoles adopted the PTSA-catalyzed protocol, the relevance of which is limited due to its strong acidity. In the case of the reaction with methanol, the yield of the desired product was 60%. In 2013, Brouet’s group reported copper-catalyzed insertion and cyclization using a dimethoxycarbene equivalent and isocyanates to afford decorated 3,3-dimethoxy-2-oxindoles (Scheme 1B) [29]. With this approach, nine different 3,3-dimethoxyisatins were obtained in 20–84% yields. However, the reactions resulted in limited products bearing a methoxy group, probably due to the troublesome accessibility of carbene precursors. In 2007, the acid- and metal-free ketalization of isatins with diols was developed by Ribeiro’s group (Scheme 1C) [30]. The key to success is the use of a combination of clay (Montrmorlillonite K10) and microwave irradiation to afford 3,3-dimethoxy-2-oxindoles. Montrmorlillonite K10 is known as an acidic clay bearing a releasable proton, which can act as a Brønsted acid [31]. This protocol requires using microwave irradiation equipment. Considering the low user-friendliness of the reported protocols, in 2020, Xie and co-workers developed the acid- and metal-free acetalization and ketalization of aldehydes and ketones using α-chymotrypsin as an enzyme (Scheme 1D) [32]. In this work, the ketalization of isatin was accomplished to afford 3,3-dimethoxy-2-oxindole in a 30% yield. In general, this type of enzyme-mediated reaction is intrinsically applicable to limited substrates. In previous reports, a transition-metal catalyst or harsh reaction conditions (using strong acids or under microwave irradiation) are indispensable to achieve acceptable product yields. Therefore, the development of a novel, mild, and metal- and acid-free protocol is highly desirable. Considering the low accessibility of the starting materials, the use of a combination of isatins and alcohol is an important component in the production of 3,3-dialkoxy-2-oxindoles. However, to the best of our knowledge, the eco- and user-friendly ketalization of isatins has not yet been established, probably due to a lack of green and efficient catalysts.
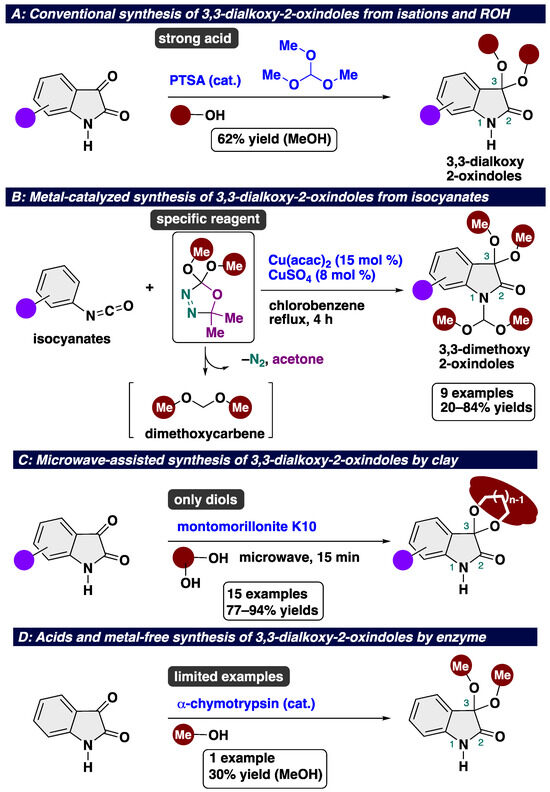
Scheme 1.
State-of-the-art in synthesis of 3,3-dialkoxy-2-oxindoles: (A) conventional synthesis of 3,3-dialkoxy-2-oxindoles using strong acid and ROH; (B) metal-catalyzed synthesis of 3,3-dimethoxy-2-oxindoles using a dimethoxycarbene equivalent; (C) microwave-assisted synthesis of isatin ketals in the presence of Montrmorlillonite K10; (D) acids and metal-free synthesis of 3,3-dimethoxy-2-oxindole.
Molecular iodine (I2) has attracted much attention from organic chemists due to its green, easily available, inexpensive, non-toxic, and highly reactive properties and user-friendly nature [33,34,35,36,37,38,39,40,41,42]. It is worth noting that the iodine-catalyzed reaction system does not require transition metals, making it attractive in sustainable green chemistry. Molecular iodine is tolerant against H2O, with easier-to-handle Lewis acid catalysts, while Lewis acids are usually deactivated by H2O. Thus, it is considered to be an efficient altanative to transition metal or Lewis or Brønsted acid catalysts. However, to the best of our knowledge, there are no reports of the ketalization of isatins using a catalytic amout of molecular iodine.
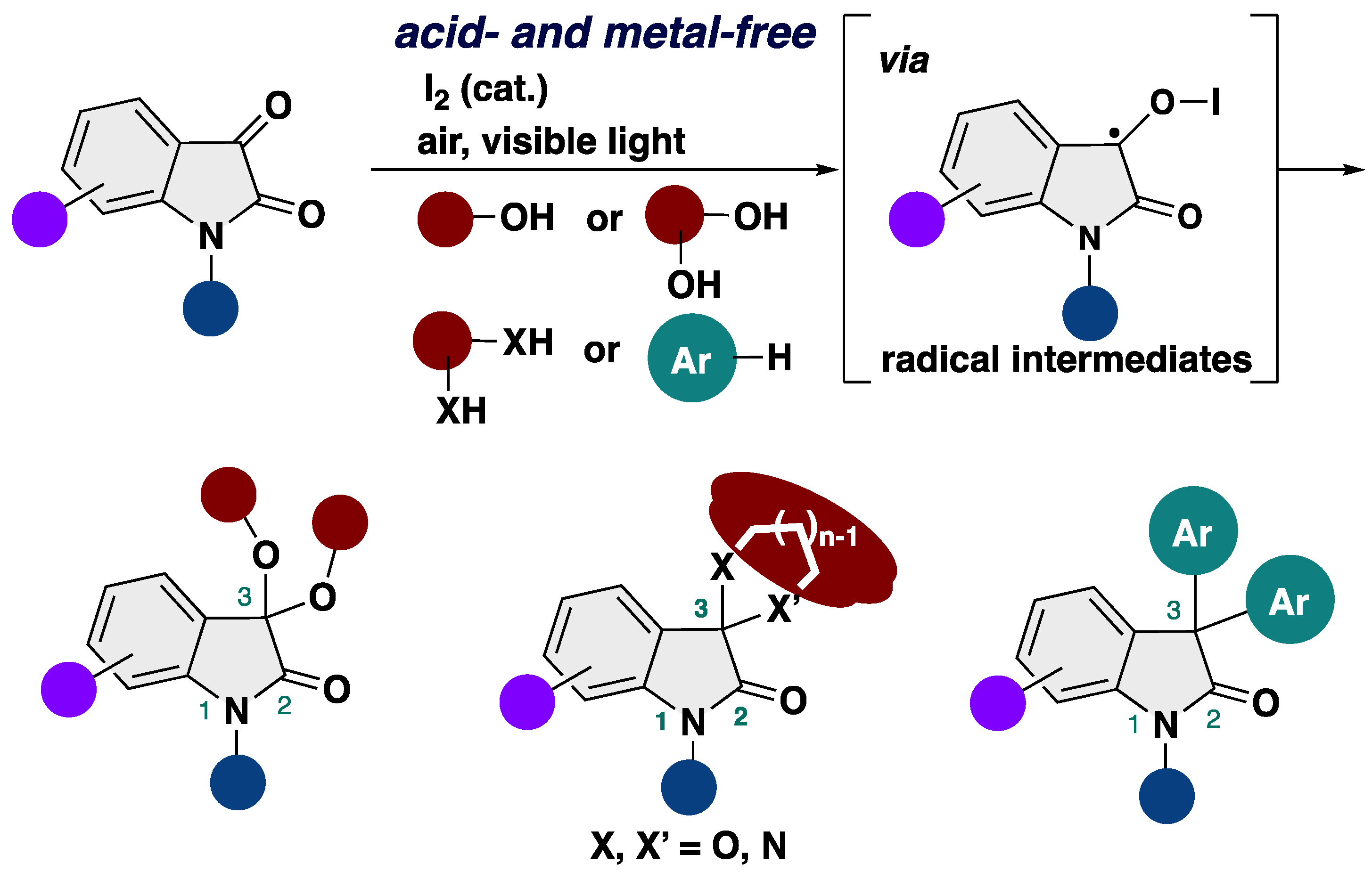
As part of our investigations into the synthesis of alkoxyindoles such as 2-alkoxyindoles [43,44,45,46], 3-alkoxyindoles [47,48,49], 2,3-dialkoxyindolines [50], and 1-alkoxyindoles [51], we were aware of the fact that a catalytic amount of molecular iodine can activate the methoxy group, producing a ketone moiety from 2,3-dialkoxyindolines [52]. Inspired by this interaction between molecular iodine and the methoxy group, we envisage that the use of iodine reagents as a Lewis acid catalyst or halogen bonding catalyst would facilitate ketalization between isatins and alcohols to afford 3,3-dialkoxy-2-oxindoles. Herein, we report on the efficient synthesis of 3,3-dialkoxy-2-oxindoles starting from isatins and alcohol through a mild ketalization by molecular iodine (Scheme 2). Our transformation paves the way for a diverse array of not only 3,3-dialkoxy-2-oxindoles but also 3,3-diaryl-2-oxindoles under metal- and acid-free conditions.
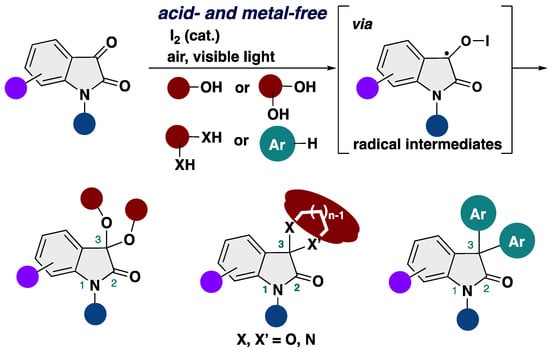
Scheme 2.
Our synthetic strategy for construction of 3,3-dialkoxy-2-oxindoles.
2. Results and Discussion
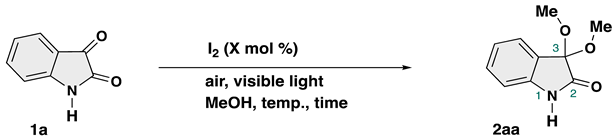
To identify the optimized reaction conditions for ketalization of isatin, we initiated the investigation with isatin and methanol using catalyst at 60 °C under air and visible light (Table 1). To our delight, PhI(OCOCF3)2 (10 mol % catalyst loading) afforded 3,3-dimethoxy-2-oxindole 2aa in 52% yield (runs 1–2). Further screening of the iodine catalysts (runs 3–8) revealed that molecular iodine (I2) is the most effective catalyst in our transformation (84% yield, run 6). Decreasing the catalyst loading to 5 mol % resulted in a slight drop in yield of the desired product to 68% (run 9). Increasement of I2 to 50 mol % catalyst loading afforded almost comparable results (run 10).

Table 1.
Optimization of reaction conditions.
The sensitivity of the transformations to key parameters such as concentration, water level, oxygen level, light intensity, and scale is crucial for increasing the uptake of new reactions and their reproducibility [53]. Following this guideline, we investigated effects of H2O levels. To our surprise, the reaction using MgSO4 or Na2SO4 as a dehydrating reagent resulted in a slightly decreased yields (63% and 75% yields, respectively, runs 12 and 13). The CaCl2-tube as a H2O-scavenger did not affect the reaction outcome (run 14). These results indicates that our protocol does not require special caution about H2O to promote the reaction with good reproducibility. Aerial oxidation against I2 catalyst was observed, as the reaction under argon atmosphere (inert atmosphere) led to decrease the product yield (runs 15 and 16), which is evidence of the crucial role of atmospheric oxygen in the open flask. Then, the influence of visible light was investigated. The control reaction was performed under dark (in the absence of light) using flask covered by aluminum-foil, a decreased yield of 2aa was observed (run 17). Combination of dark and inert conditions interrupted transformation to afford a desired product in a decreased yield (run 18). This highlights the necessity of light for our transformation to take place. Next, the reaction temperature was investigated. When the reaction temperature was decreased to rt, a decreased yield of 2aa was observed (run 20). No 2aa was isolated in the reaction at 0 °C (run 21). The 0.1 M (mol/L, molar concentration) or 0.5 M concentration of the reaction mixtures shows low conversion (runs 22 and 23). The ketalization was conducted in the absence of I2 catalyst, resulting in the unchanged starting material recovery (run 24). This suggests that an I2 catalyst proved indispensable for ensuring the success of the ketaization of isatin using MeOH. Therefore, we determined that the optimal reaction conditions were 10 mol % of I2-catalyzed ketalization in MeOH (0.2 M) under air and visible light for 24 h at 60 °C (Table 1, run 6).
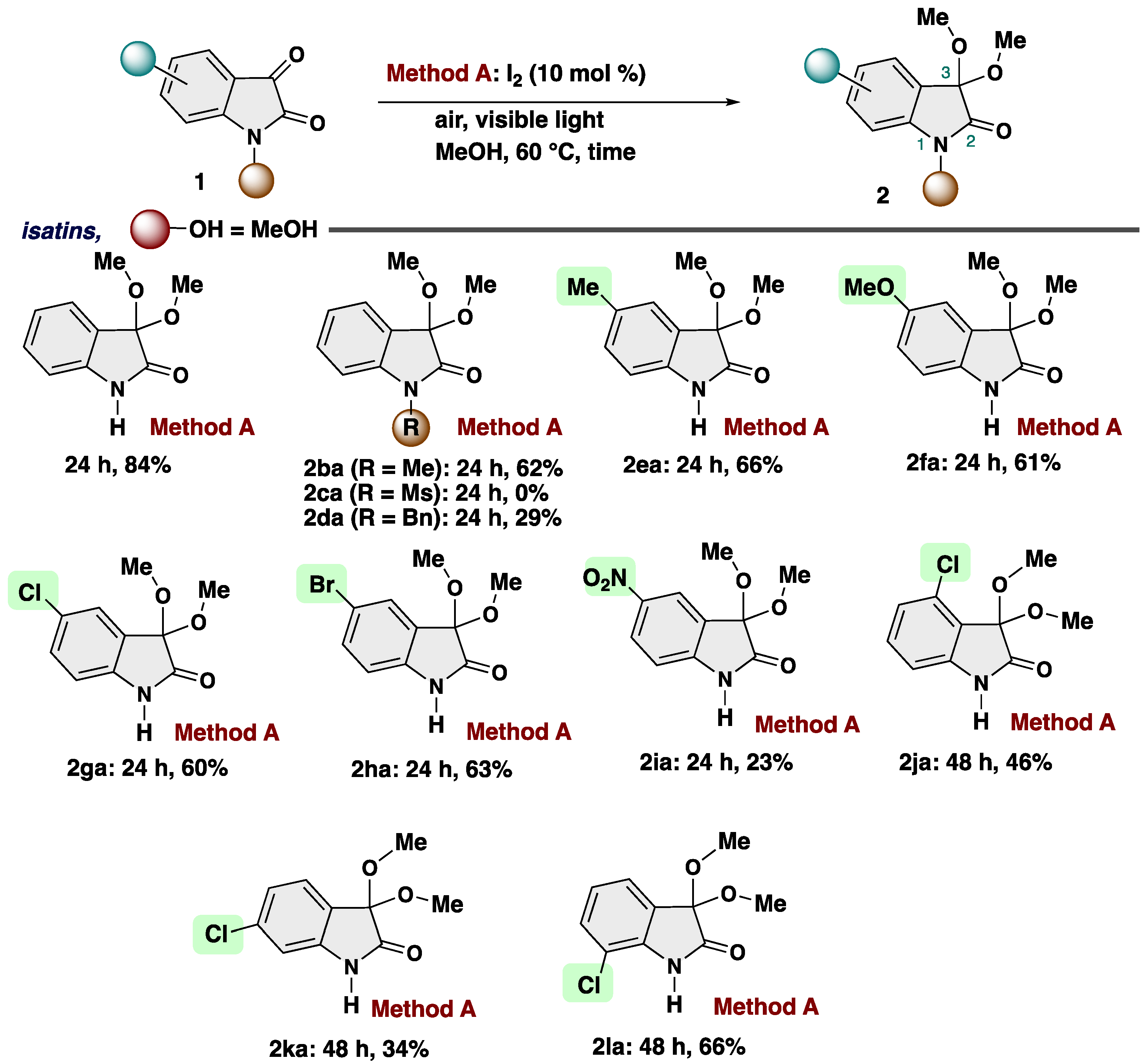
With the optimized reaction conditions in hand, we examined the scope of reactions (Scheme 3). First, we evaluated the applicability of substituents at the isatin. N-Substituted isatins could undergo the reaction to afford corresponding products 2ba (R = Me), and 2da (R = Bn) in 62%, and 29% yields, respectively. Ms-substituted substrate did not participitate into our transformation, resulting no reaction (2ca: R = Ms). 5-Substituted isatins featuring such as methyl, methoxy, chloro, and bromo group can also be successfully converted into the isatin ketals 2ea (R = Me), 2fa (R = OMe), 2ga (R = Cl), and 2ha (R = Br) in good yields. Either electron-donating or electron-withdrawing groups had little impact on the reaction efficiency. Chloro substituents at various positions of the phenyl ring (2ja: 4-Cl, 2ka: 6-Cl, and 2la: 7-Cl), yielded the desired products in moderate to good yields.
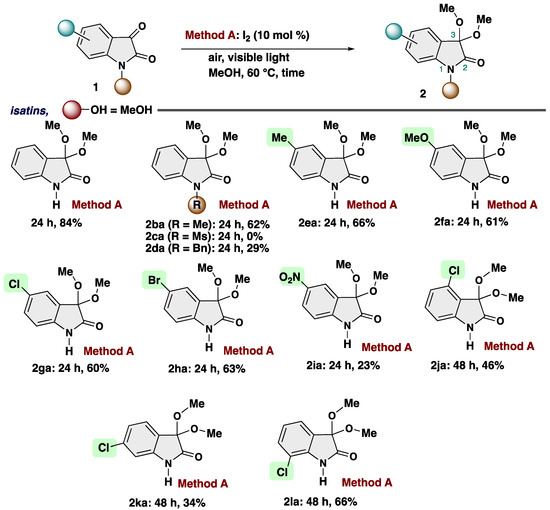
Scheme 3.
Substrate scope of isatins.
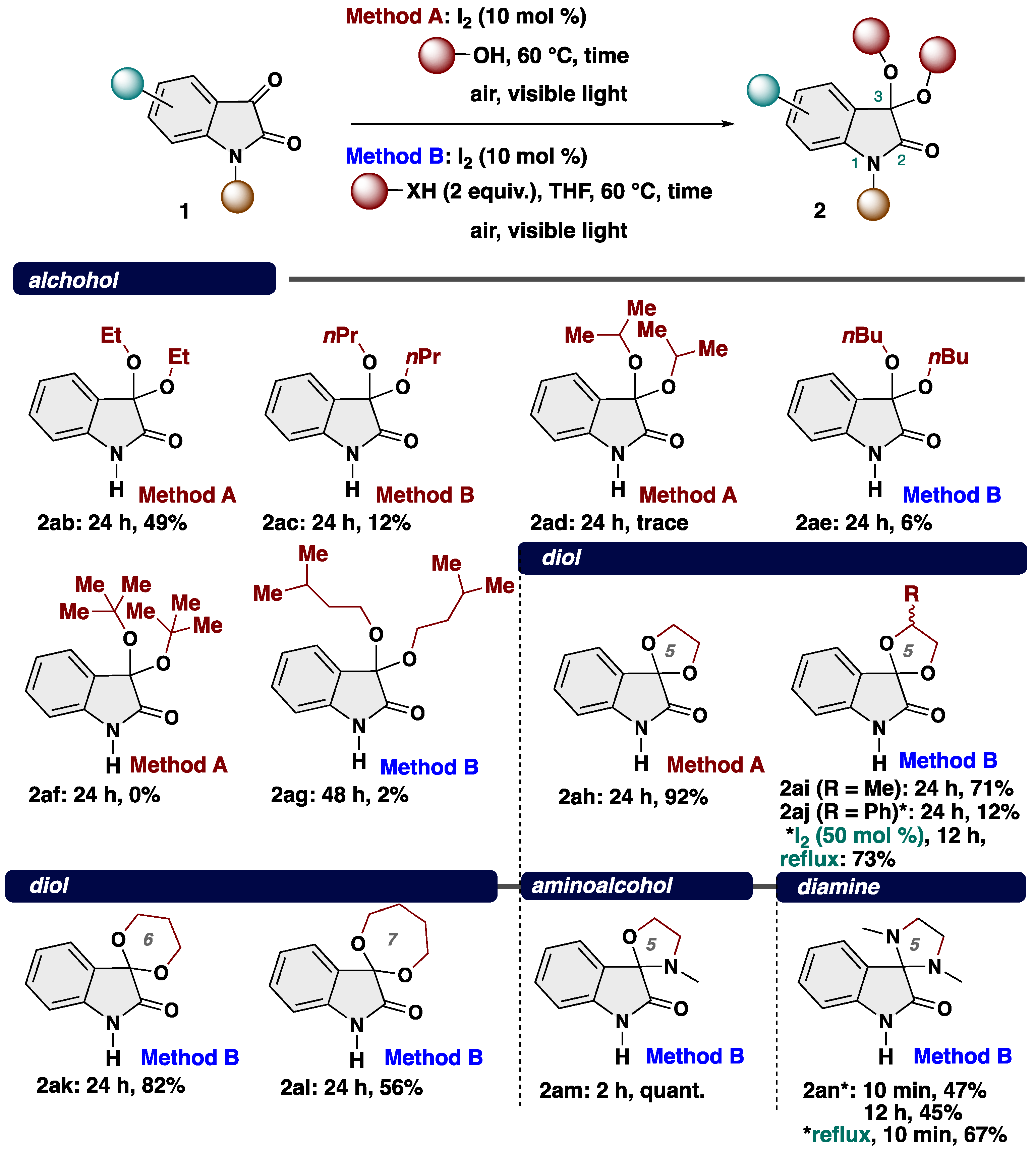
Next, to showcase the potentially broad applicability of our ketalization system, we explored the reaction scope with respect to nucleophiles such as alcohol, diol, aminoalcohol, and diamine (Scheme 4). Various alcohol including both linear and blanched alkyl alcohol were found to be suitable and gave the corresponding ketals 2ab (ethanol), 2ac (normal-propanol), 2ad (iso-propanol), 2ae (normal-butanol), and 2ag (isoamyl alcohol) in low yields. In the case of the reaction using sterically hindered tert-butanol, the desired reaction did not proceed at all (2af, 0%). To show the robustness of our protocol, we employed ethylene glycol as a dinucleophile, successfully delivering the spiroketal 2ah in 92% yield. We further explored the variation in the diol moiety, including 1,2-propanediol (2ai) and 1-phenyl-1,2-ethanediol (2aj), resulting in the production of the corresponding spiroketals as mixtures of diastereomers. Modified reaction conditions (50 mol % of I2, 2 equiv. of alcohol, reflux, 12 h) make the product yield improved (73% yield). The reaction was also amenable for 1,3-propanediol (2ak) and 1,4-butanediol (2al), which gave rise to the spiroketals bearing six-, and seven-membered rings (2ak: 82% yield; 2al: 56% yield). In addition to diols, aminoalcohol (2am) and diamine (2an) were amenable to undergo our transformation with low yields. 3,3-Diamino-2-oxindole is known as indole alkaloid isolated from a native orchid Cephalanceropsis gracilis [54].
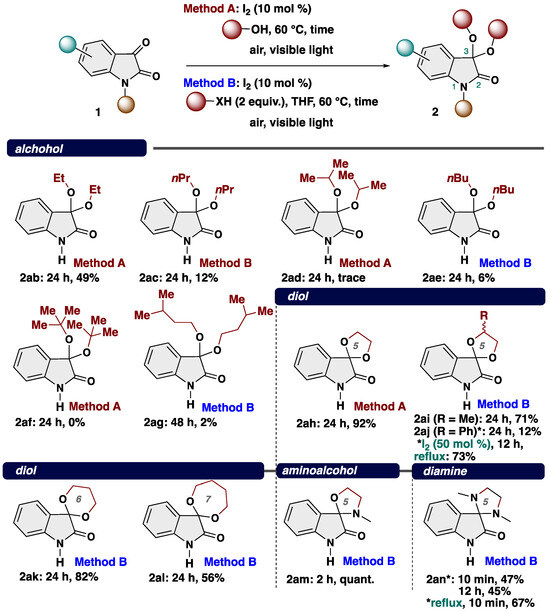
Scheme 4.
Substrate scope of nucleophile. * Changing conditions.
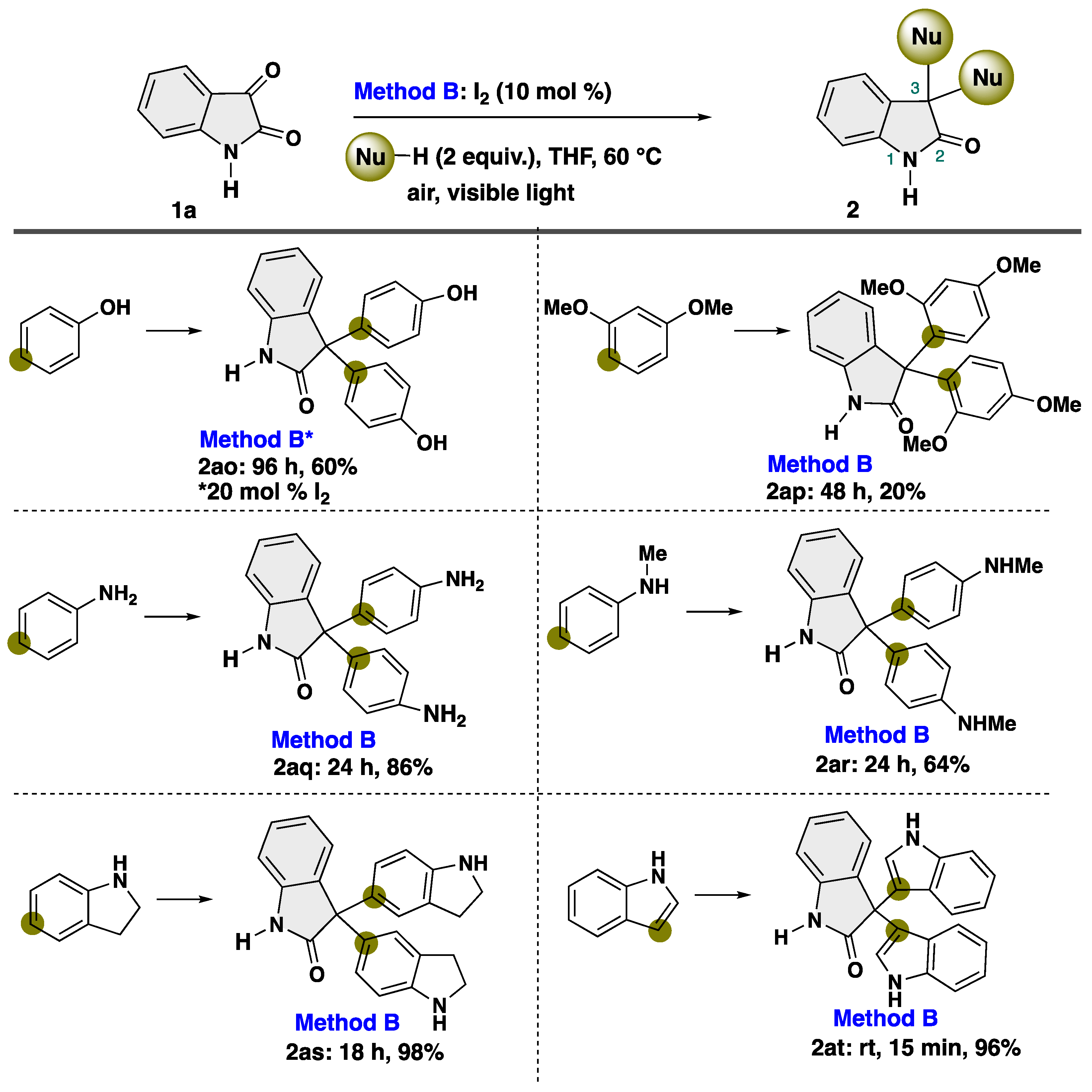
To further demonstrate the applicability of our protocol, we turned our attention to the reaction with heteroatom-substituted aryl compounds under modified conditions, which play an important synthetic intermediate for organic and medicinal chemistry [55,56,57,58,59,60] (Scheme 5). After intensive investigations, we found that a diverse array of heteroatoms-substituted aryl compounds proved to be suitable nucleophiles with isatin. The reaction with phenol proceeded smoothly, resulting in production of unexpected product 2ao in 60% yield, albeit using increased loading of I2 catalyst. The diacetate derivative of 2ao is known as a laxative [47]. Notably, the reaction proceeds at the para-position of the heteroatom-substituted aromatic rings. When the reaction was performed with 1,3-dimethoxybenzene, para-substituted product 2ap was obtained in 20% yield. In the case of aniline derivatives, the substituent at the nitrogen atom affected the efficiency of the desired transformation. Notably, aniline demonstrated efficacy as nucleophiles, yielding para-disubstituted product 2aq in 86% yield. Meanwhile, N-methylaniline showed a slight drop yield (2ar, 64% yield). Indole participated into our transformation to give 3-substituted product 2at in quantitative yield, which are known as indole alkaloid trisindoline [61,62,63], while the reaction with indoline afforded 5-substituted product 2as.
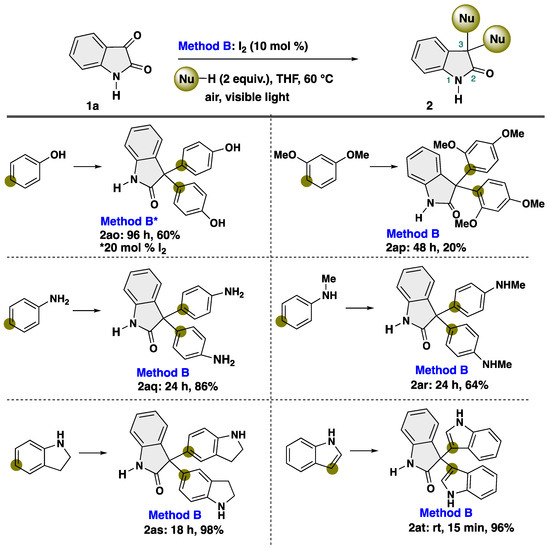
Scheme 5.
Substrate scope of aromatic compounds as a nucleophile. * Changing conditions.
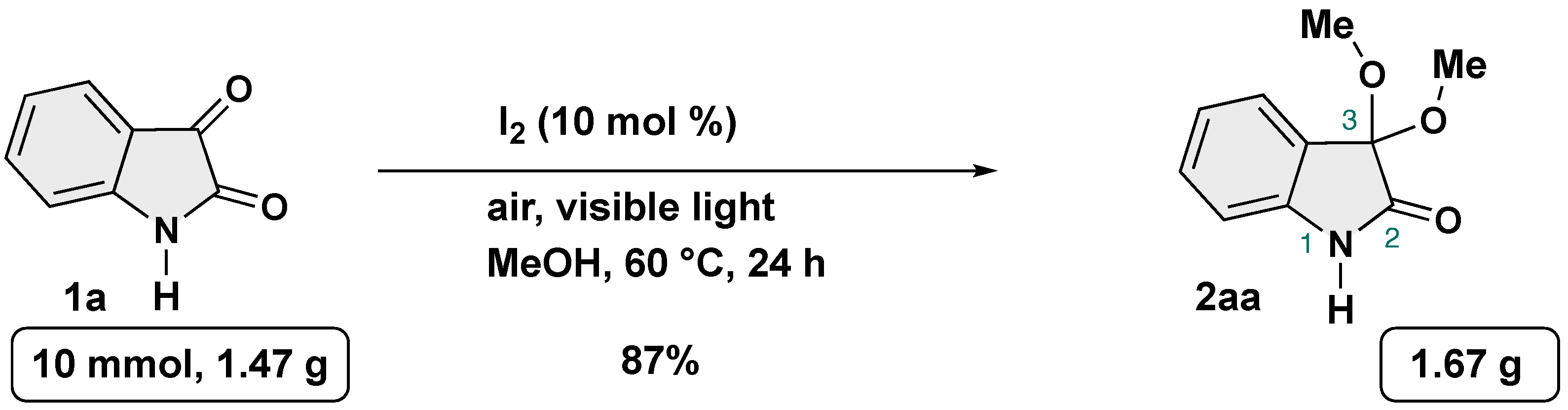
To showcase the robustness and practicality of our protocol, gram-scale reaction was conducted by using 1a as the substrate under the optimal reaction conditions (Scheme 6). To our delight, gram quantity of target product could be obtained without a slight loss of efficiency.
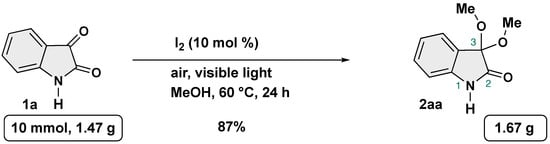
Scheme 6.
Gram-scale synthesis of 3,3-dimethoxy-2-oxindole.
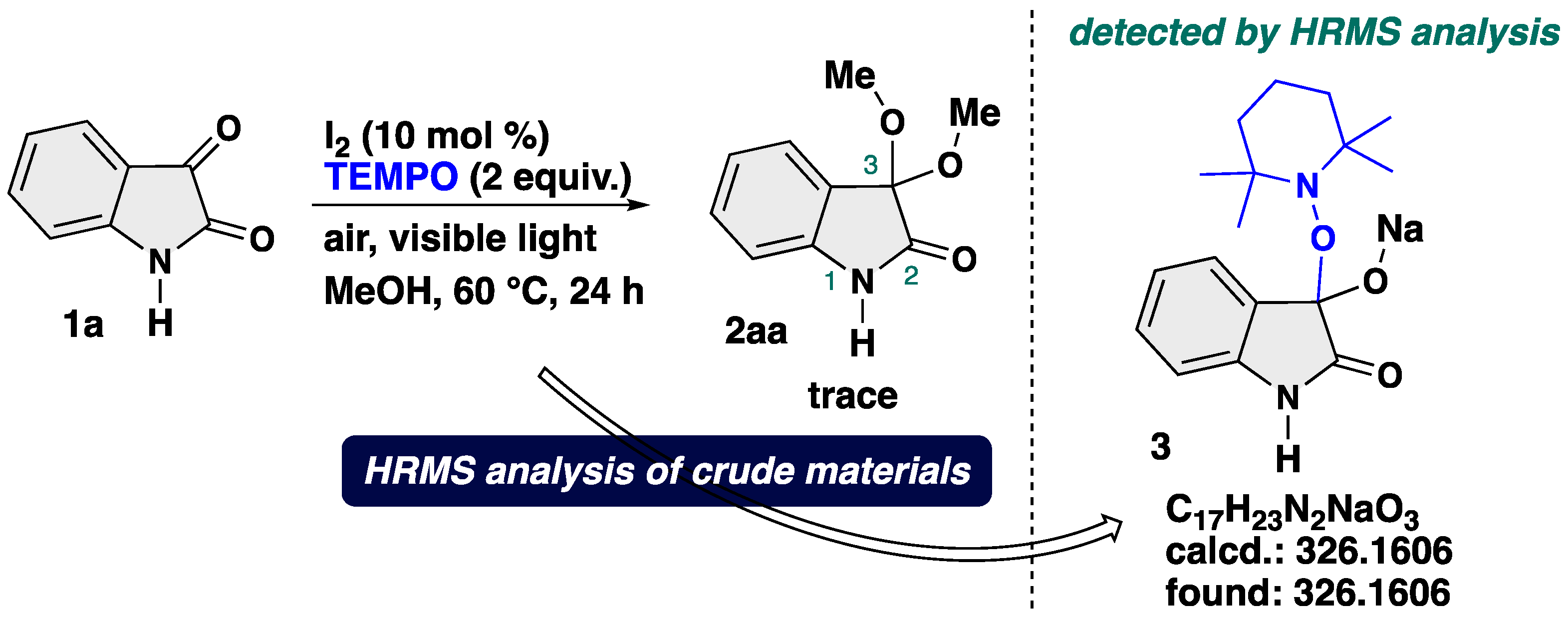
In order to shed light on the possible reaction mechanism, mechanistic experiment was conducted (Scheme 7). We carried out the reaction of 1a under the optimal reaction conditions in the presence of TEMPO (2,2,6,6-tetramethylpiperidine 1-oxyl) as a radical scavenger. With 2 equiv. of TEMPO as a radical scavenger, a trace amount of the target product 2aa was obtained, and an TEMPO-adduct 3 was observed by ESI-TOF (electrospray ionization time-of-flight) HRMS (high resolution mass spectrometry) analysis of the crude reaction mixtures. The TEMPO-adduct could not be isolated by column chromatography probably due to its instability. These results suggest that the reaction involves a radical species, which play an important role to operate smooth transformation.
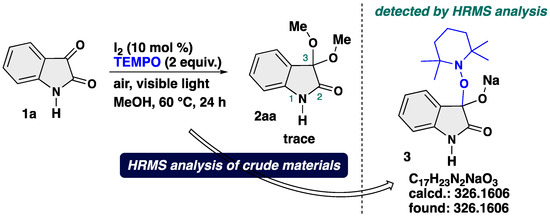
Scheme 7.
Control experiment using TEMPO as a radical scavenger.
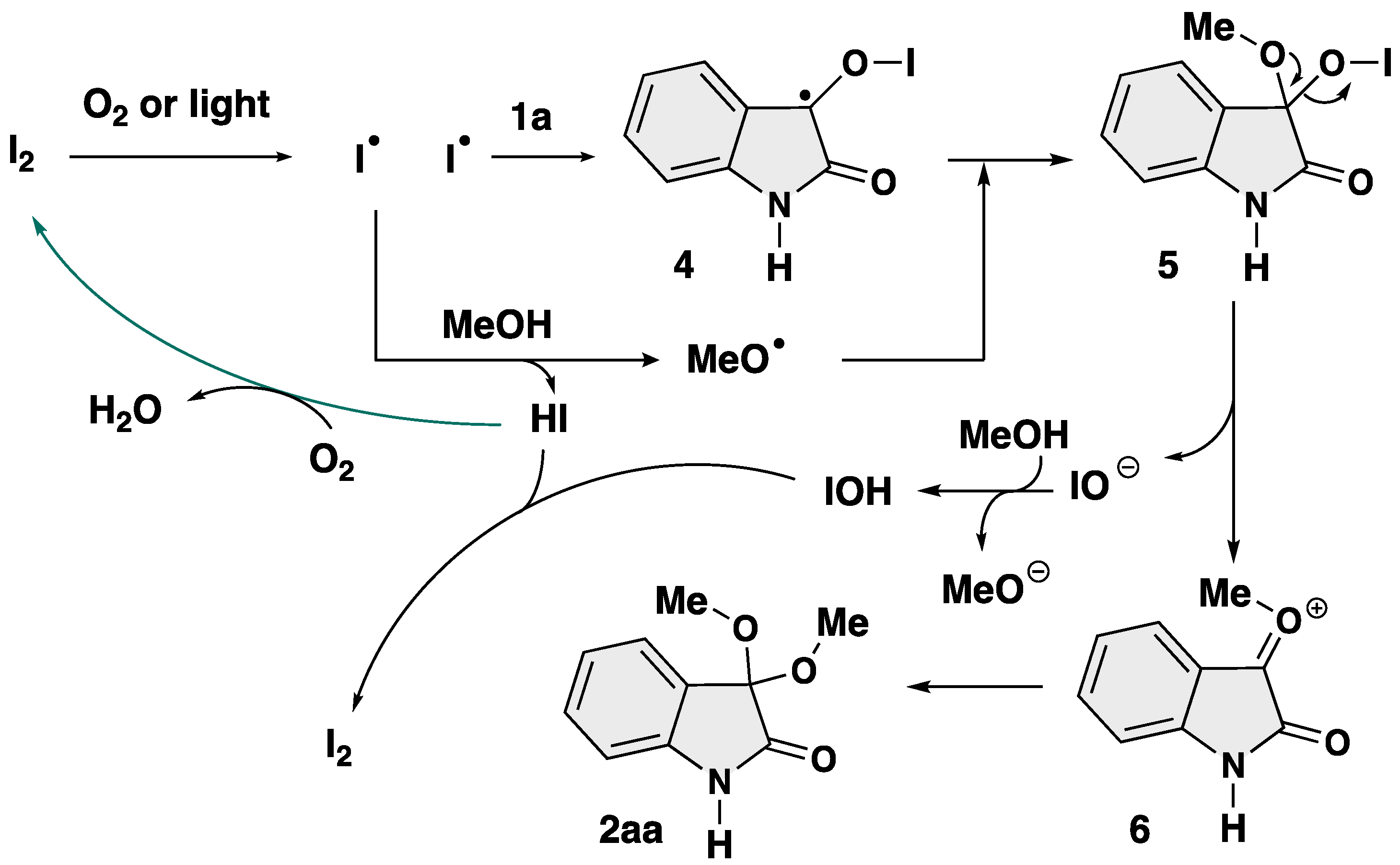
Based on the above experiments and previous reports [64,65,66,67,68], a plausible mechanism has been proposed (Scheme 8). First, the iodine radical is generated from molecular iodine by O2 and/or light [65]. Then, the carbonyl moiety of isatin (1a) was trapped by the iodine radical, yielding 3-isatin monoradical intermediate 4. This hypothesis was confirmed by a detection of TEMPO-adduct 3 derived from in situ generated monoradical species. At the same time, the iodine radical undergoes hydrogen abstraction from MeOH to give a methoxy radical (MeO•). The combination of MeO• and 4 afford 3,3-oxifunctionalized 2-oxindole 5. Finally, elimination of IO− affords oxonium intermediate 6, which further undergoes addition by MeO− to afford the final product 2aa. The catalyst I2 can be reproduced from the resultant HI and/or IOH [68]. However, the detail in the reaction mechanism is unclear at this stage.
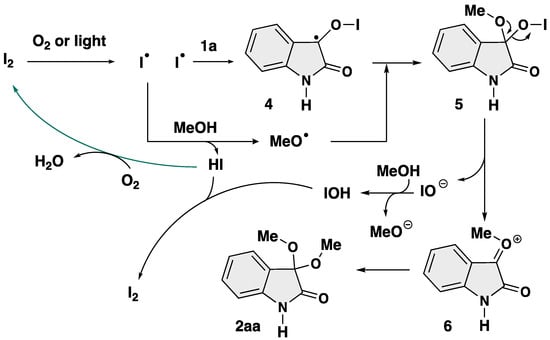
Scheme 8.
Plausible reaction mechanism.
3. Conclusions
We disclosed an acid- and metal-free approach to 3,3-dialkoxy-2-oxindoles from isatins through an iodine-catalyzed ketalization under air and visible light. This photochemical protocol does not require the use of any specific reagents including metal-catalysts. Based on the control experiments, our ketalization involves a radical intermediate. Furthermore, the total synthesis of the unprecedented 2-oxindole alkaloid bearing 3,3-dimethoxy moiety has been achieved. Further synthetic studies utilizing 3,3-dialkoxy-2-oxindoles as a synthetic intermediate are ongoing in our laboratory.
4. Materials and Methods
General: High-resolution MS spectra were recorded with Brucker micrOTOF mass spectrometers (ESI-TOF-MS). NMR experiments were performed with JEOL JNM-ECZ600R (1H NMR: 600 MHz, 13C NMR: 151 MHz) spectrometer. Chemical shifts are expressed in δ (parts per million, ppm) values, and coupling constants are expressed in hertz (Hz). 1H NMR spectra were referenced to a solvent signal (CDCl3: 7.26 ppm, DMSO-d6: 2.49 ppm). 13C NMR spectra were referenced to a solvent signal (CDCl3: 77.1 ppm, DMSO-d6: 39.5 ppm). Signal multiplicities are abbreviated as follows: singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), doublet of doublets (dd), doublet of triplet (dt) septet (sept), broad (br). Melting points (mp) were recorded with a Yamato melting point apparatus model MP-21 and are uncorrected. IR spectra were measured with a HORIBA Fourier transform infrared (FTIR) spectrometer FT-720, and absorbance frequencies are reported in reciprocal centimeters (cm−1). Reactions were monitored by thin layer chromatography (TLC) carried out on silica gel plates (60F-254) and visualized under UV illumination at 254 or 365 nm depending on the compound. Column chromatography was performed on silica gel (WAKO Gel 75–150 mesh, WAKO Co., Ltd.). Preparative tin-layer chromatography was performed with silica gel plates (60F-254). All substrates were used as received from commercial suppliers (Sigma-Aldrich, Kanto Chemical, TCI and Wako) and all reagents were weighed and handled in air at room temperature. The 1H- and 13C-NMR-spectra of the synthesized compounds can be downloaded as Supplementary Materials.
4.1. Synthesis of N-Substituted Isatins (1) [69,70]
N-Substituted isatins (1c and 1d) were prepared by the reported method [69,70]. Analytical data are in accordance with the literature values (1c [69], 1d [70]).
4.2. General Procedure for the Synthesis of 3,3-Dialcohoxyindolin-2-one (2)
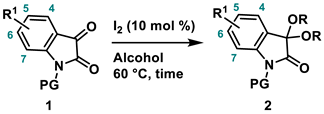
A mixture of isatin 1 (1.0 mmol) and iodine (25.4 mg, 0.10 mmol, 10 mol %) was dissolved in alcohol (5.0 mL, 0.2 M). The mixture was stirred at 60 °C in an oil bath. After the whole was quenched with sat. Na2S2O3, H2O (10 mL) was added to the mixture and extracted with AcOEt (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica-gel column chromatography.
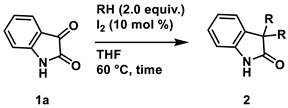
To a mixture of isatin 1a (1.0 mmol), nucleophile (2.0 mmol. 2.0 equiv.) and iodine (25.4 mg, 0.10 mmol, 10 mol %) was dissolved in THF (5.0 mL, 0.2 M). The mixture was stirred at 60 °C in oil bath. After the whole was quenched with sat. Na2S2O3, H2O (10 mL) was added to the mixture and extracted with AcOEt (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica-gel column chromatography.
4.3. Substrate Scope of Isatins and Alcohol (Scheme 3 and Scheme 4)
4.3.1. Synthesis of 3,3-Dimethoxyindolin-2-one (2aa) [17]
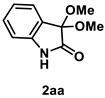
The reaction was performed according to the general procedure A using 147 mg (1.0 mmol) of 1a for 24 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (4/1–2/1 [v/v]) to give 2aa (163 mg, 0.84 mmol, 84% yield) as a colorless solid.
Colorless solid (163 mg, 0.84 mmol, 84% yield; mp 80–82 °C); IR (KBr) ν: 3405, 3097, 2967, 2942, 2894, 1727, 1473, 1060, 759 cm−1; 1H NMR (600 MHz, CDCl3) δ 9.52 (br s, 1H), 7.37 (d, J = 7.2 Hz, 1H), 7.26 (dt, J = 7.2, 1.2 Hz, 1H), 7.03 (dt, J = 7.8, 1.2 Hz, 1H), 6.91 (d, J = 7.8 Hz, 1H), 3.55 (s, 6H); 13C{1H} NMR (151 MHz, CDCl3) δ 173.6, 140.8, 130.8, 125.0, 124.9, 122.7, 111.3, 97.6, 50.9; HRMS (ESI) m/z: [M+Na]+ Calcd for C10H11NO3Na 216.0637; Found 216.0639.
4.3.2. Synthesis of 3,3-Dimethoxy-1-methylindolin-2-one (2ba) [71]
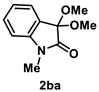
The reaction was performed according to the general procedure A using 161 mg (1.0 mmol) of 1b for 24 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (4/1–2/1 [v/v]) to give 2ba (129 mg, 0.62 mmol, 62% yield) as a colorless solid.
Colorless solid (129 mg, 0.62 mmol, 62% yield; mp 80–82 °C); IR (KBr) ν: 3442, 3027, 2994, 2940, 2830, 1727, 1614, 1475, 1245, 746 cm−1; 1H NMR (600 MHz, CDCl3) δ 7.40 (dd, J = 7.2, 0.6 Hz, 1H), 7.34 (dt, J = 7.8, 1.2 Hz, 1H), 7.09 (dt, J = 7.8, 0.6 Hz, 1H), 6.83 (d, J = 8.4 Hz, 1H), 3.56 (s, 6H), 3.16 (s, 3H); 13C{1H} NMR (151 MHz, CDCl3) δ 170.8, 143.4, 130.8, 125.0, 124.8, 122.8, 108.9, 97.1, 50.9, 25.9; HRMS (ESI) m/z: [M+Na]+ Calcd for C11H13NO3Na 230.0793; Found 230.0792.
4.3.3. Synthesis of 1-Benzyl-3,3-dimethoxyindolin-2-one (2da) [13]
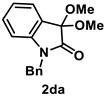
The reaction was performed according to the general procedure A using 237 mg (1.0 mmol) of 1d for 24 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (4/1–2/1 [v/v]) to give 2da (80.8 mg, 0.29 mmol, 29% yield) as an orange oil.
Orange oil (80.8 mg, 0.29 mmol, 29% yield); IR (KBr) ν: 3448, 3062, 3031, 2975, 2944, 2911, 2832, 1727, 1614, 1469, 1365, 1180, 1070, 757 cm−1; 1H NMR (600 MHz, CDCl3) δ 7.41 (dd, J = 7.2, 1.2 Hz, 1H), 7.32–7.25 (m, 5H), 7.23 (dt, J = 7.8, 1.2 Hz, 1H), 7.04 (dt, J = 7.8, 0.6 Hz, 1H), 6.70 (d, J = 7.8 Hz, 1H), 4.86 (s, 2H), 3.59 (s, 6H); 13C{1H} NMR (151 MHz, CDCl3) δ 171.0, 142.5, 135.4, 130.7, 129.0, 127.8, 127.3, 125.0, 124.9, 122.9, 109.9, 7.1, 51.0, 43.5; HRMS (ESI) m/z: [M+Na]+ Calcd for C17H17NO3Na 306.1106; Found 306.1104.
4.3.4. Synthesis of 3,3-Dimethoxy-5-methylindolin-2-one (2ea)
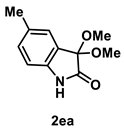
The reaction was performed according to the general procedure A using 161 mg (1.0 mmol) of 1e for 24 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (4/1–2/1 [v/v]) to give 2ea (137 mg, 0.66 mmol, 66% yield) as a yellow solid.
Yellow solid (137 mg, 0.66 mmol, 66% yield; mp 104–106 °C); IR (KBr) ν: 3305, 3037, 2975, 2937, 2832, 1754, 1494, 1083, 740 cm−1; 1H NMR (600 MHz, CDCl3) δ 8.72 (br s, 1H), 7.21 (s, 1H), 7.10 (d, J = 7.8 Hz, 1H), 6.79 (d, J = 7.8 Hz, 1H), 3.57 (s, 6H), 2.33 (s, 3H); 13C{1H} NMR (151 MHz, CDCl3) δ 173.3, 138.1, 132.5, 131.2, 125.9, 125.2, 110.8, 97.6, 51.0, 21.2; HRMS (ESI) m/z: [M+Na]+ Calcd for C11H13NO3Na 230.0793; Found 230.0792.
4.3.5. Synthesis of 3,3-Dimethoxy-5-methoxylindolin-2-one (2fa)
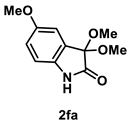
The reaction was performed according to the general procedure A using 177 mg (1.0 mmol) of 1f for 24 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (4/1–2/1 [v/v]) to give 2fa (135 mg, 0.61 mmol, 61% yield) as a colorless solid.
Colorless solid (135 mg, 0.61 mmol, 61% yield; mp 155 °C); IR (KBr) ν: 3324, 3006, 2969, 2942, 2904, 2834, 1737, 1481, 1199, 1060, 748 cm−1; 1H NMR (600 MHz, CDCl3) δ 9.26 (br s, 1H), 6.98 (s, 1H), 6.83 (d, J = 7.8 Hz, 1H), 6.81 (d, J = 7.8 Hz, 1H), 3.77 (s, 3H), 3.55 (s, 6H); 13C{1H} NMR (151 MHz, CDCl3) δ 173.6, 155.9, 134.0, 126.2, 115.4, 112.1, 111.7, 98.0, 55.9, 51.0; HRMS (ESI) m/z: [M+Na]+ Calcd for C11H13NO4Na 246.0742; Found 246.0745.
4.3.6. Synthesis of 5-Chloro-3,3-dimethoxyindolin-2-one (2ga) [72]
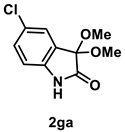
The reaction was performed according to the general procedure A using 182 mg (1.0 mmol) of 1g for 24 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (4/1–2/1 [v/v]) to give 2ga (137 mg, 0.60 mmol, 60% yield) as a pale-yellow solid.
Pale-yellow solid (137 mg, 0.60 mmol, 60% yield; mp 153–154 °C); IR (KBr) ν: 3324, 3091, 3006, 2975, 2940, 2906, 2834, 1743, 1479, 1068, 754 cm−1; 1H NMR (600 MHz, CDCl3) δ 9.29 (br s, 1H), 7.37 (d, J = 1.8 Hz, 1H), 7.28 (dd, J = 8.4, 2.4 Hz, 1H), 6.86 (d, J = 8.4 Hz, 1H), 3.56 (s, 6H); 13C{1H} NMR (151 MHz, CDCl3) δ 173.3, 139.2, 130.8, 128.3, 126.8, 125.6, 112.3, 97.4, 51.0; HRMS (ESI) m/z: [M+Na]+ Calcd for C10H10ClNO3Na 250.0247, 252.0217; Found 250.0245, 252.0220.
4.3.7. Synthesis of 5-Bromo-3,3-dimethoxyindolin-2-one (2ha)
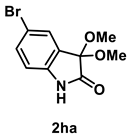
The reaction was performed according to the general procedure A using 226 mg (1.0 mmol) of 1h for 24 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (4/1–2/1 [v/v]) to give 2ha (171 mg, 0.63 mmol, 63% yield) as a colorless solid.
Colorless solid (171 mg, 0.63 mmol, 63% yield; mp 164–165 °C); IR (KBr) ν: 3338, 3081, 3002, 2971, 2940, 2910, 2834, 1754, 1477, 1066, 750 cm−1; 1H NMR (600 MHz, CDCl3) δ 9.38 (br s, 1H), 7.50 (d, J = 1.8 Hz, 1H), 7.42 (dd, J = 8.4, 1.8 Hz, 1H), 6.82 (d, J = 7.8 Hz, 1H), 3.56 (s, 6H); 13C{1H} NMR (151 MHz, CDCl3) δ 173.2, 139.7, 128.3, 127.1, 115.6, 112.8, 97.3, 51.0; HRMS (ESI) m/z: [M+Na]+ Calcd for C10H10BrNO3Na 295.9721, 293.9742; Found 295.9725, 293.9740.
4.3.8. Synthesis of 5-Nitro-3,3-dimethoxyindolin-2-one (2ia)
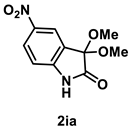
The reaction was performed according to the general procedure A using 192 mg (1.0 mmol) of 1i for 24 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (4/1–2/1 [v/v]) to give 2ia (54 mg, 0.23 mmol, 23% yield) as a colorless solid.
Colorless solid (54 mg, 0.23 mmol, 23% yield; mp 162 °C); IR (KBr) ν: 3324, 2989, 2950, 2838, 1756, 1722, 1608, 1517, 1344, 1116, 750 cm−1; 1H NMR (600 MHz, CDCl3) δ 8.97 (br s, 1H), 8.31 (d, J = 8.4 Hz, 1H), 8.30 (s, 1H), 7.04 (d, J = 8.4 Hz, 1H), 3.62 (s, 6H); 13C{1H} NMR (151 MHz, CDCl3) δ 173.0, 146.1, 143.6, 127.7, 126.3, 121.3, 111.0, 96.4, 51.1; HRMS (ESI) m/z: [M+Na]+ Calcd for C10H10N2O5Na 261.0487; Found 261.0483.
4.3.9. Synthesis of 4-Chloro-3,3-dimethoxyindolin-2-one (2ja)
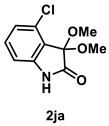
The reaction was performed according to the general procedure A using 182 mg (1.0 mmol) of 1j for 48 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (4/1–2/1 [v/v]) to give 2ja (105 mg, 0.46 mmol, 46% yield) as a colorless solid.
Colorless solid (105 mg, 0.46 mmol, 46% yield; mp 86–88 °C); IR (KBr) ν: 3168, 3016, 1989, 1948, 2830, 1743, 1704, 1450, 1243, 1095, 782 cm−1; 1H NMR (600 MHz, CDCl3) δ 9.23 (br s, 1H), 7.24 (t, J = 8.4 Hz, 1H), 7.03 (d, J = 8.4 Hz, 1H), 6.83 (dd, J = 7.8, 0.6 Hz, 1H), 3.51 (s, 6H); 13C{1H} NMR (151 MHz, CDCl3) δ 173.4, 142.8, 132.32, 132.25, 124.8, 121.7, 109.6, 99.7, 52.2; HRMS (ESI) m/z: [M+Na]+ Calcd for C10H10ClNO3Na 250.0247, 252.0217; Found 250.0243, 252.0216.
4.3.10. Synthesis of 6-Chloro-3,3-dimethoxyindolin-2-one (2ka)
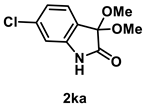
The reaction was performed according to the general procedure A using 182 mg (1.0 mmol) of 1k for 48 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (4/1–2/1 [v/v]) to give 2ka (78 mg, 0.34 mmol, 34% yield) as a yellow solid.
Yellow solid (78 mg, 0.34 mmol, 34% yield; mp 173–174 °C); IR (KBr) ν: 3305, 3089, 2979, 2944, 2913, 2834, 1754, 1720,1614, 1130, 1058, 748 cm−1; 1H NMR (600 MHz, CDCl3) δ 8.43 (br s, 1H), 7.31 (d, J = 7.8 Hz, 1H), 7.06 (dd, J = 7.8, 1.8 Hz, 1H), 6.92 (d, J = 1.2 Hz, 1H), 3.56 (s, 6H); 13C{1H} NMR (151 MHz, CDCl3) δ 172.8, 141.7, 136.7, 126.3, 123.7, 122.9, 111.7, 96.9, 51.0; HRMS (ESI) m/z: [M+Na]+ Calcd for C10H10ClNO3Na 250.0247, 252.0217; Found 250.0242, 252.0215.
4.3.11. Synthesis of 7-Chloro-3,3-dimethoxyindolin-2-one (2la)
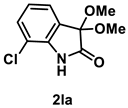
The reaction was performed according to the general procedure A using 182 mg (1.0 mmol) of 1l for 48 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (4/1–2/1 [v/v]) to give 2la (150 mg, 0.66 mmol, 66% yield) as a pale-brown oil.
Pale-brown oil (150 mg, 0.66 mmol, 66% yield); IR (KBr) ν: 3253, 3097, 2996, 2946, 2910, 2834, 1739, 1621, 1475, 1326, 1180, 781 cm−1; 1H NMR (600 MHz, CDCl3) δ 8.89 (br s, 1H), 7.27–7.26 (m, 2H), 6.99 (dd, J = 8.4, 7.2 Hz, 1H), 3.54 (s, 6H); 13C{1H} NMR (151 MHz, CDCl3) δ 172.2, 138.4, 130.7, 126.7, 123.6, 123.3, 116.3, 97.8, 51.0; HRMS (ESI) m/z: [M+Na]+ Calcd for C10H10ClNO3Na 250.0247, 252.0217; Found 250.0247, 252.0219.
4.3.12. Synthesis of 3,3-Diethoxyindolin-2-one (2ab) [17]
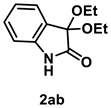
The reaction was performed according to the general procedure A using 147 mg (1.0 mmol) of 1a for 24 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (4/1–2/1 [v/v]) to give 2ab (109 mg, 0.49 mmol, 49% yield) as a yellow solid.
Yellow solid (109 mg, 0.49 mmol, 49% yield; mp 93–95 °C); IR (KBr) ν: 3208, 3099, 2979, 2935, 2898, 1727, 1619, 1473, 1340, 1213, 1056, 767 cm−1; 1H NMR (600 MHz, CDCl3) δ 9.40 (br s, 1H), 7.38 (d, J = 7.2 Hz, 1H), 7.27 (dt, J = 7.8, 1.2 Hz, 1H), 7.04 (dt, J = 7.2, 0.6 Hz, 1H), 6.92 (d, J = 7.2 Hz, 1H), 3.96–3.91 (m, 2H), 2.82–3.77 (m, 2H), 1.24 (t, J = 7.2 Hz, 6H); 13C{1H} NMR (151 MHz, CDCl3) δ 174.2, 140.7, 130.6, 125.9, 125.1, 122.7, 111.2, 97.6, 59.0, 15.3; HRMS (ESI) m/z: [M+Na]+ Calcd for C12H15NO3Na 244.0950; Found 244.0954.
4.3.13. Synthesis of 3,3-Dipropoxyindolin-2-one (2ac) [73]
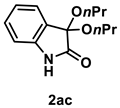
The reaction was performed according to the general procedure B using 147 mg (1.0 mmol) of 1a and 0.150 mL (2.0 mmol) of 1-propanol for 24 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (4/1–3/1 [v/v]) to give 2ac (29 mg, 0.12 mmol, 12% yield) as a pale-yellow oil.
Pale-yellow oil (29 mg, 0.12 mmol, 12% yield); IR (KBr) ν: 3307, 2964, 2937, 2877, 1743, 1621, 1471, 1205, 1087, 754 cm−1; 1H NMR (600 MHz, CDCl3) δ 7.71 (br s, 1H), 7.40 (d, J = 7.2 Hz, 1H), 7.29 (dt, J = 7.8, 1.2 Hz, 1H), 7.06 (dt, J = 7.8, 0.6 Hz, 1H), 6.85 (d, J = 7.2 Hz, 1H), 3.83–3.80 (m, 2H), 3.71–3.67 (m, 2H), 1.63 (dq, J = 7.2, 1.2 Hz, 4H), 0.93 (t, J = 7.8 Hz, 6H); 13C{1H} NMR (151 MHz, CDCl3) δ 173.0, 140.3, 130.6, 126.3, 125.4, 122.8, 110.6, 97.1, 65.0, 23.2, 10.7; HRMS (ESI) m/z: [M+Na]+ Calcd for C14H19NO3Na 272.1263; Found 272.1265.
4.3.14. Synthesis of 3,3-Dibutoxyindolin-2-one (2ae)
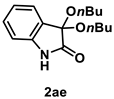
The reaction was performed according to the general procedure B using 147 mg (1.0 mmol) of 1a and 0.183 mL (2.0 mmol) of 1-butanol for 24 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (6/1–3/1 [v/v]) to give 2ae (16 mg, 0.06 mmol, 6% yield) as a yellow oil.
Yellow oil (16 mg, 0.06 mmol, 6% yield); IR (KBr) ν: 3305, 2958, 2935, 2873, 1731, 1623, 1473, 1205, 1089, 754 cm−1; 1H NMR (600 MHz, CDCl3) δ 8.69 (br s, 1H), 7.28 (dt, J = 7.8, 1.2 Hz, 1H), 7.05 (dt, J = 7.8, 0.6 Hz, 1H), 6.89 (d, J = 7.8 Hz, 1H), 3.86–3.82 (m, 2H), 3.74–3.71 (m, 2H), 1.62–1.57 (m, 4H), 1.42–1.36 (m, 4H), 0.90 (t, J = 7.2 Hz, 6H); 13C{1H} NMR (151 MHz, CDCl3) δ 173.8, 140.6, 130.6, 126.2, 125.3, 122.8, 110.9, 97.4, 63.2, 32.0, 19.4, 14.0; HRMS (ESI) m/z: [M+Na]+ Calcd for C16H23NO3Na 300.1576; Found 300.1578.
4.3.15. Synthesis of 3,3-Bis(isopentyloxy)indolin-2-one (2ag)
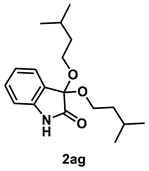
The reaction was performed according to the general procedure B using 147 mg (1.0 mmol) of 1a and 0.217 mL (2.0 mmol) of 3-methyl-1-butanol for 48 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (6/1–3/1 [v/v]) to give 2ag (6.4 mg, 0.02 mmol, 2% yield) as a brown oil.
Brown oil (6.4 mg, 0.02 mmol, 2% yield); IR (KBr) ν: 3305, 2956, 2931, 2871, 1731, 1623, 1473, 1205, 754 cm−1; 1H NMR (600 MHz, CDCl3) δ 7.98 (br s, 1H), 7.39 (d, J = 6.6 Hz, 1H, 7.29 (dt, J = 7.8, 1.2 Hz, 1H), 7.06 (dt, J = 7.8, 1.2 Hz, 1H), 6.86 (d, J = 7.8 Hz, 1H), 3.87–3.83 (m, 2H), 3.77–3.73 (m, 2H), 1.72 (sept, J = 6.6 Hz, 2H), 1.50 (q, J = 7.2 Hz, 4H), 0.88 (d, J = 6.6 Hz, 12H); 13C{1H} NMR (151 MHz, CDCl3) δ 173.2, 140.4, 130.6, 126.2, 125.3, 122.8, 110.7, 97.3, 61.9, 38.7, 25.1, 22.8; HRMS (ESI) m/z: [M+Na]+ Calcd for C18H27NO3Na 328.1889; Found 328.1891.
4.4. Substrate Scope of Diol, Aminoalcohol, and Diamine (Scheme 4)
4.4.1. Synthesis of Spiro[1,3-dioxolane-2,3′-indolin]-2′-one (2ah) [17]
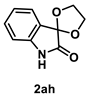
The reaction was performed according to the general procedure A using 147 mg (1.0 mmol) of 1a. The residue was purified by silica-gel column chromatography using hexane/AcOEt (4/1–2/1 [v/v]) to give 2ah (176 mg, 0.92 mmol, 92% yield) as a white solid.
White solid (176 mg, 0.92 mmol, 92% yield; mp 131–132 °C); IR (KBr) ν: 3214, 3041, 2998, 2969, 2896, 1743, 1621, 1473, 1216, 1085, 759 cm−1; 1H NMR (600 MHz, CDCl3) δ 8.90 (br s, 1H), 7.34 (d, J = 7.2 Hz, 1H), 7.28 (dt, J = 7.8, 1.2 Hz, 1H), 7.04 (dt, J = 7.8, 0.6 Hz, 1H), 6.82 (d, J = 8.4 Hz, 1H), 4.59–4.54 (m, 2H), 4.35–4.30 (m, 2H); 13C{1H} NMR (151 MHz, CDCl3) δ 176.0, 142.0, 131.7, 125.1, 124.5, 123.4, 111.0, 102.5, 65.9; HRMS (ESI) m/z: [M+Na]+ Calcd for C10H9NO3Na 214.0480; Found 214.0476.
4.4.2. Synthesis of 4′-Methylspiro[indoline-3,2′-[1,3]dioxolan]-2-one (2ai) (dr = 62:38)
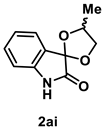
The reaction was performed according to the general procedure B using 147 mg (1.0 mmol) of 1a and 0.146 mL (2.0 mmol) of propylene glycol for 24 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (4/1–2/1 [v/v]) to give 2ai (145 mg, 0.71 mmol, 71% yield) as a yellow oil.
Yellow oil (145 mg, 0.71 mmol, 71% yield); IR (KBr) ν: 3272, 2977, 2931, 2894, 1743, 1623, 1473, 1199, 1025, 755 cm−1; 1H NMR (600 MHz, CDCl3) δ 8.71 (br s, 1H), 7.35 (d, J = 7.2 Hz, 0.6H), 7.34 (d, J = 7.8 Hz, 0.4H), 7.30–7.26 (m, 1H), 7.10 (dt, J = 7.8, 1.2 Hz, 0.6H), 7.04 (t, J = 7.8 Hz, 0.4H), 6.81 (d, J = 7.2 Hz, 1H), 4.99 (q, J = 6.6 Hz, 0.6H), 4.62–4.58 (m, 0.4H), 4.60 (t, J = 6.6 Hz, 0.6H), 4.42 (dd, J = 8.4, 6.0 Hz, 0.4H), 4.19 (t, J = 8.4 Hz, 0.4H), 3.79 (t, J = 7.2 Hz, 0.6H), 1.51 (d, J = 6.0 Hz, 1.2H), 1.45 (d, J = 6.0 Hz, 1.8H); 13C{1H} NMR (151 MHz, CDCl3) δ 176.09, 176.09, 141.9, 141.8, 131.7, 131.6, 125.5, 125.2, 124.9, 124.8, 123.40, 123.37, 110.88, 110.88, 103.0, 102.9, 74.8, 73.7, 72.2, 72.0, 18.9, 18.4; HRMS (ESI) m/z: [M+Na]+ Calcd for C11H11NO3Na 228.0637; Found 228.0638.
4.4.3. Synthesis of 4′-Phenylspiro[indoline-3,2′-[1,3]dioxolan]-2-one (2aj) (dr = 61:39)
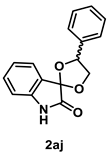
To a mixture of isatin 1a (147 mg, 1.0 mmol), 1-phenylethane-1,2-diol (276 mg, 2.0 mmol. 2.0 equiv.) and iodine (127 mg, 0.50 mmol, 50 mol %) were dissolved in THF (5.0 mL, 0.2 M). The mixture was stirred at reflux for 24 h in an oil bath. After the whole was quenched with sat. Na2S2O3, H2O (10 mL) was added to the mixture and extracted with AcOEt (3 × 10 mL). The combined organic layer was washed with brine (10 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica-gel column chromatography using hexane/AcOEt (4/1–2/1 [v/v]) to give 2aj (194 mg, 0.73 mmol, 73% yield) as a colorless oil.
Colorless oil (194 mg, 0.73 mmol, 73% yield); IR (KBr) ν: 3218, 3033, 2940, 2894, 1727, 1625, 1473, 1201, 1078, 752 cm−1; 1H NMR (600 MHz, CDCl3) δ 8.08 (br s, 0.6H), 8.05 (br s, 0.4H), 7.65 (d, J = 7.8 Hz, 0.6H), 7.50–7.48 (m, 2H), 7.45 (d, J = 7.2 Hz, 0.4H), 7.43–7.40 (m, 2H), 7.38–7.31 (m, 2H), 7.12–7.09 (m, 1H), 6.86 (d, J = 7.8 Hz, 0.6H), 6.85 (d, J = 7.8 Hz, 0.4H), 5.86 (t, J = 7.2 Hz, 0.6H), 5.39 (dd, J = 10.2, 6.0 Hz, 0.4H), 4.90 (t, J = 7.8 Hz, 0.6H), 4.61 (dd, J = 6.0, 9.0 Hz, 0.4H), 4.51 (t, J = 9.6 Hz, 0.4H), 4.13 (t, J = 7.8 Hz, 0.6H); 13C{1H} NMR (151 MHz, CDCl3) δ 175.5, 175.4, 141.9, 141.7, 138.5, 136.7, 131.9, 131.8, 128.9, 128.84, 128.77, 128.7, 127.6, 126.5, 125.6, 125.4, 125.3, 124.5, 123.56, 123.56, 110.9, 110.8, 103.5, 103.0, 80.5, 78.7, 73.0, 72.7; HRMS (ESI) m/z: [M+Na]+ Calcd for C16H13NO3Na 290.0793; Found 290.0795.
4.4.4. Synthesis of Spiro[1,3-dioxane-2,3′-indolin]-2′-one (2ak) [30]
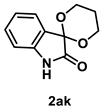
The reaction was performed according to the general procedure B using 147 mg (1.0 mmol) of 1a and 0.145 mL (2.0 mmol) of 1,3-propanediol for 24 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (4/1–2/1 [v/v]) to give 2ak (169 mg, 0.82 mmol, 82% yield) as a pale-yellow solid.
Pale-yellow solid (169 mg, 0.82 mmol, 82% yield; mp 148–150 °C); IR (KBr) ν: 3388, 3091, 2965, 2927, 2892, 1708, 1623, 1473, 1243, 1087, 748 cm−1; 1H NMR (600 MHz, CDCl3) δ 8.86 (br s, 1H), 7.43 (d, J = 7.8 Hz, 1H), 7.24 (dt, J = 7.8, 1.2 Hz, 1H), 7.03 (dt, J = 7.8, 1.2 Hz, 1H), 6.73 (d, J = 7.8 Hz, 1H), 4.98 (dt, J = 12.0, 2.4 Hz, 2H), 4.01 (ddd, J = 12.0, 4.8, 1.8 Hz, 2H), 2.41–2.33 (m, 1H), 1.67 (ddd, J = 13.8, 2.4, 2.4 Hz, 1H); 13C{1H} NMR (151 MHz, CDCl3) δ 174.2, 140.3, 130.9, 127.7, 124.2, 123.2, 110.3, 93.9, 61.2, 25.3; HRMS (ESI) m/z: [M+Na]+ Calcd for C11H11NO3Na 228.0637; Found 228.0636.
4.4.5. Synthesis of Spiro[1,3-dioxepene-2,3′-indolin]-2′-one (2al)
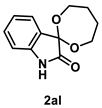
The reaction was performed according to the general procedure B using 147 mg (1.0 mmol) of 1a and 0.177 mL (2.0 mmol) of 1,4 butanediol for 24 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (4/1–2/1 [v/v]) to give 2al (122 mg, 0.56 mmol, 56% yield) as an orange solid.
Orange solid (122 mg, 0.56 mmol, 56% yield; mp 176–178 °C); IR (KBr) ν: 3326, 2983, 2952, 2911, 2836, 1727, 1619, 1471, 1199, 1124, 1051, 769 cm−1; 1H NMR (600 MHz, CDCl3) δ 9.11 (br s, 1H), 7.45 (d, J = 7.2 Hz, 1H), 7.25 (dt, J = 7.8, 1.2 Hz, 1H), 7.02 (dt, J = 7.8, 0.6 Hz. 1H), 6.86 (d, J = 7.2 Hz, 1H), 4.45–4.42 (m, 2H), 4.10–4.07 (m, 2H), 1.90–1.78 (m, 4H); 13C{1H} NMR (151 MHz, CDCl3) δ 175.3, 140.4, 130.5, 128.4, 124.0, 122.9, 111.1, 96.4, 65.0, 29.8; HRMS (ESI) m/z: [M+Na]+ Calcd for C12H13NO3Na 242.0793; Found 242.0796.
4.4.6. Synthesis of 3′-Methylspiro[indoline-3,2′-oxazolidin]-2-one (2am)
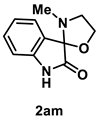
The reaction was performed according to the general procedure B using 147 mg (1.0 mmol) of 1a and 0.160 mL (2.0 mmol) of 2-(methylamino)ethanol for 2 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (4/1–2/1 [v/v]) to give 2am (204 mg, 1.0 mmol, quant.) as a yellow oil.
Yellow oil (204 mg, 1.0 mmol, quant.); IR (KBr) ν: 3255, 2969, 2948, 2896, 2856, 1733, 1621, 1471, 1211, 1068, 757 cm−1; 1H NMR (600 MHz, CDCl3) δ 8.33 (br s, 1H), 7.35 (d, J = 7.2 Hz, 1H), 7.28 (dt, J = 7.8, 1.2 Hz, 1H), 7.06 (dt, J = 7.8, 0.6 Hz, 1H), 6.82 (d, J = 7.8 Hz, 1H), 4.41 (dt, J = 7.8, 3.0 Hz, 1H), 4.31 (q, J = 7.2 Hz, 1H), 3.58 (q, J = 8.4 Hz, 1H), 3.38–3.35 (m, 1H), 2.30 (s, 3H); 13C{1H} NMR (151 MHz, CDCl3) δ 177.4, 142.2, 131.0, 125.9, 125.7, 123.4, 110.4, 94.5, 66.6, 51.6, 34.6; HRMS (ESI) m/z: [M+Na]+ Calcd for C11H12N2O2Na 227.0796; Found 227.0795.
4.4.7. Synthesis of 1,3-Dimethylspiro[imidazolidine-2,3′-indolin]-2′-one (2an) [74]
A mixture of isatin 1a (147 mg, 1.0 mmol) and iodide (25.4 mg, 0.10 mmol, 10 mol %) was dissolved in THF (5.0 mL, 0.2 M). The mixture was stirred at reflux in an oil bath and 0.215 mL (2.0 mmol, 2.0 equiv.) of N,N’-dimethylethylenediamine was dropped into the mixture. The resulting mixture was stirred for a further 10 min. After the whole was quenched with sat. Na2S2O3, H2O (10 mL) was added to the mixture and extracted with AcOEt (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by recrystallization using MeCN to give 2an (145 mg, 0.67 mmol, 67% yield) as a pale-yellow solid.
Pale-yellow solid (145 mg, 0.67 mmol, 67% yield; mp 155 °C (decomposition); IR (KBr) ν: 3421, 2969, 2938, 2865, 1716, 1617, 1473, 1195, 1049, 750 cm−1; 1 H NMR (600 MHz, CDCl3) δ 8.64 (br s, 1H), 7.31 (d, J = 7.2 Hz, 1H), 7.25 (dt, J = 7.8, 1.2 Hz, 1H), 7.05 (dt, J = 7.2, 1.2 Hz, 1H), 6.83 (d, J = 7.8 Hz, 1H), 3.40–3.37 (m, 2H), 3.33–3.30 (m, 2H), 2.22 (s, 6H); 13 C{1H} NMR (151 MHz, CDCl3) δ 178.4, 142.6, 130.2, 126.4, 126.1, 123.1, 110.0, 86.5, 51.2, 35.8; HRMS (ESI) m/z: [M+Na] + Calcd for C12H15N3ONa 240.1113; Found 240.1111.
4.5. Substrate Scope of Aromatic Compounds (Scheme 5)
4.5.1. Synthesis of 3,3-Bis(4-hydroxyphenyl)indolin-2-one (2ao) [55]
To a mixture of isatin 1a (1.0 mmol), phenol (188 mg, 2.0 mmol. 2.0 equiv.) and iodine (25.4 mg, 0.10 mmol, 10 mol %) were dissolved in THF (5.0 mL, 0.2 M). The mixture was stirred at 60 °C in an oil bath for 48 h and iodine (25.4 mg, 0.10 mmol, 10 mol %) was added to the mixture. After stirring the mixture further 48 h, the whole was quenched with sat. Na2S2O3 (10 mL) and extracted with AcOEt (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica-gel column chromatography using hexane/AcOEt (3/1 [v/v]) to give 2ao (193 mg, 0.60 mmol, 60% yield) as a white solid.
White solid (193 mg, 0.60 mmol, 60% yield; mp 258–260 °C); IR (KBr) ν: 3345, 3255, 3019, 2883, 1673, 1612, 1511, 1236, 827, 746 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 10.57 (br s, 1H), 9.38 (br s, 2H), 7.19 (dt, J = 7.8, 1.2 Hz, 1H), 7.15 (d, J = 7.2 Hz, 1H), 6.97 (dt, J = 7.8, 1.2 Hz, 1H), 6.92 (d, J = 8.4 Hz, 4H), 6.90 (d, J = 7.8 Hz, 1H), 6.66 (d, J = 9.0 Hz, 4H); 13C{1H} NMR (151 MHz, DMSO-d6) δ 179.0, 156.3, 141.2, 134.2, 132.5, 129.1, 127.9, 125.8, 121.7, 115.0, 109.8, 60.7; HRMS (ESI) m/z: [M+Na]+ Calcd for C20H15NO3Na 340.0950; Found 340.0950.
4.5.2. Synthesis of 3,3-Bis(2,4-dimethoxyphenyl)indolin-2-one (2ap) [55]
The reaction was performed according to the general procedure B using 147 mg (1.0 mmol) of 1a and 258 mg (2.0 mmol) of 1,3-dimethoxybenzene for 48 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (3/1 [v/v]) to give 2ap (81 mg, 0.20 mmol, 20% yield) as a pale-yellow solid.
Pale-yellow solid (81 mg, 0.20 mmol, 20% yield; mp 173–175 °C); IR (KBr) ν: 3183, 2987, 2935, 2902, 2836, 1708, 1610, 1211, 1133, 1035, 759 cm−1; 1H NMR (600 MHz, CDCl3) δ 8.50 (br s, 1H), 7.22 (d, J = 9.0 Hz, 1H), 7.21 (d, J = 7.2 Hz, 1H), 7.11 (dt, J = 7.8, 1.2 Hz, 1H), 6.89 (dt, J = 7.8, 1.2 Hz, 1H), 6.82 (d, J = 7.2 Hz, 1H), 6.81 (d, J = 7.8 Hz, 1H), 6.463 (s, 1H), 6.455 (s, 1H), 6.43 (d, J = 9.0 Hz, 1H), 6.37 (d, J = 8.4 Hz, 1H), 3.78 (s, 3H), 3.77 (s, 3H), 3.64 (s, 3H), 3.46 (s, 3H); 13C{1H} NMR (151 MHz, CDCl3) δ 181.3, 160.4, 160.2, 159.1, 158.7, 140.9, 135.6, 131.0, 129.8, 127.2, 125.6, 121.8, 120.8, 120.4, 109.1, 104.4, 104.3, 100.3, 99.9, 59.5, 55.8, 55.44, 55.40, 55.3; HRMS (ESI) m/z: [M+Na]+ Calcd for C24H23NO5Na 428.1474; Found 428.1471.
4.5.3. Synthesis of 3,3-Bis(4-aminophenyl)indolin-2-one (2aq)
The reaction was performed according to the general procedure B using 147 mg (1.0 mmol) of 1a and 183 mg (2.0 mmol) of aniline for 24 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (1/1 [v/v]) to give 2aq (270 mg, 0.86 mmol, 86% yield) as a white solid.
White solid (270 mg, 0.86 mmol, 86% yield; mp 227–229 °C (decomposition)); IR (KBr) ν: 3397, 3326, 3224, 3027, 2998, 1704, 1616, 1511, 1184, 1099, 761 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 10.44 (br s, 1H), 7.16 (t, J = 7.8 Hz, 1H), 7.10 (d, J = 7.2 Hz, 1H), 6.95 (t, J = 7.8 Hz, 1H), 6.87 (d, J = 7.8 Hz, 1H), 6.78 (d, J = 9.0 Hz, 4H), 6.45 (d, J = 8.4 Hz, 4H), 5.12 (br s, 4H); 13C{1H} NMR (151 MHz, DMSO-d6) δ 179.5, 147.1, 141.2, 134.8, 129.6, 128.6, 127.5, 125.7, 121.5, 113.6, 109.6, 60.6; HRMS (ESI) m/z: [M+Na]+ Calcd for C20H17N3ONa 338.1269; Found 338.1270.
4.5.4. Synthesis of 3,3-Bis(4-(methylamino)phenyl)indolin-2-one (2ar) [55]
The reaction was performed according to the general procedure B using 147 mg (1.0 mmol) of 1a and 217 mg (2.0 mmol) of N-methylaniline for 24 h. The residue was purified by silica-gel column chromatography using hexane/AcOEt (1/1 [v/v]) to give 2ar (220 mg, 0.64 mmol, 64% yield) as a white solid.
White solid (220 mg, 0.64 mmol, 64% yield; mp 237–239 °C); IR (KBr) ν: 3419, 3384, 3018, 2979, 2881, 2811, 1708, 1612, 1521, 1471, 1186, 813, 750 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 9.50 (br s, 1H), 7.19 (d, J = 7.2 Hz, 1H), 7.16 (dt, J = 7.8, 1.2 Hz, 1H), 7.14 (d, J = 8.4 Hz, 4H), 7.01 (dt, J = 7.8 Hz, 1H), 6.89 (d, J = 8.4 Hz, 1H), 6.53 (d, J = 9.0 Hz, 4H), 3.73 (br s, 2H), 2.78 (s, 6H); 13C{1H} NMR (151 MHz, DMSO-d6) δ 181.8, 148.4, 140.5, 135.0, 130.5, 129.3, 127.7, 125.9, 122.5, 112.3, 110.4, 61.8, 30.8; HRMS (ESI) m/z: [M+Na]+ Calcd for C22H21N3ONa 366.1582; Found 366.1578.
4.5.5. Synthesis of 3,3-Bis[1H-indolin-5-yl]indolin-2-one (2as)
The reaction was performed according to the general procedure B using 147 mg (1.0 mmol) of 1a and 238 mg (2.0 mmol) of indoline for 18 h. The residue was purified by recrystallization using hexane/AcOEt to give 2as (362 mg, 0.98 mmol, 98% yield) as a pale-brown solid.
Pale-brown solid (362 mg, 0.98 mmol, 98% yield; mp 191–193 °C (decomposition)); IR (KBr) ν: 3367, 3023, 2935, 2848, 1704, 1616, 1471, 1251, 1105, 748 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 10.47 (br s, 1H), 7.16 (dt, J = 7.8, 1.2 Hz, 1H), 7.13 (d, J = 7.8 Hz, 1H), 6.95 (dt, J = 7.8, 1.2 Hz, 1H), 6.88 (d, J = 7.2 Hz, 1H), 6.82 (s, 2H), 6.69 (dd, J = 7.8, 1.8 Hz, 2H), 6.38 (d, J = 8.4 Hz, 2H), 5.54 (br s, 2H), 3.35 (t, J = 8.4 Hz, 4H), 2.80 (t, J = 9.0 Hz, 4H); 13C{1H} NMR (151 MHz, DMSO-d6) δ 179.5, 151.4, 141.1, 134.9, 130.9, 128.7, 127.5, 126.8, 125.8, 124.0, 121.5, 109.6, 107.6, 61.1, 46.6, 29.2; HRMS (ESI) m/z: [M+Na]+ Calcd for C24H21N3ONa 390.1582; Found 390.1584.
4.5.6. Synthesis of 3,3-Bis[1H-indol-3-yl]indolin-2-one (2at) [55]
To a mixture of isatin 1a (1.0 mmol) in THF (5.0 mL, 0.2 M), indole (234 mg, 2.0 mmol. 2.0 equiv.) and iodine (25.4 mg, 0.10 mmol, 10 mol %) were added. The mixture was stirred at room temperature for 15 min. After the whole was quenched with sat. Na2S2O3 (10 mL), H2O (10 mL) was added to the mixture and extracted with AcOEt (3 × 10 mL). The combined organic layers were washed with brine (10 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by recrystallization using hexane/AcOEt/CHCl3 to give 2at (349 mg, 0.96 mmol, 96% yield) as a pale-brown solid.
Pale-brown solid (349 mg, 0.96 mmol, 96% yield; mp >250 °C); IR (KBr) ν: 3554, 3396, 3340, 3058, 2965, 2929, 1685, 1616, 1469, 1338, 1099, 752 cm−1; 1H NMR (600 MHz, DMSO-d6) δ 10.94 (br d, J = 2.4 Hz, 1H), 10.58 (br s, 1H), 7.33 (d, J = 8.4 Hz, 2H), 7.23–7.20 (m, 4H), 7.00 (dt, J = 7.2, 0.6 Hz, 2H), 6.97 (d, J = 8.4 Hz, 1H), 6.91 (dt, J = 7.8, 1.2 Hz, 1H), 6.83 (d, J = 2.4 Hz, 2H), 6.78 (t, J = 7.8 Hz, 2H); 13C{1H} NMR (151 MHz, DMSO-d6) δ 178.7, 141.3, 136.9, 134.6, 127.8, 125.7, 124.9, 124.3, 121.4, 120.9, 120.8, 118.2, 114.3, 111.6, 109.5, 52.5; HRMS (ESI) m/z: [M+Na]+ Calcd for C24H17N3ONa 386.1269; Found 386.1269.
4.6. Gram Scale Synthesis of 3,3-Dimethoxyisatin (Scheme 6)
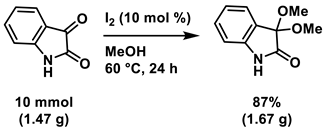
A mixture of isatin 1a (10 mmol, 1.47 g) and iodine (254 mg, 1.0 mmol, 10 mol %) was dissolved in MeOH (50 mL, 0.2 M). The mixture was stirred at 60 °C in an oil bath for 24 h. After the whole was quenched with sat. Na2S2O3, H2O (100 mL) was added to the mixture and extracted with AcOEt (3 × 100 mL). The combined organic layer was washed with brine (100 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The residue was purified by silica-gel column chromatography using hexane/AcOEt (4/1–2/1 [v/v]) to give 2aa (1.67 g, 8.7 mmol, 87% yield).
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/chemistry7020043/s1. The Supplementary Materials contain analytical data including 1H- and 13C-NMR spectra.
Author Contributions
Conceptualization, T.A.; investigation, T.A.; resources, T.A.; visualization, T.A.; experiments, K.T. and S.A.; writing—original draft preparation, T.A.; writing—review and editing, K.T., S.A. and T.A. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partly supported by JSPS KAKENHI (Grant Number 22K06503). This work was also supported by JST OU-SPRING, Grant Number JPMJSP2126 (K.T.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and Supplementary Materials.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jao, C.-W.; Hung, T.-H.; Chang, C.-F.; Chuang, T.-H. Chemical Constituents of Phaius mishmensis. Molecules 2016, 21, 1605. [Google Scholar] [CrossRef] [PubMed]
- Wuts, P.G.M.; Michigan, K. Protection for the Carbonyl Group. Greene’s Protective Groups in Organic Synthesis, 5th ed.; Academic Press: New York, NY, USA, 2014; pp. 554–558. [Google Scholar]
- Rajopadhye, M.; Popp, F.D. Potential Anticonvulsants. 11. Synthesis and Anticonvulsant Activity of Spiro[1,3-dioxolane-2,3′-indolin]-2′-ones and Structural Analogues. J. Med. Chem. 1988, 31, 1001–1005. [Google Scholar] [CrossRef] [PubMed]
- Zapata-Sudo, G.; Pontes, L.B.; Gabriel, D.; Mendes, T.C.F.; Ribeiro, N.M.; Pinto, A.C.; Trachez, M.M.; Sudo, R.T. Sedative-hypnotic profile of novel isatin ketals. Pharmacol. Biochem. Behav. 2007, 86, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Geronikaki, A.; Babaev, E.; Dearden, J.; Dehaen, W.; Filimonov, D.; Galaeva, I.; Krajneva, V.; Lagunin, A.; Macaev, F.; Molodavkin, G.; et al. Design, synthesis, computational and biological evaluation of new anxiolytics. Bioorg. Med. Chem. 2004, 12, 6559–6568. [Google Scholar] [CrossRef]
- Marques, C.S.; González-Bakkker, A.; Padrón, J.M.; Burke, A.J. Easy access to Ugi-derived isatin-peptoids and their potential as small-molecule anticancer reagents. New J. Chem. 2023, 47, 743–750. [Google Scholar] [CrossRef]
- Dung, D.T.M.; Dung, P.T.P.; Oanh, D.T.K.; Vu, T.K.; Hahn, H.; Han, B.W.; Pyo, M.; Kim, Y.G.; Han, S.-B.; Nam, N.-H. Exploration of novel 5′(7′)-substituted-2′-oxospiro[1,3]dioxolane-2,3′-indoline-based N-hydroxypropenamides as histone deacetylase inhibitors and antitumor agents. Arab. J. Chem. 2017, 10, 465–472. [Google Scholar] [CrossRef]
- Marques, C.S.; López, O.; Leitzbach, L.; Fernández-Bolaños, J.G.; Stark, H.; Burke, A.J. Survey of New, Small-Molecule Isatin-Based Oxindole Hybrids as Multi-Targeted Drugs for the Treatment of Alzheimer’s Disease. Synthesis 2022, 54, 4304–4319. [Google Scholar] [CrossRef]
- Singh, G.S.; Desta, Z.Y. Isatins As Privileged Molecules in Design and Synthesis of Spiro-Fused Cyclic Frameworks. Chem. Rev. 2012, 112, 6104–6155. [Google Scholar] [CrossRef]
- Pinder, J.L.; Weinreb, S.M. Preliminary feasibility studies on total synthesis of the unusual marine bryozoan alkaloids chartellamide A and B. Tetrahedron Lett. 2003, 44, 4141–4143. [Google Scholar] [CrossRef]
- Castaldi, M.P.; Troast, D.M.; Porco, J.A., Jr. Stereoselective Synthesis of Spirocyclic Oxindoles via Prins Cyclization. Org. Lett. 2009, 11, 3362–3365. [Google Scholar] [CrossRef]
- Zhang, Y.; Panek, J.S. Stereocontrolled Synthesis of Spirooxindoles through Lewis Acid-Promoted [5 + 2]-Annulation of Chiral Silyl Alcohols. Org. Lett. 2009, 11, 3366–3369. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Crane, E.A.; Scheidt, K.A. Highly Stereoselective Brønsted Acid Catalyzed Synthesis of Spirooxindole Pyrans. Org. Lett. 2011, 13, 3086–3089. [Google Scholar] [CrossRef] [PubMed]
- Wenkert, E.; Hudlicky, T. Reactions of Isatin Dimethyl Ketal and Its Ethyl Imino Ether with Methyllithium. Synth. Commun. 1977, 7, 541–547. [Google Scholar] [CrossRef]
- Li, H.; Bonderoff, S.A.; Cheng, B.; Padwa, A. Model Studies Directed toward the Alkaloid Mersicarpine Utilizing a Rh(II)-Catalyzed Insertion/Cycloaddition Sequence. J. Org. Chem. 2014, 79, 392–400. [Google Scholar] [CrossRef]
- Marques, C.S.; González-Bakker, A.; Padrón, J.M. The Ugi4CR as effective tool to access promising anticancer isatin-based α-acetamide carboxamide oxindole hybrids. Beilstein J. Org. Chem. 2024, 20, 1213–1220. [Google Scholar] [CrossRef]
- Dou, X.; Yao, W.; Jiang, C.; Lu, Y. Enantioselective N-alkylation of isatins and synthesis of chiral N-alkylated indoles. Chem. Commun. 2014, 50, 11354–11357. [Google Scholar] [CrossRef]
- Marques, C.S.; McArdle, P.; Erxleben, A.; Burke, A.J. Accessing New 5-a-(3,3-Disubstituted Oxindole)-Benzylamine Derivatives from Isatin: Stereoselective Organocatalytic Three Component Petasis Reaction. Eur. J. Org. Chem. 2020, 2020, 3622–3634. [Google Scholar] [CrossRef]
- Vine, K.L.; Locke, J.M.; Ranson, M.; Pyne, S.G.; Bremner, J.B. In vitro cytotoxicity evaluation of some substituted isatin derivatives. Bioorg. Med. Chem. 2007, 15, 931–938. [Google Scholar] [CrossRef]
- Dong, J.-L.; Yu, L.-S.-H.; Xie, J.-W. A Simple and Versatile Method for the Formation of Acetals/Ketals Using Trace Conventional Acids. ACS Omega 2018, 3, 4975–4984. [Google Scholar] [CrossRef]
- Azzena, U.; Carraro, M.; Corrias, M.; Crisafulli, R.; de Luca, L.; Gaspa, S.; Nuvoli, L.; Pintus, S.; Polese, R.; Sanna, M.; et al. Ammonium Salts Catalyzed Acetalization Reactions in Green Ethereal Solvents. Catalysts 2020, 10, 1108. [Google Scholar] [CrossRef]
- Banik, B.K.; Chapa, M.; Marquez, J.; Cardona, M. A remarkable iodine-catalyzed protection of carbonyl compounds. Tetrahedron Lett. 2005, 46, 2341–2343. [Google Scholar] [CrossRef]
- Khan, M.A.; Teixeira, I.F.; Li, M.J.; Koito, Y.; Tsang, S.C.E. Graphitic carbon nitride catalysed photoacetalization of aldehydes/ketones under ambient conditions. Chem. Commun. 2016, 52, 2772–2775. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.W.; Jeong, K.; Seo, J.Y.; Baek, K.-Y. Facile Synthesis of Biomass-Derived Degradable Poly(ketal-ester) Elastomers Using Dual-Acidic Catalysts. ACS Appl. Polym. Mater. 2024, 6, 3507–3516. [Google Scholar] [CrossRef]
- Putro, W.S.; Iijima, S.; Matsumoto, S.; Hamura, S.; Yabushita, M.; Tomishige, K.; Fukaya, N.; Choi, J.-C. Sustainable synthesis of diethyl carbonate from carbon dioxide and ethanol featuring acetals as regenerable dehydrating agents. RSC Sustain. 2024, 2, 1613–1620. [Google Scholar] [CrossRef]
- Steuernagel, D.; Wagenknecht, H.-A. Photocatalytic Synthesis of Acetals and Ketals from Aldehydes and Silylenolethers without the use of Acids. Chem. Eur. J. 2023, 29, e202203767. [Google Scholar] [CrossRef]
- Alzard, R.H.; Alsaedi, S.; Alseiari, S.; Aljasmi, S.; El-Maghraby, H.F.; Poulose, V.; Hassan, A.; Kamel, M.; Ali, A.; Abdel-Hafiez, M.; et al. Heterogeneous Acetalization of Benzaldehyde over Lanthanoide Oxalate Metal-Organic Frameworks. ACS Omega 2024, 9, 37386–37395. [Google Scholar] [CrossRef]
- Webber, S.E.; Tikhe, J.; Worland, S.T.; Fuhrman, S.A.; Hendrickson, T.F.; Matthews, D.A.; Love, R.A.; Patick, A.K.; Meador, J.W.; Ferre, R.A.; et al. Design, Synthesis, and Evaluation of Nonpeptidic Inhibitors of Human Rhinovirus 3C Protease. Med. Chem. 1996, 39, 5072–5082. [Google Scholar] [CrossRef]
- Rigby, J.H.; Brouet, S.A. Metal-mediated reactions of aryl isocyanates with dimethoxycarbene to form isatin derivatives. Tetrahedron Lett. 2013, 54, 2542–2545. [Google Scholar] [CrossRef]
- Ribeiro, N.M.; da Chunha Pinto, A.; da Silva, B.V.; de Almeida Violante, F.; Dias, M.O. A fast, efficient and eco-friendly procedure to prepare isatin ketals. Cat. Commun. 2007, 8, 2130–2136. [Google Scholar] [CrossRef]
- Shimizu, K.; Higuchi, T.; Takasugi, E.; Hatamachi, T.; Kodama, T.; Satsuma, A. Characterization of Lewis acidity of cation-exchanged montmorillonite K-10 clay as effective heterogeneous catalyst for acetylation of alcohol. J. Mol. Catal. A Chem. 2008, 284, 89–96. [Google Scholar] [CrossRef]
- Lan, J.; Jiang, G.; Yang, J.; Zhu, H.; Le, Z.; Xie, Z. α-Chymotrypsin-Induced Acetalization of Aldehydes and Ketones with Alcohols. Synthesis 2020, 52, 2121–2126. [Google Scholar]
- Yusubov, M.S.; Zhdankin, V.V. Iodine catalysis: A green alternative to transition metals in organic chemistry and technology. Resour.-Effic. Technol. 2015, 1, 49–67. [Google Scholar] [CrossRef]
- Ren, Y.-M.; Cai, C.; Yang, R.-C. Molecular iodine-catalyzed multicomponent reactions: An efficient catalyst for organic synthesis. RSC Adv. 2013, 3, 7182–7204. [Google Scholar] [CrossRef]
- Jereb, M.; Vrazic, D.; Zupan, M. Iodine-catalyzed transformation of molecules containing oxygen functional groups. Tetrahedron 2011, 67, 1355–1387. [Google Scholar] [CrossRef]
- Finkbeiner, P.; Nachtsheim, B.J. Iodine in Modern Oxidation Catalysis. Synthesis 2013, 45, 979–999. [Google Scholar]
- Jadhav, P.M.; Rode, A.B.; Kótai, L.; Pawar, R.P.; Tekale, S.U. Revisiting applications of molecular iodine in organic synthesis. New J. Chem. 2021, 45, 16389–16425. [Google Scholar] [CrossRef]
- Wang, J.-Q.; Zuo, Z.-Y.; He, W. Recent Advances of Green Catalytic System I2/DMSO in C–C and C–Heteroatom Bonds Formation. Catalysts 2022, 12, 821. [Google Scholar] [CrossRef]
- Chander, M.; Ram, S.; Sharma, P.K. A review on Molecular Iodine Catalyzed/Mediated Multicomponent Reactions. Asian J. Org. Chem. 2023, 12, e202200616. [Google Scholar]
- Minakata, S.; Kano, D.; Oderaotoshi, Y.; Komatsu, M. Silica-Water Reaction Media: Its Application to the Formation and Ring Opening of Aziridines. Angew. Chem. Int. Ed. 2003, 43, 79–81. [Google Scholar] [CrossRef]
- Minakata, S.; Miwa, H.; Yamamoto, K.; Hirayama, A.; Okumura, S. Diastereodivergent Intramolecular 1,2-Diamination of Unactivated Alkenes Enabled by Iodine Catalysis. J. Am. Chem. Soc. 2021, 143, 4112–4118. [Google Scholar] [CrossRef]
- Kitamura, T.; Oyamada, J.; Higashi, M.; Kishikawa, Y. Molecular Iodine as a Catalyst for Alkene Difluorination. J. Org. Chem. 2024, 89, 5896–5900. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, T.; Yamada, K.; Yoshida, H.; Tomisaka, Y.; Nishi, T.; Abe, T. Silver-Mediated Intramolecular Friedel–Crafts-Type Cyclizations of 2-Benzyloxy-3-bromoindolines: Synthesis of Isochromeno[3,4-b]indolines and 3-Arylindoles. Synlett 2019, 30, 2247–2252. [Google Scholar] [CrossRef]
- Abe, T.; Kosaka, Y.; Asano, M.; Harasawa, N.; Mishina, A.; Nagasue, M.; Sugimoto, Y.; Katakawa, K.; Sueki, S.; Anada, M.; et al. Direct C4-Benzylation of Indoles via Tandem Benzyl Claisen/Cope Rearrangements. Org. Lett. 2019, 21, 826–829. [Google Scholar] [CrossRef]
- Yamashiro, T.; Abe, T.; Tanioka, M.; Kamino, S.; Sawada, D. cis-3-Azido-2-methoxyindolines as safe and stable precursors to overcome the instability of fleeting 3-azidoindoles. Chem. Commun. 2021, 57, 13381–13384. [Google Scholar] [CrossRef]
- Yamashiro, T.; Abe, T.; Sawada, D. Synthesis of 2-monosubstituted indolin-3-ones by cine-substitution of 3-azido-2-methoxyindolines. Org. Chem. Front. 2022, 9, 1897–1903. [Google Scholar] [CrossRef]
- Abe, T.; Kosaka, Y.; Kawasaki, T.; Ohata, Y.; Yamashiro, T.; Yamada, K. Revisiting 2-Alkoxy-3-bromoindolines: Control C-2 vs. C-3 Elimination for Regioselective Synthesis of Alkoxyindoles. Chem. Pharm. Bull. 2020, 68, 555–558. [Google Scholar] [CrossRef]
- Sugitate, K.; Yamashiro, T.; Takahashi, I.; Yamada, K.; Abe, T. Oxytrofalcatin Puzzle: Total Synthesis and Structural Revision of Oxytrofalcatins B and C. J. Org. Chem. 2023, 88, 9920–9926. [Google Scholar] [CrossRef]
- Kimata, M.; Abe, T. Total Synthesis of the Proposed Structure of Indolyl 1,2-Propanediol Alkaloids, 1-(1H-Indol-3-yloxy)propan-2-ol. Chemistry 2023, 5, 2772–2784. [Google Scholar] [CrossRef]
- Hirao, S.; Yamashiro, T.; Kohira, K.; Mishima, N.; Abe, T. 2,3-Dimethoxyindolines: A latent electrophile for SNAr reactions triggered by indium catalysts. Chem. Commun. 2020, 56, 5139–5142. [Google Scholar] [CrossRef]
- Tokushige, K.; Yamashiro, T.; Hirao, S.; Abe, T. Aluminum-Catalyzed Cross Selective C3–N1’ Coupling Reactions of N-Methoxyindoles with Indoles. Chemistry 2023, 5, 452–462. [Google Scholar] [CrossRef]
- Abe, T.; Hirao, S. Rapid access to indole-fused bicyclo[2.2.2]octanones by merging the umpolung strategy and molecular iodine as a green catalyst. Org. Biomol. Chem. 2020, 18, 4193–4197. [Google Scholar] [CrossRef] [PubMed]
- Pitzer, L.; Schäfers, F.; Glorius, F. Rapid Assessment of the Reaction-Condition-Based Sensitivity of Chemical Transformations. Angew. Chem. Int. Ed. 2019, 58, 8572–8576. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.-L.; Hsu, Y.-L.; Jao, C.-W. Indole Alkaloids from Cephalanceropsis gracilis. J. Nat. Prod. 2006, 69, 1467–1470. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.; Tyagi, A.; Yadav, N.; Mahato, R.; Hazra, C.K. Lambert Salt-Initiated Development of Friedel–Crafts Reaction on Isatin to Access Distinct Derivatives of Oxindoles. J. Org. Chem. 2021, 86, 17833–17847. [Google Scholar] [CrossRef]
- Garrido, F.; Ibanez, J.; Gonalons, E.; Giraldez, A. Synthesis and laxative properties of some derivative esters of 3,3-bis-(4-hydroxyphenyl)-2-indolinone. Eur. J. Med. Chem. 1975, 10, 143–146. [Google Scholar] [CrossRef]
- Song, H.M.; Lee, H.J.; Kim, H.R.; Ryu, K.; Kim, J.N. Friedel–Crafts Type Reactions of Some Activated Cyclic Ketones with Phenol Derivatives. Synth. Commun. 1999, 29, 3303–3311. [Google Scholar] [CrossRef]
- Paira, P.; Hazra, A.; Kumar, S.; Paira, R.; Sahu, K.B.; Naskar, S.; Saha, P.; Mondal, S.; Maity, A.; Banerjee, S.; et al. Efficient synthesis of 3,3-diheteroaromatic oxindole analogues and their in vitro evaluationsfor spermicidal potential. Bioorg. Med. Chem. Lett. 2009, 19, 4786–4789. [Google Scholar] [CrossRef]
- Uddin, M.K.; Reignier, S.G.; Coulter, T.; Montalbetti, C.; Grånäs, C.; Butcher, S.; Krog-Jensen, C.; Felding, J. Syntheses and antiproliferative evaluation of oxyphenisatin derivatives. Bioorg. Med. Chem. Lett. 2007, 17, 2854–2857. [Google Scholar] [CrossRef]
- Karu, S.K.; Pilli, N.; Malapaka, C. An Efficient Multi-Component Double Friedel–Crafts Alkylation of Isatin: Access to Unsymmetrical and Symmetrical 3,3-Diaryl oxindoles. ChestrySelects 2024, 9, e202403328. [Google Scholar] [CrossRef]
- Kobayashi, M.; Aoki, S.; Gato, K.; Matsunami, K.; Kurosu, M.; Kitagawa, I. Marine Natural Products. XXXIV. Trisindoline, a New Antibiotic Indole Trimer, produced by a Bacterium of Vibrio sp. Separated from the Marine Sponge Hyrtios altum. Chem. Pharm. Bull. 1994, 12, 2449–2451. [Google Scholar] [CrossRef]
- Abe, T.; Nakamura, S.; Yanada, R.; Choshi, T.; Hibino, S.; Ishikura, M. One-Pot Construction of 3,3′-Bisindolylmethanes through Bartoli Indole Synthesis. Org. Lett. 2013, 15, 3622–3625. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.V.S.; Rajeswari, N.; Sarangapani, M.; Prashanthi, Y.; Ganji, R.J. Iodine-catalyzed condensation of isatin with indoles: A facile synthesis of di(indolyl)indolin-2-ones and evaluation of their cytotoxicity. Bioorg. Med. Chem. Lett. 2012, 22, 2460–2463. [Google Scholar] [CrossRef] [PubMed]
- Duhamel, T.; Stein, C.J.; Martínez, C.; Reiher, M.; Muñiz, K. Engineering Molecular Iodine Catalysis for AlkylNitrogen Bond Formation. ACS Catal. 2018, 8, 3918–3925. [Google Scholar] [CrossRef]
- Lu, Y.; Chen, C.; Zhu, H.; Luo, Z.; Zhang, Y. Highly efficient and fast synthesis of di-iodinated succinimide derivatives from 1,6-enyne and I2 under air at room temperature. Green Chem. 2022, 24, 8021–8028. [Google Scholar] [CrossRef]
- Meng, Z.; Shi, M.; Wie, Y. Iodine radical mediated cascade [3 + 2] carbocyclization of ene-vinylidecyclopropanes with thiols and selenols via photoredox catalysis. Org. Chem. Front. 2024, 11, 1395–1403. [Google Scholar] [CrossRef]
- Huang, H.-Y.; Wu, H.-R.; Wei, F.; Wang, D.; Liu, L. Iodine-Catalyzed Direct Olefination of 2-Oxindoles and Alkenes via Cross-Dehydrogenative Coupling (CDC) in Air. Org. Lett. 2015, 17, 3702–3705. [Google Scholar] [CrossRef]
- Huang, H.-M.; Li, Y.-J.; Ye, Q.; Han, L.; Jia, J.-H.; Gao, J.-R. Iodine-Catalyzed 1,3-Dipolar Cycloaddition/Oxidation/Aromatization Cascade with Hydrogen Peroxide as the Terminal Oxidant: General Route to Pyrrolo[2,1-a]isoquinolines. J. Org. Chem. 2014, 79, 1084–1092. [Google Scholar] [CrossRef]
- Sohail, M.; Tanaka, F. Dynamic Kinetic Asymmetric Transformation of Racemic Diastereomers: Diastereo-and Enantioconvergent Michael-Henry Reactions to Afford Spirooxindoles Bearing Furan-Fused Rings. Angew. Chem. Int. Ed. 2021, 60, 21256–21260. [Google Scholar] [CrossRef]
- Wee, X.K.; Yeo, W.K.; Zhang, B.; Tan, V.B.C.; Lim, K.M.; Tay, T.E.; Go, M.-L. Synthesis and evaluation of functionalized isoindigos as antiproliferative agents. Bioorg. Med. Chem. 2009, 17, 7562–7571. [Google Scholar] [CrossRef]
- Muschalek, B.; Weidner, I.; Butenschön, H.J. Synthesis of tricarbonyl (N-methylisatin) chromium (0) and an unanticipated transformation of a N-MEM to a N-MOM group. Organomet. Chem. 2007, 692, 2415–2424. [Google Scholar] [CrossRef]
- Hans, R.H.; Gut, J.; Rosenthal, P.J.; Chibale, K. Comparison of the antiplasmodial and falcipain-2 inhibitory activity of β-amino alcohol thiolactone-chalcone and isatin-chalcone hybrids. Bioorg. Med. Chem. Lett. 2010, 20, 2234–2237. [Google Scholar] [CrossRef] [PubMed]
- Mohanta, N.; Chaudhari, M.B.; Digrawal, N.K.; Gnanaprakasam, B. Rapid and Multigram Synthesis of Vinylogous Esters under Continuous Flow: An Access to Transetherification and Reverse Reaction of Vinylogous Esters. Org. Pross. Res. Dev. 2019, 23, 1034–1045. [Google Scholar] [CrossRef]
- Bergman, J.; Stålhandske, C.; Vallberg, H. Studies of the reaction between indole-2,3-diones (isatins) and secondary aliphatic amines. Acta Chem. Scand. 1997, 51, 753–759. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).