
Chemical Space of Fluorinated Nucleosides/Nucleotides in Biomedical Research and Anticancer Drug Discovery


Abstract
:1. Introduction
2. Fluorinating Reagents
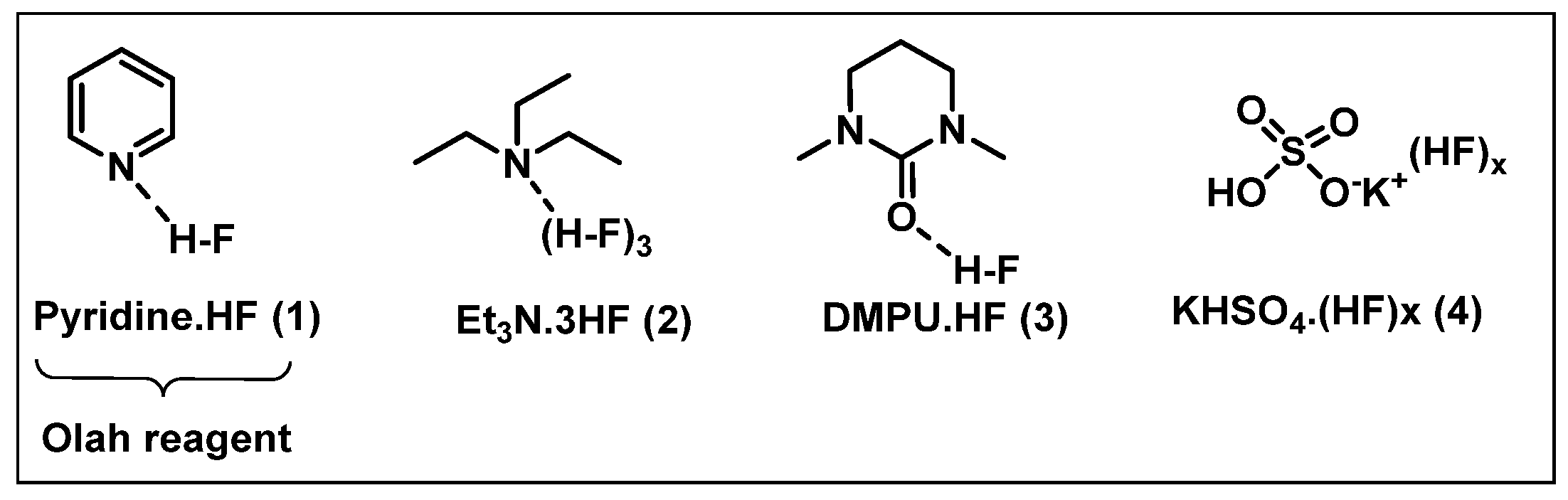
2.1. Nucleophilic Fluorinating Agents
- (i)
- HF-based nucleophilic reagents
- (ii)
- Metal Fluorides
- (iii)
- Quaternary Ammonium Fluoride
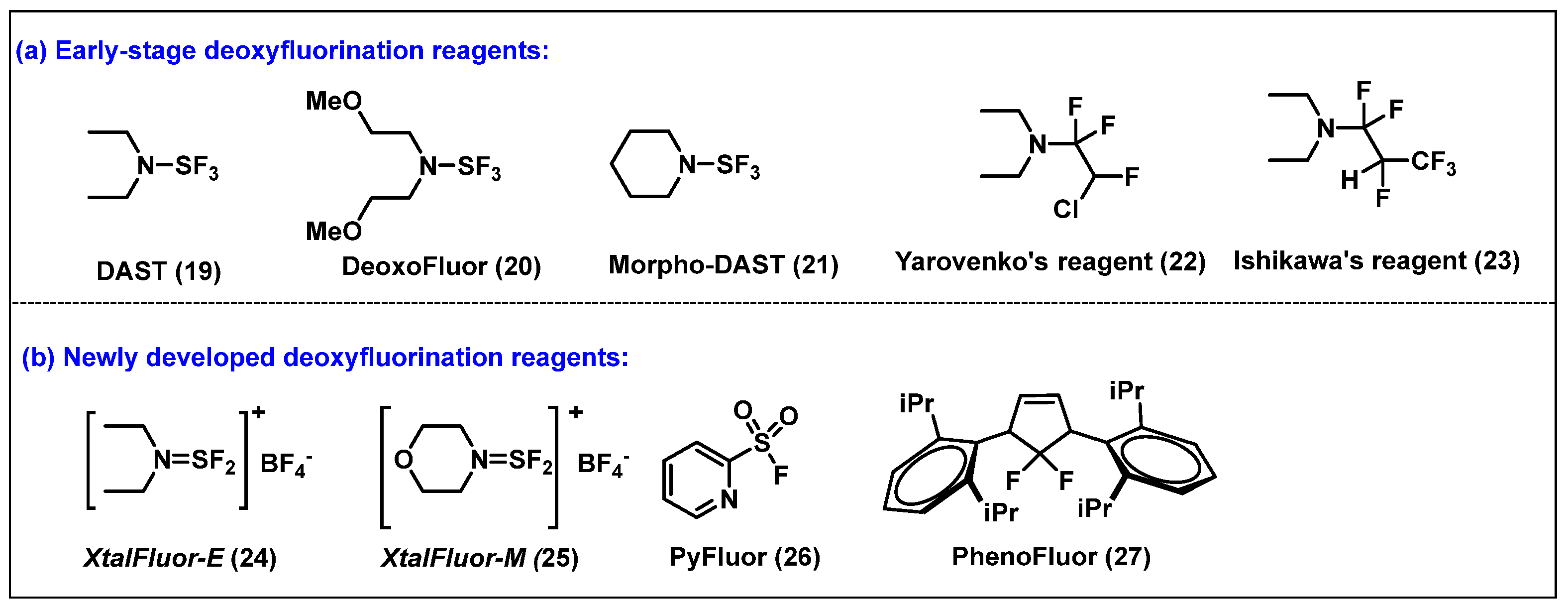
- (iv) Deoxy fluorinating reagents
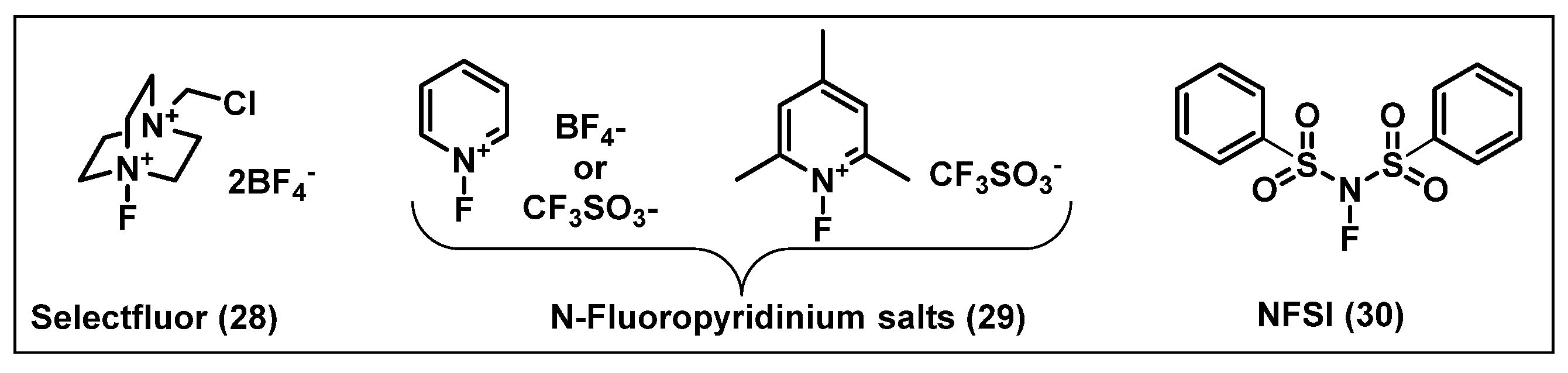
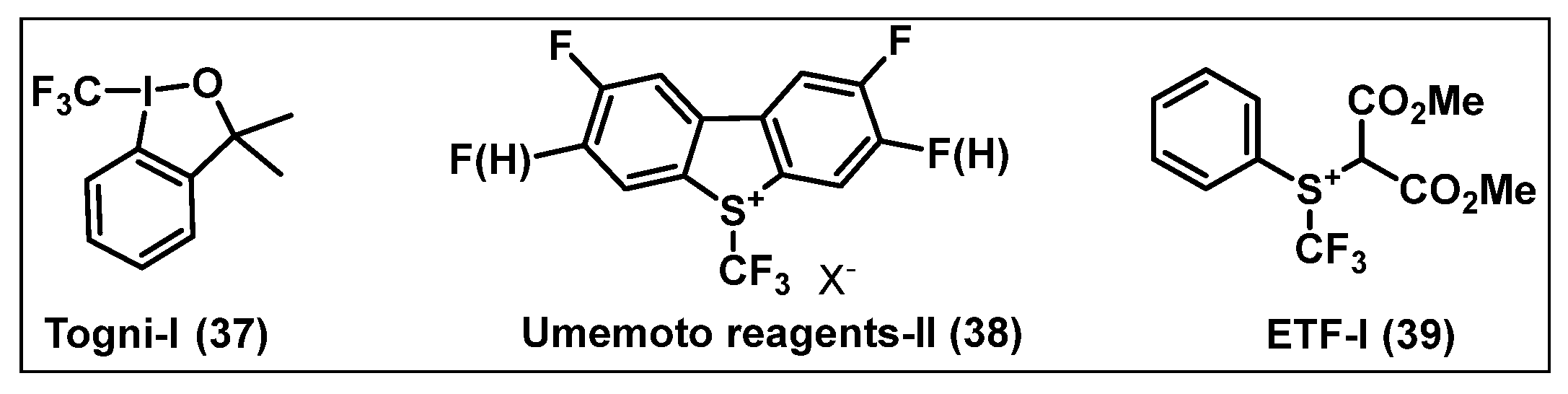
2.2. Electrophilic Fluorinating Agents
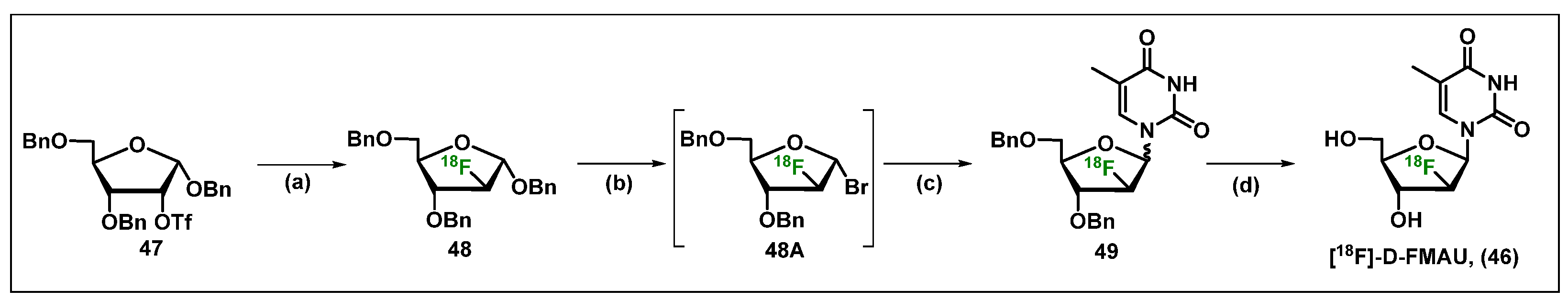
3. [18F]-Radiolabeled Nucleosides in Biomedical Research and Their Applications
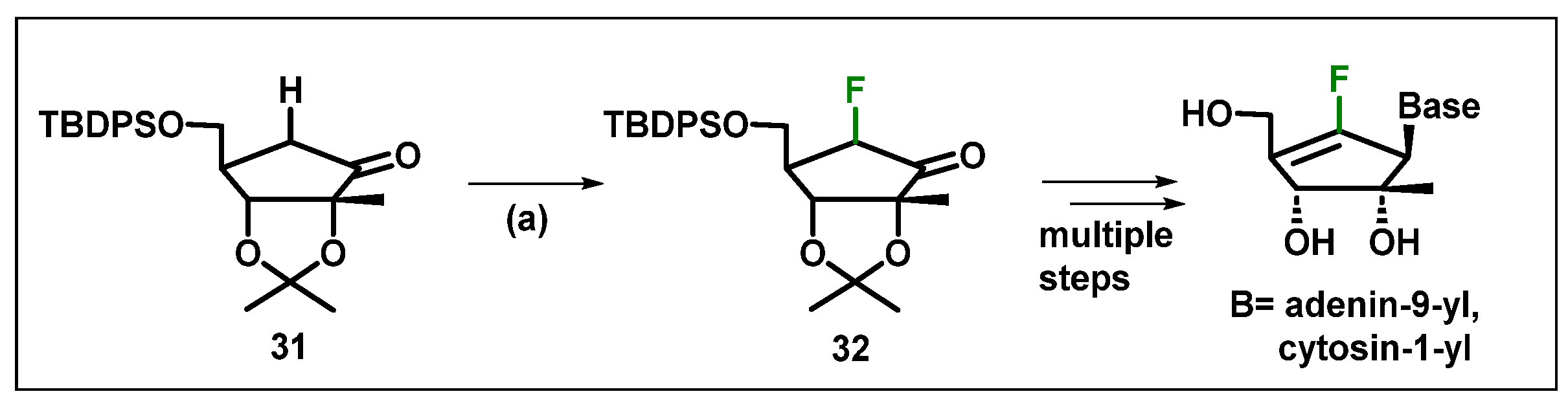
4. Synthesis and Anticancer Activity of Fluorinated Nucleos(t)ides
4.1. 5-Fluorouracil (5-FU, 50)
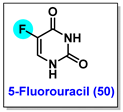
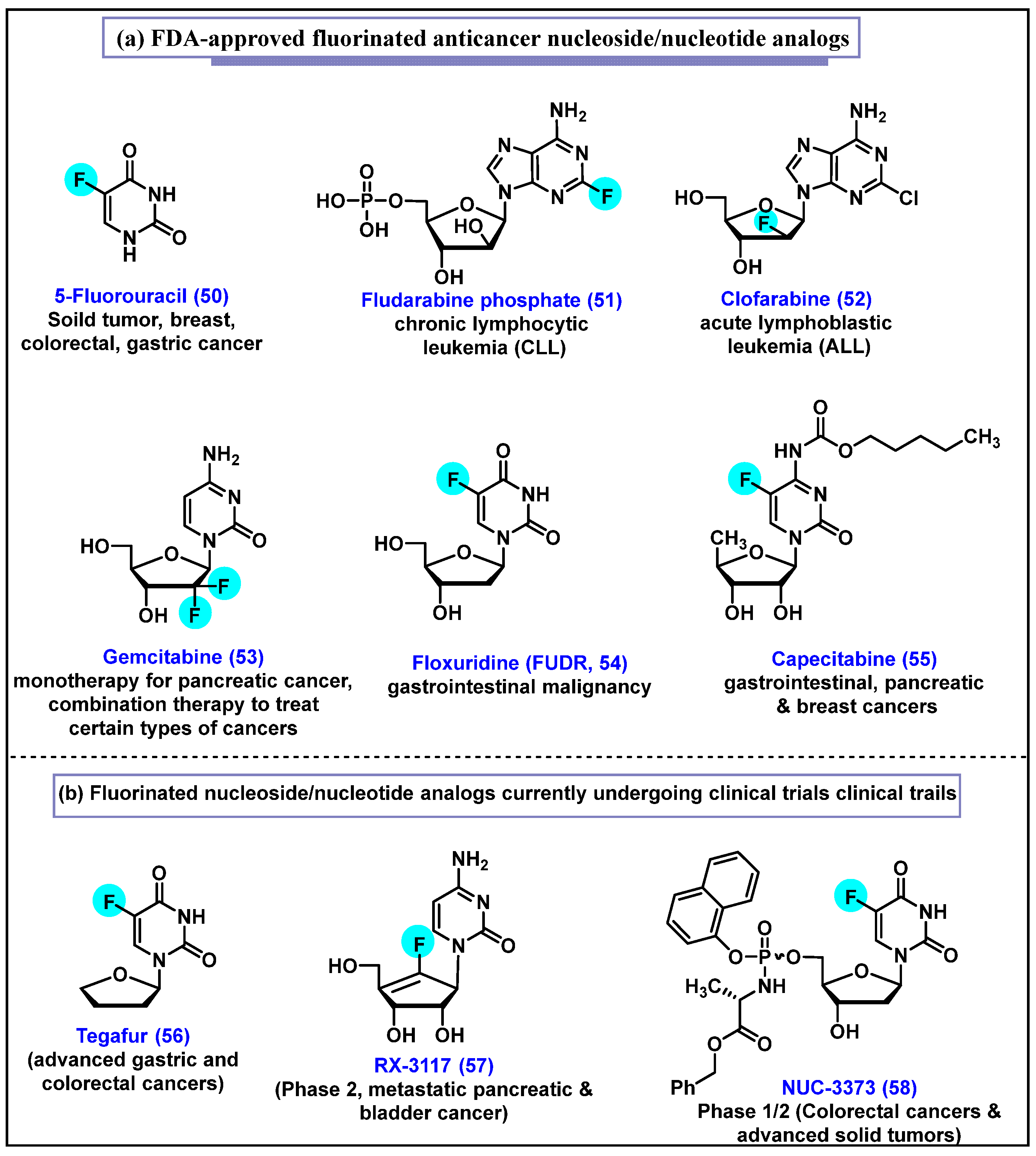
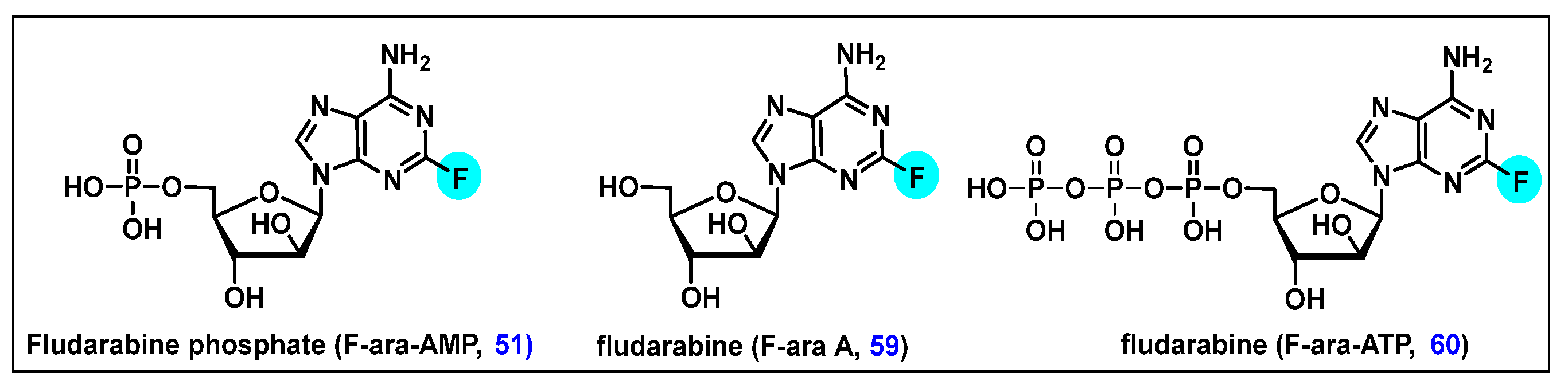
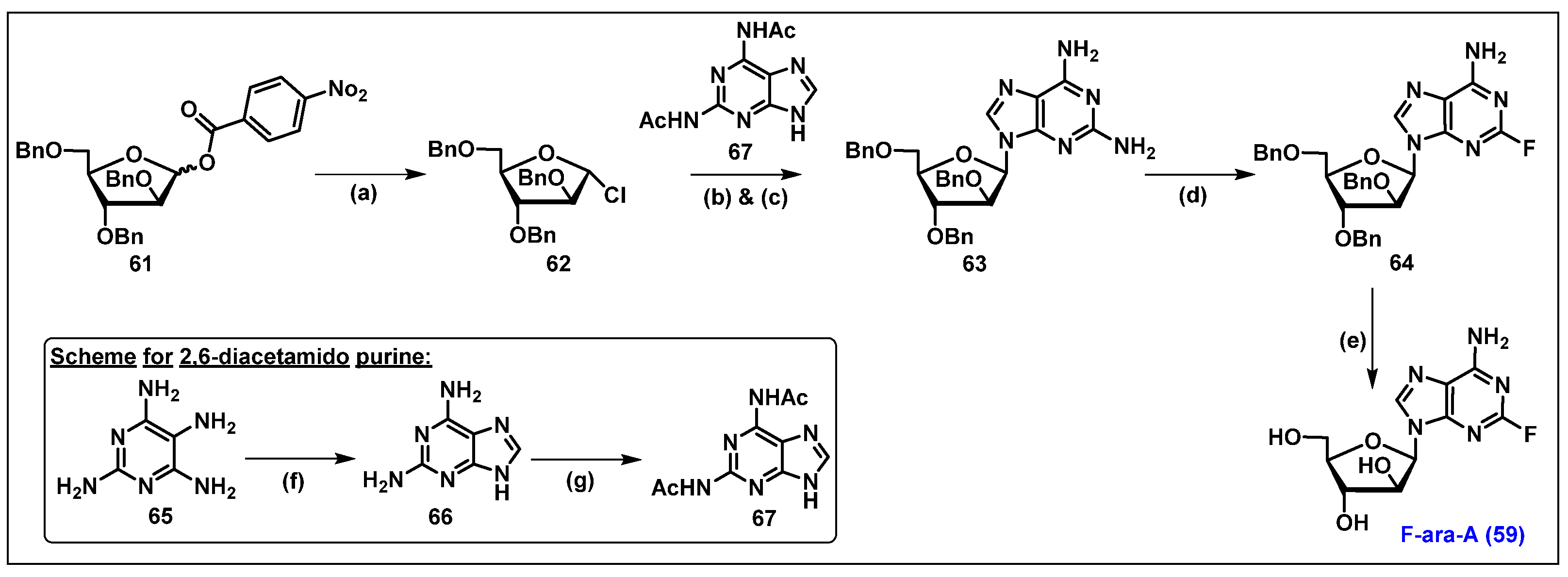
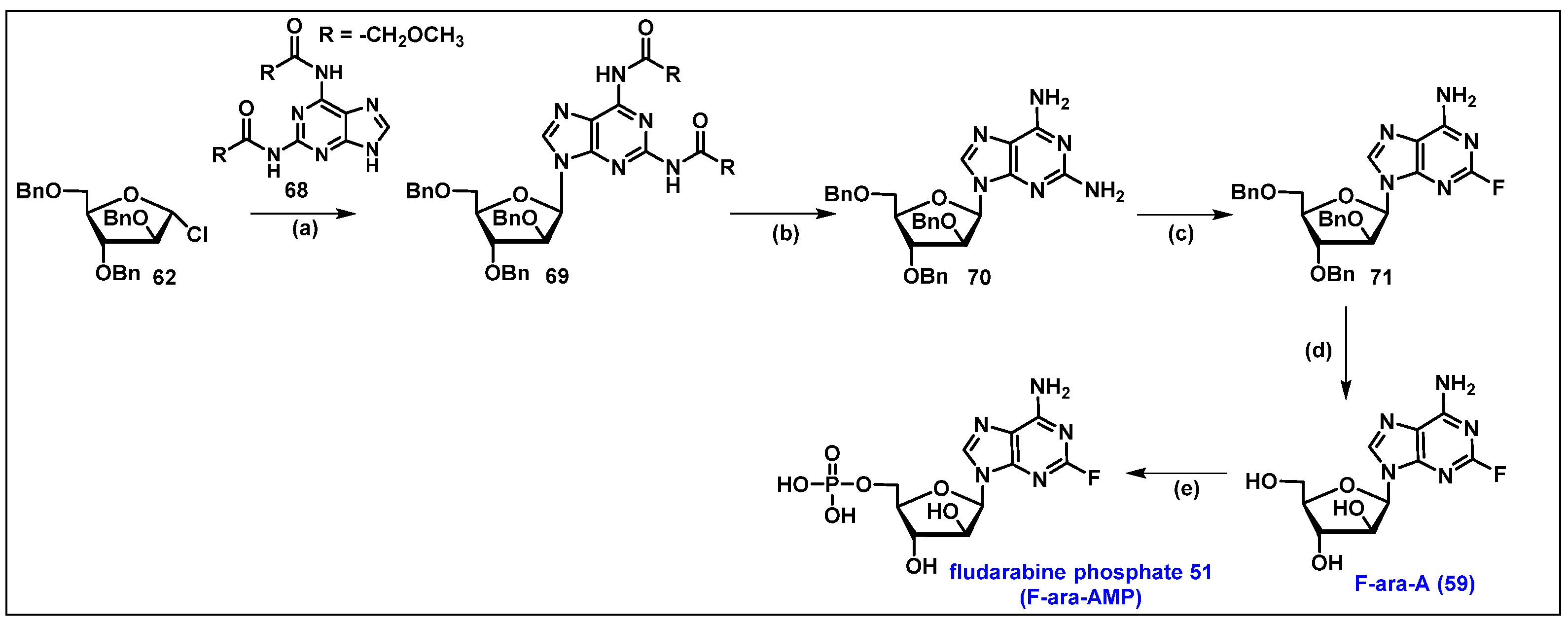
4.2. Fludarabine Phosphate (F-ara-AMP, 51)
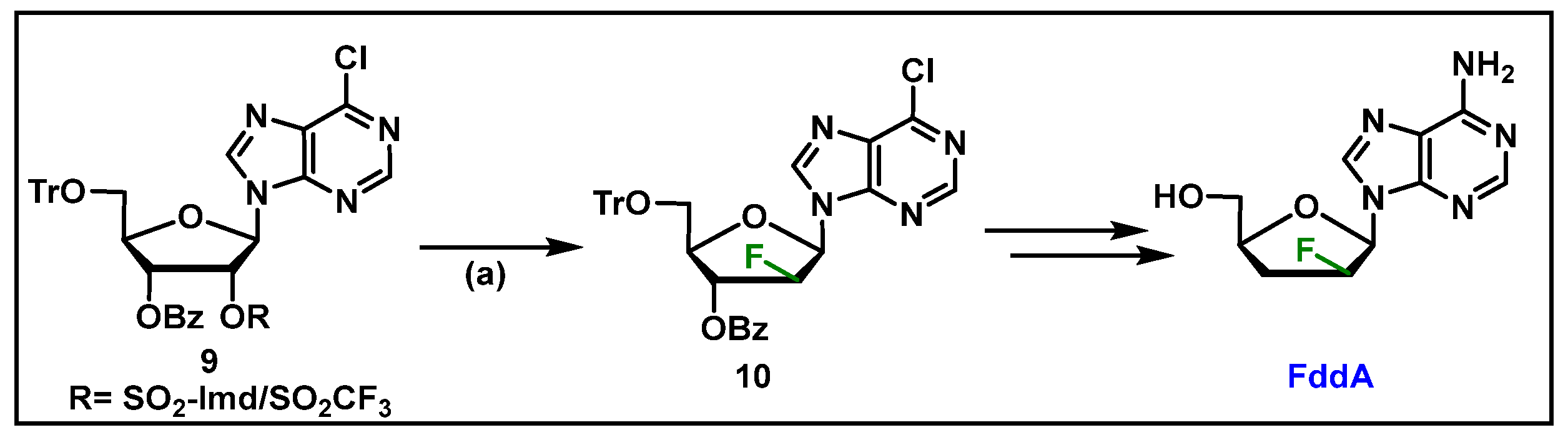
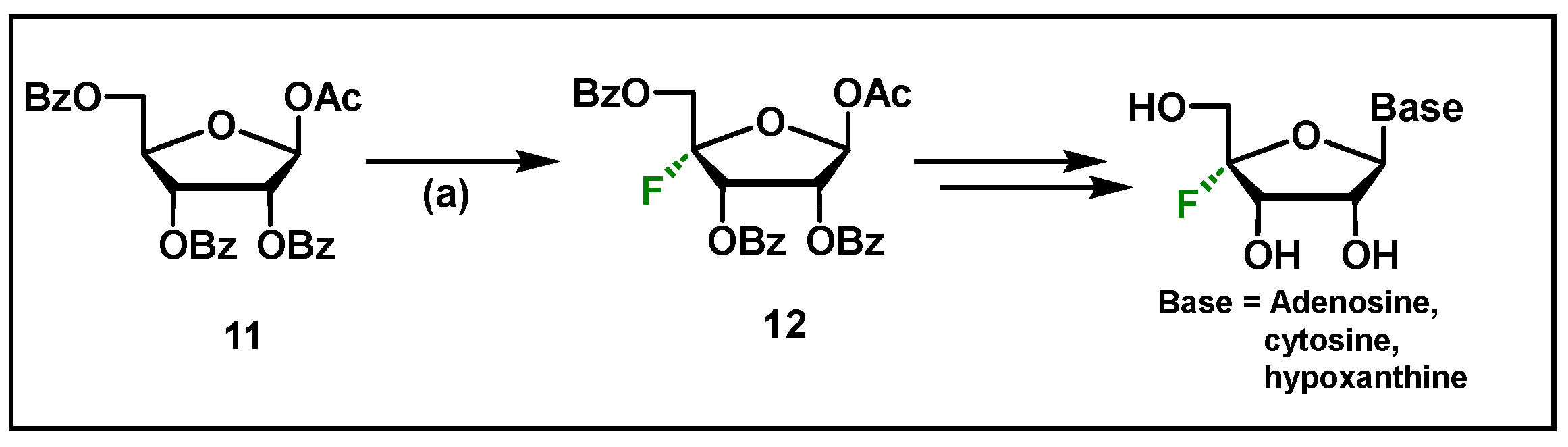
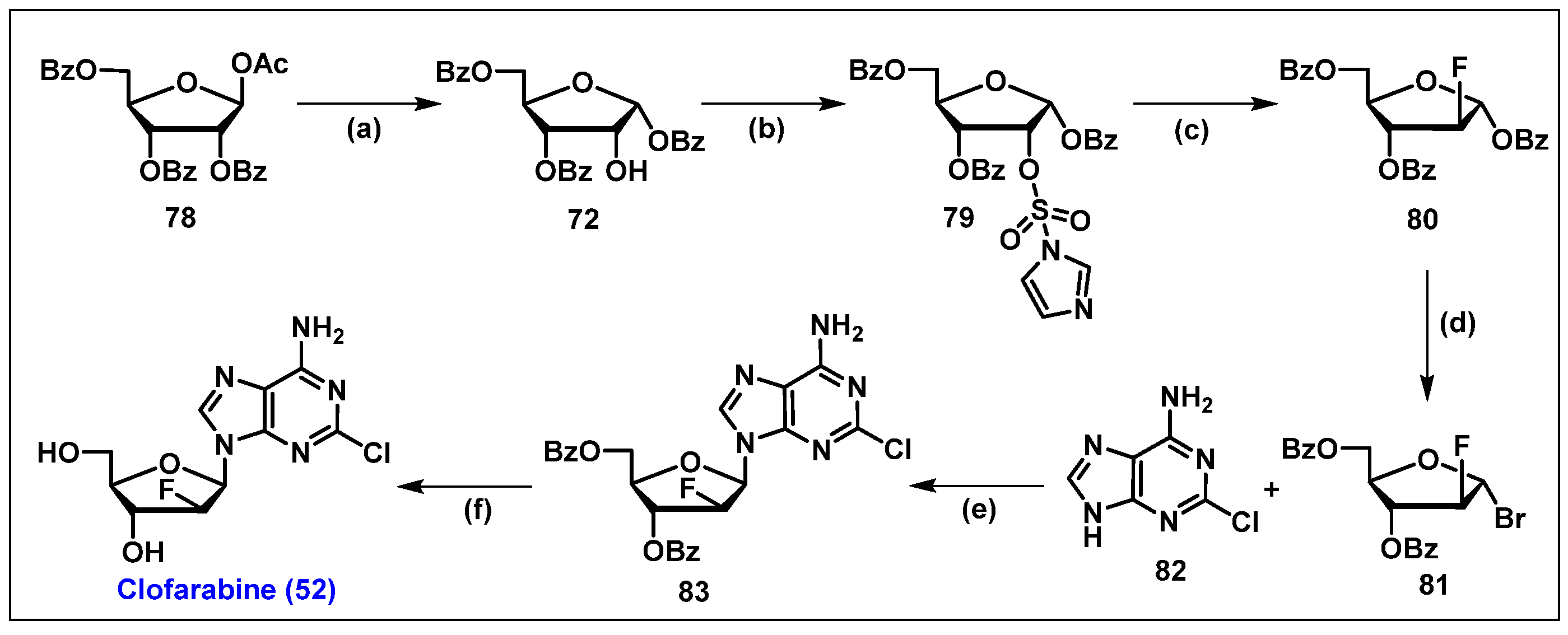
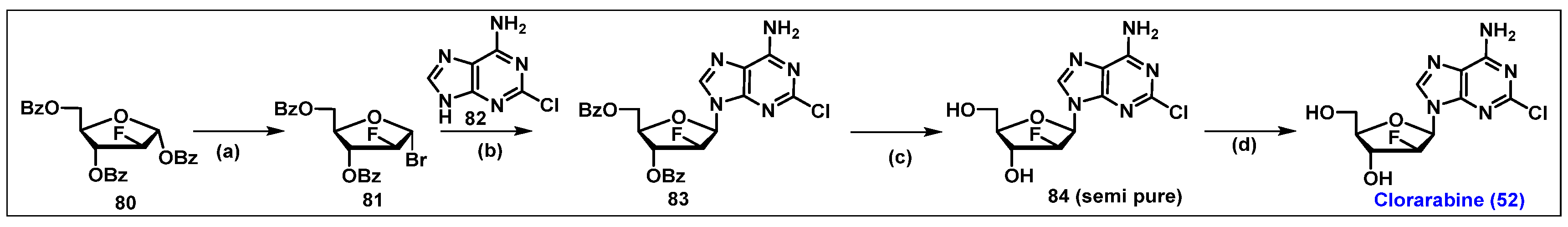
4.3. Clofarabine, 52

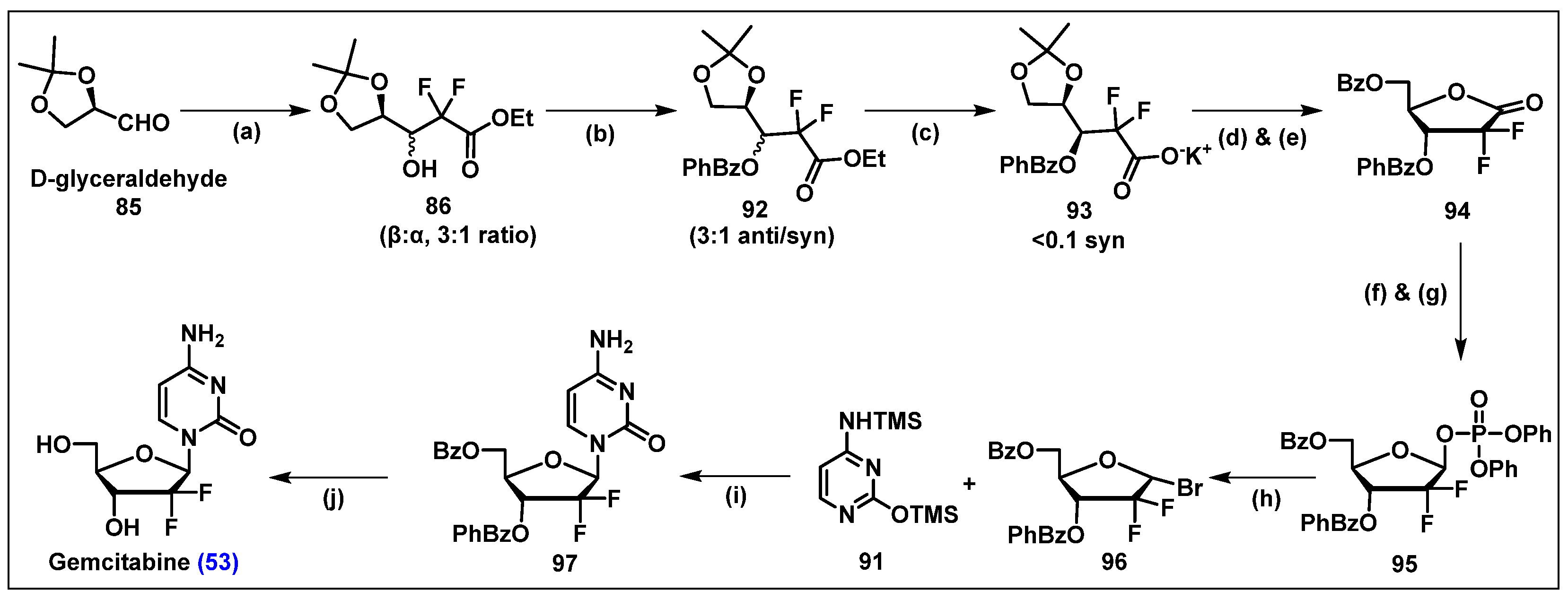
4.4. Gemcitabine, 53


4.4.1. Drug Resistance of Gemcitabine (53)
4.4.2. Implementation of Prodrug Strategies to Overcome Gemcitabine (53) Resistance and Cellular Uptake

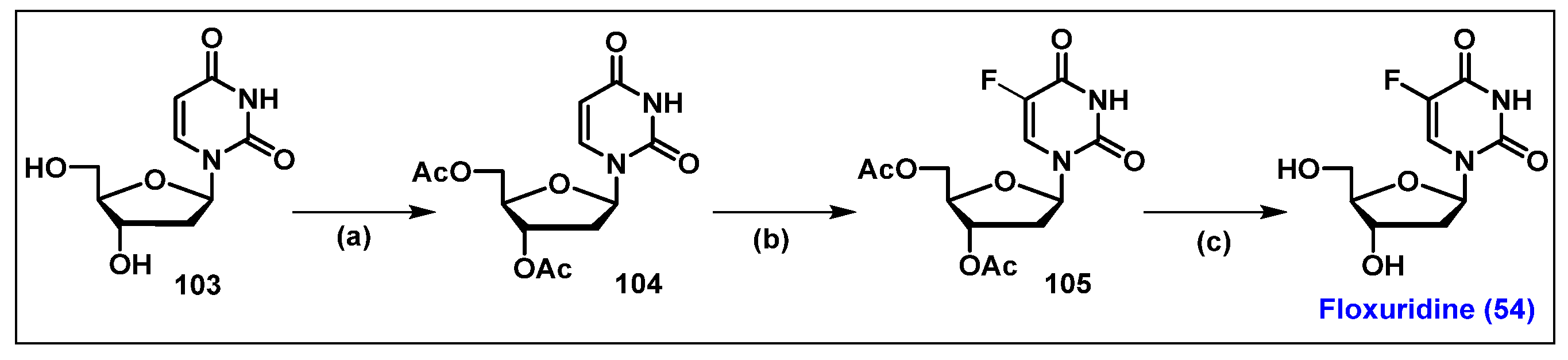
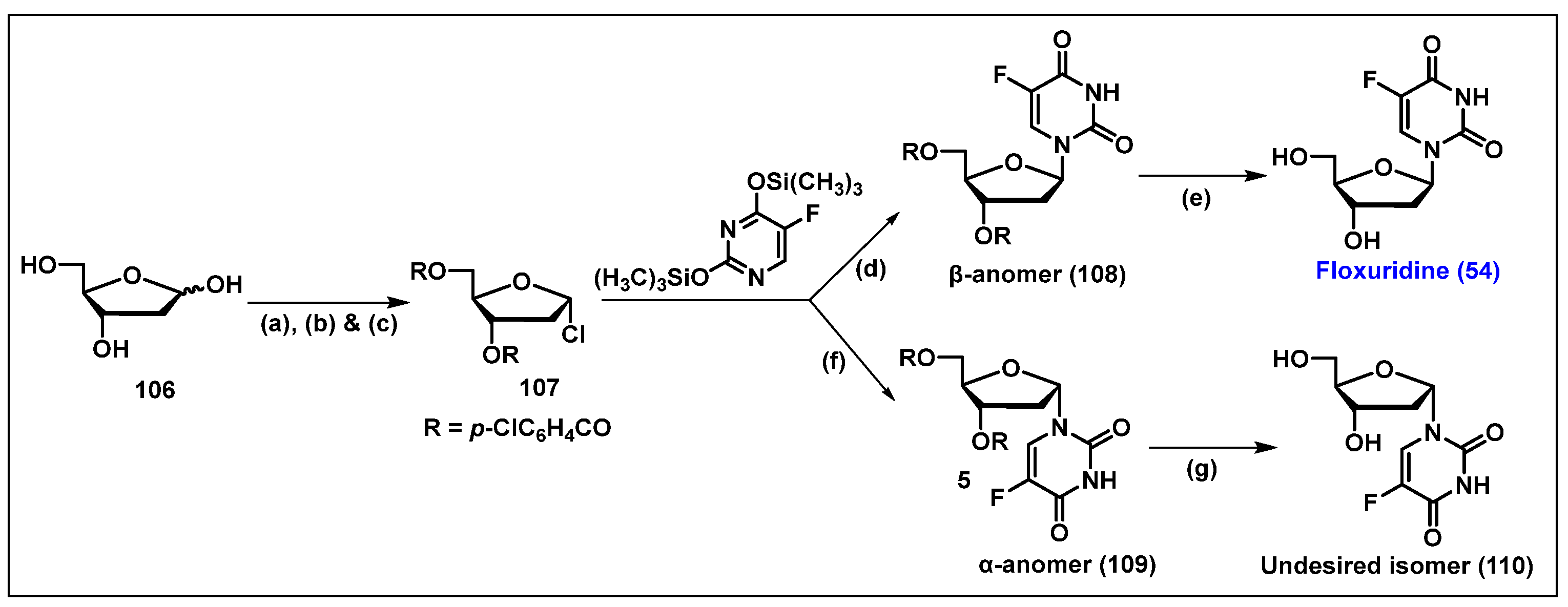
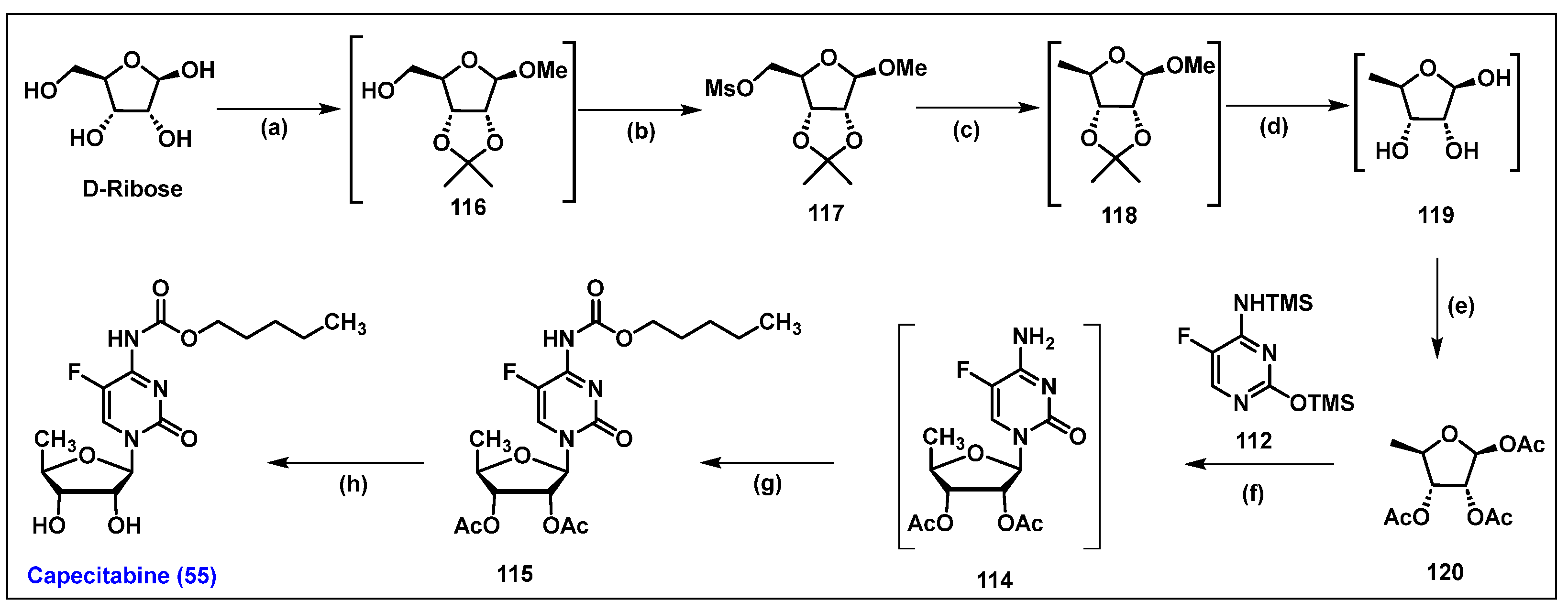
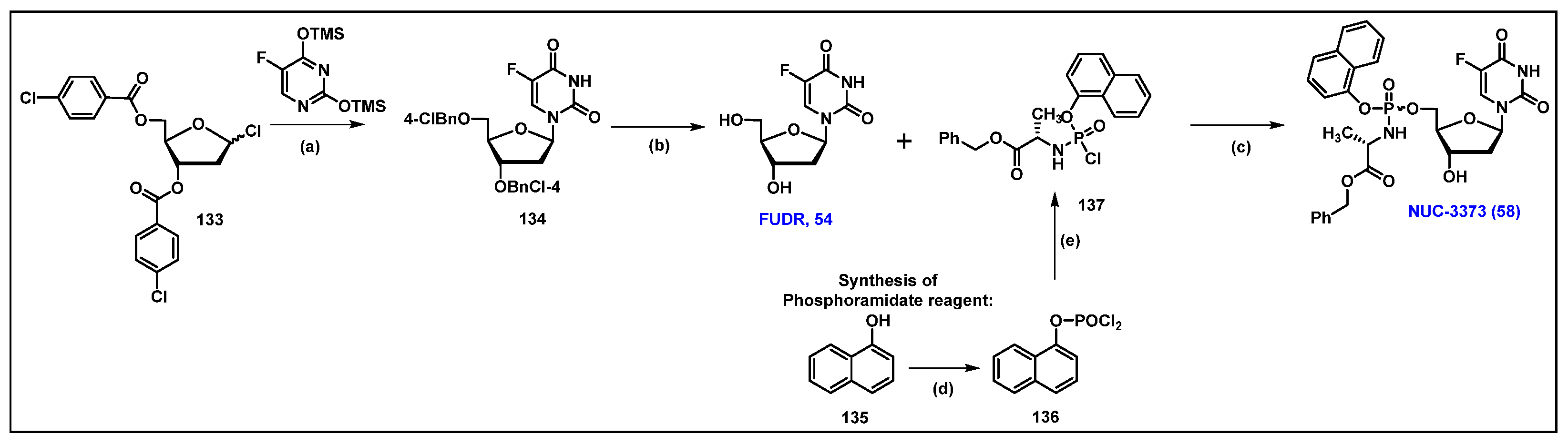
4.5. Floxuridine (FUDR, 54)

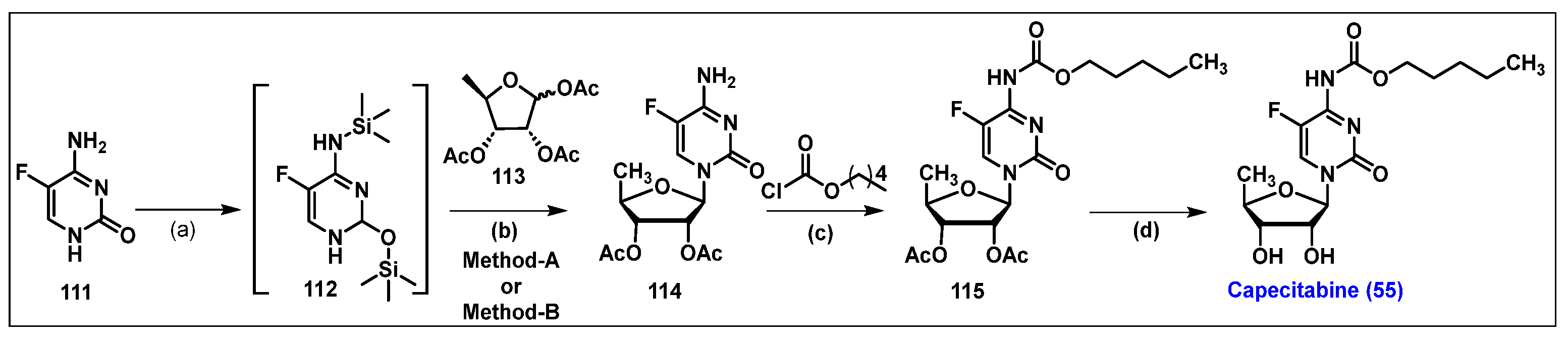
4.6. Capecitabine, 55
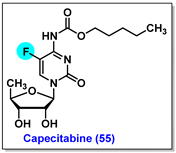
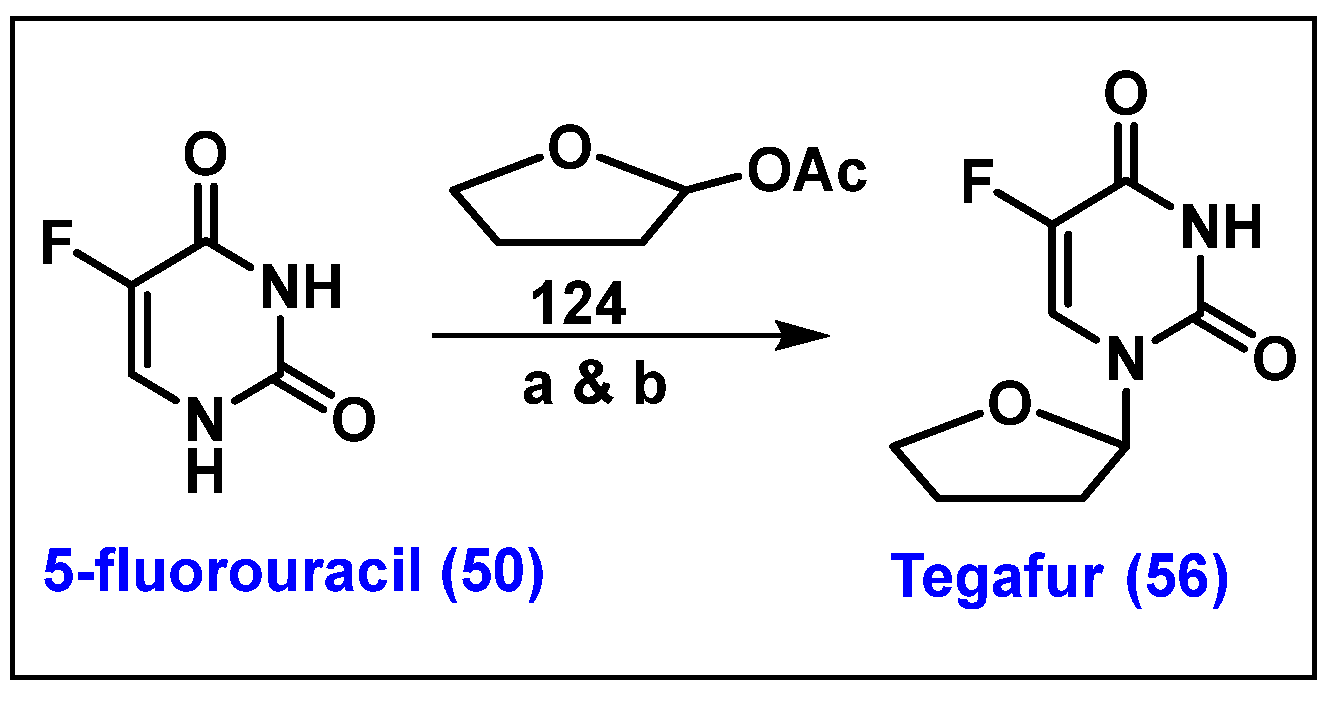
4.7. Tegafur, 56


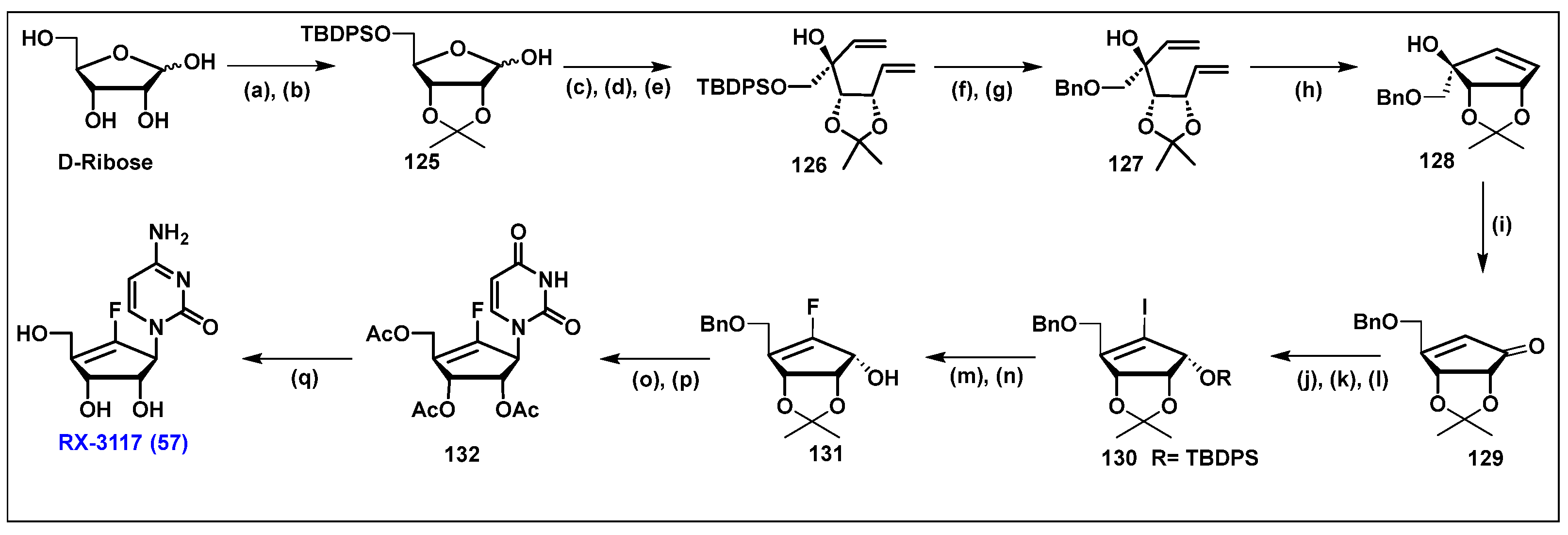
4.8. RX-3117, 57
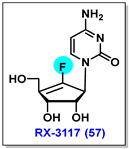
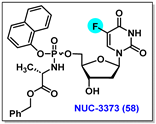
4.9. NUC-3373, 58
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hruba, L.; Das, V.; Hajduch, M.; Dzubak, P. Nucleoside-based anticancer drugs: Mechanism of action and drug resistance. Biochem. Pharmacol. 2023, 215, 115741. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Ramos, A.A.; Marchetti-Laurent, C.; Poindessous, V.; Antonio, S.; Laurent-Puig, P.; Bortoli, S.; Loriot, M.A.; Pallet, N. 6-mercaptopurine promotes energetic failure in proliferating T cells. Oncotarget 2017, 8, 43048–43060. [Google Scholar] [CrossRef]
- Munshi, P.N.; Lubin, M.; Bertino, J.R. 6-thioguanine: A drug with unrealized potential for cancer therapy. Oncologist 2014, 19, 760–765. [Google Scholar] [CrossRef] [PubMed]
- DeVita, V.T., Jr.; Chu, E. A History of Cancer Chemotherapy. Cancer Res. 2008, 68, 8643–8653. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yin, Y.; Xu, S.J.; Chen, W.S. 5-Fluorouracil: Mechanisms of resistance and reversal strategies. Molecules 2008, 13, 1551–1569. [Google Scholar] [CrossRef] [PubMed]
- Henary, E.; Casa, S.; Dost, T.L.; Sloop, J.C.; Henary, M. The Role of Small Molecules Containing Fluorine Atoms in Medicine and Imaging Applications. Pharmaceuticals 2024, 17, 281. [Google Scholar] [CrossRef]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef] [PubMed]
- Clercq, E.D.; Li, G. Approved Antiviral Drugs over the Past 50 Years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [PubMed]
- Anand, U.; Dey, A.; Chandel, A.K.S.; Sanyal, R.; Mishra, A.; Pandey, D.K.; De Falco, V.; Upadhyay, A.; Kandimalla, R.; Chaudhary, A.; et al. Cancer chemotherapy and beyond: Current status, drug candidates, associated risks and progress in targeted therapeutics. Genes Dis. 2023, 10, 1367–1401. [Google Scholar] [CrossRef]
- Shelton, J.; Lu, X.; Hollenbaugh, J.A.; Cho, J.H.; Amblard, F.; Schinazi, R.F. Metabolism, Biochemical Actions, and Chemical Synthesis of Anticancer Nucleosides, Nucleotides, and Base Analogs. Chem. Rev. 2016, 116, 14379–14455. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.G.; Sun, N.N.; Liang, L.; Yu, B.; Chang, J.B. 2′-Fluorinated nucleoside chemistry for new drug discovery: Achievements and prospects. Natl. Sci. Rev. 2024, 11, nwae331. [Google Scholar] [CrossRef]
- Qing, F.-L.; Liu, X.-Y.; Ma, J.-A.; Shen, Q.; Song, Q.; Tang, P. A Fruitful Decade of Organofluorine Chemistry: New Reagents and Reactions. CCS Chem. 2022, 4, 2518–2549. [Google Scholar] [CrossRef]
- Bajraktarova-Valjakova, E.; Korunoska-Stevkovska, V.; Georgieva, S.; Ivanovski, K.; Bajraktarova-Misevska, C.; Mijoska, A.; Grozdanov, A. Hydrofluoric Acid: Burns and Systemic Toxicity, Protective Measures, Immediate and Hospital Medical Treatment. Open Access Maced. J. Med. Sci. 2018, 6, 2257–2269. [Google Scholar] [CrossRef]
- Pal, S.; Chandra, G.; Patel, S.; Singh, S. Fluorinated Nucleosides: Synthesis, Modulation in Conformation and Therapeutic Application. Chem. Rec. 2022, 22, e202100335. [Google Scholar] [CrossRef] [PubMed]
- Klose, I.; Patel, C.; Mondal, A.; Schwarz, A.; Pupo, G.; Gouverneur, V. Fluorspar to fluorochemicals upon low-temperature activation in water. Nature 2024, 635, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Okoromoba, O.E.; Han, J.; Hammond, G.B.; Xu, B. Designer HF-based fluorination reagent: Highly regioselective synthesis of fluoroalkenes and gem-difluoromethylene compounds from alkynes. J. Am. Chem. Soc. 2014, 136, 14381–14384. [Google Scholar] [CrossRef]
- Gouverneur, V. Chemistry. A new departure in fluorination chemistry. Science 2009, 325, 1630–1631. [Google Scholar] [CrossRef] [PubMed]
- Dolan, J.P.; Benckendorff, C.M.M.; Field, R.A.; Miller, G.J. Fluorinated Nucleosides, Nucleotides and Sugar Nucleotides. Future Med. Chem. 2023, 15, 1111–1114. [Google Scholar] [CrossRef] [PubMed]
- Pulido, J.; Sobczak, A.J.; Balzarini, J.; Wnuk, S.F. Synthesis and Cytostatic Evaluation of 4-N-Alkanoyl and 4-N-Alkyl Gemcitabine Analogues. J. Med. Chem. 2014, 57, 191–203. [Google Scholar] [CrossRef]
- Izawa, K.; Takamatsu, S.; Katayama, S.; Hirose, N.; Kozai, S.; Maruyama, T. An Industrial Process for Synthesizing Lodenosine (FddA). Nucleosides Nucleotides Nucleic Acids 2003, 22, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Uttamapinant, C.; Verdine, G.L. A concise synthesis of 4′-fluoro nucleosides. Org. Lett. 2007, 9, 5007–5009. [Google Scholar] [CrossRef]
- Liang, S.Z.; Hammond, G.B.; Xu, B. Hydrogen Bonding: Regulator for Nucleophilic Fluorination. Chem. Eur. J. 2017, 23, 17850–17861. [Google Scholar] [CrossRef]
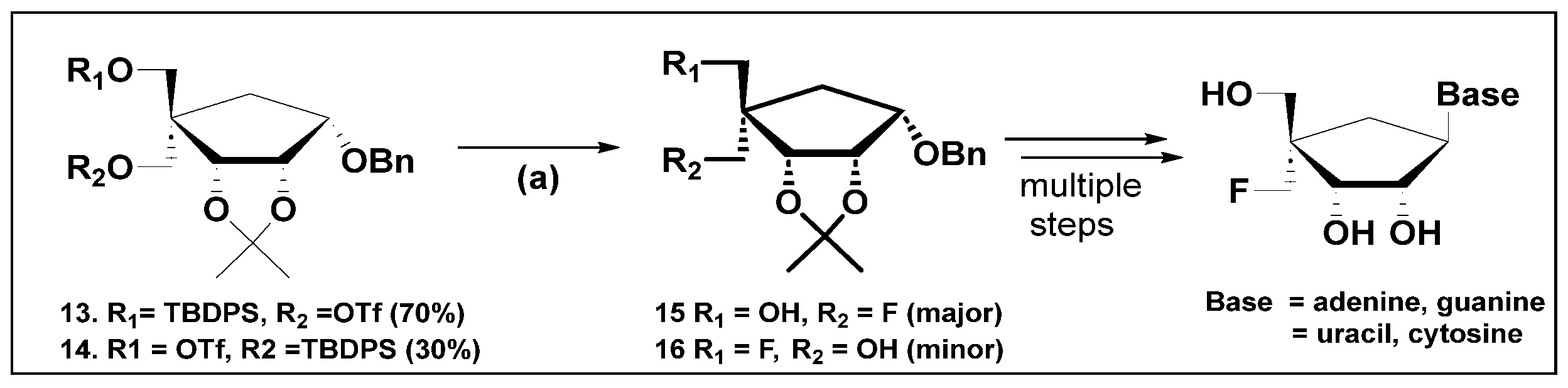
- Singh, U.S.; Jones, R.A.; Kothapalli, Y.; Mulamoottil, V.A.; Wei, P.; Chu, C.K. A Stereoselective Synthesis of 4′-α-Fluoro-methyl Carbocyclic Nucleoside Analogs. Chemistryselect 2023, 8, e202302043. [Google Scholar] [CrossRef]
- Elias, S.; Karton-Lifshin, N.; Yehezkel, L.; Ashkenazi, N.; Columbus, I.; Zafrani, Y. Synthesis, Characterization, and Reactivity of Thermally Stable Anhydrous Quaternary Ammonium Fluorides. Org. Lett. 2017, 19, 3039–3042. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Sharon, A.; Chu, C.K. Fluorinated nucleosides: Synthesis and biological implication. J. Fluor. Chem. 2008, 129, 743–766. [Google Scholar] [CrossRef] [PubMed]
- Markovskij, L.N.; Pashinnik, V.E.; Kirsanov, A.V. Application of Dialkylaminosulfur Trifluorides in the Synthesis of Fluoroorganic Compounds. Synthesis 1973, 1973, 787–789. [Google Scholar] [CrossRef]
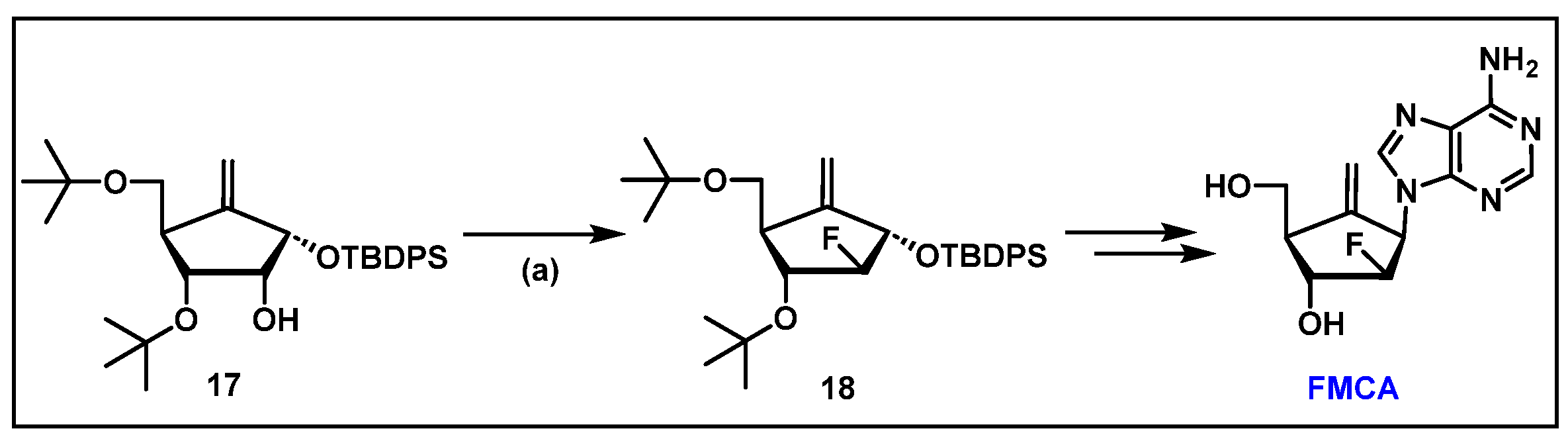
- Singh, U.S.; Mulamoottil, V.A.; Chu, C.K. Synthesis of an Anti-hepatitis B Agent, 2′-Fluoro-6′-methylene-carbocyclic Adenosine (FMCA) and Its Phosphoramidate (FMCAP). J. Org. Chem. 2019, 84, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Messina, P.A.; Mange, K.C.; Middleton, W.J. Aminosulfur trifluorides: Relative thermal stability [1]. J. Fluor. Chem. 1989, 42, 137–143. [Google Scholar] [CrossRef]
- Commare, B.; Schmitt, E.; Aribi, F.; Panossian, A.; Vors, J.P.; Pazenok, S.; Leroux, F.R. Fluoroalkyl Amino Reagents (FARs): A General Approach towards the Synthesis of Heterocyclic Compounds Bearing Emergent Fluorinated Substituents. Molecules 2017, 22, 977. [Google Scholar] [CrossRef]
- L’Heureux, A.; Beaulieu, F.; Bennett, C.; Bill, D.R.; Clayton, S.; LaFlamme, F.; Mirmehrabi, M.; Tadayon, S.; Tovell, D.; Couturier, M. Aminodifluorosulfinium Salts: Selective Fluorination Reagents with Enhanced Thermal Stability and Ease of Handling. J. Org. Chem. 2010, 75, 3401–3411. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.K.; Ugaz, C.R.; Li, W.; Doyle, A.G. PyFluor: A Low-Cost, Stable, and Selective Deoxyfluorination Reagent. J. Am. Chem. Soc. 2015, 137, 9571–9574. [Google Scholar] [CrossRef]
- Jelen, J.; Tavčar, G. Deoxyfluorination: A Detailed Overview of Recent Developments. Synthesis 2024, 56, A–Y. [Google Scholar]
- Lal, G.S. Fluorination at C2′, C3′ and C5′ of Nucleosides with 1-Chloromethyl-4-fluoro-1,4-diazabicyclo [2.2.2]octane bis(Tetrafluoroborate) SelectfluorTM Reagent. Synth. Commun. 1995, 25, 725–737. [Google Scholar] [CrossRef]
- Lal, G.S.; Pastore, W.; Pesaresi, R. A Convenient Synthesis of 5-Fluoropyrimidines Using 1-(Chloromethyl)-4-fluoro-1, 4-diazabicyclo [2. cntdot. 2. cntdot. 2] octane Bis (tetrafluoroborate)-SELECTFLUOR Reagent. J. Org. Chem. 1995, 60, 7340–7342. [Google Scholar] [CrossRef]
- Umemoto, T.; Yang, Y.H.; Hammond, G.B. Development of N-F fluorinating agents and their fluorinations: Historical perspective. Beilstein J. Org. Chem. 2021, 17, 1752–1813. [Google Scholar] [CrossRef]
- Kumamoto, H.; Kobayashi, M.; Kato, N.; Balzarini, J.; Tanaka, H. Synthesis of the 5′-Fluoro-2′β-methyl Analogues of Neplanocin. Eur. J. Org. Chem. 2011, 2011, 2685–2691. [Google Scholar] [CrossRef]
- Wang, X.J.; Seth, P.P.; Ranken, R.; Swayze, E.E.; Migawa, M.T. Synthesis and biological activity of 5-fluorotubercidin. Nucleosides Nucleotides Nucleic Acids 2004, 23, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Burkart, M.D.; Zhang, Z.Y.; Hung, S.C.; Wong, C.H. A new method for the synthesis of fluoro-carbohydrates and glycosides using selectfluor. J. Am. Chem. Soc. 1997, 119, 11743–11746. [Google Scholar] [CrossRef]
- Li, Y.F.; Mao, S.; Hager, M.W.; Becnel, K.D.; Schinazi, R.F.; Liotta, D.C. Synthesis and evalution of 2′-substituted cyclobutyl nucleosides and nucleotides as potential anti-HIV agents. Bioorg. Med. Chem. Lett. 2007, 17, 3398–3401. [Google Scholar] [CrossRef]
- Umemoto, T.; Fukami, S.; Tomizawa, G.; Harasawa, K.; Kawada, K.; Tomita, K. Power and Structure-Variable Fluorinating Agents—The N-Fluoropyridinium Salt System. J. Am. Chem. Soc. 1990, 112, 8563–8575. [Google Scholar] [CrossRef]
- Meanwell, N.A. The Influence of Bioisosteres in Drug Design: Tactical Applications to Address Developability Problems. In Tactics in Contemporary Drug Design; Springer: Berlin/Heidelberg, Germany, 2014; Volume 9, pp. 283–381. [Google Scholar]
- Salvetti, R.; Pregnolato, M.; Verri, A.; Focher, F.; Spadari, S.; Marchand, A.; Mathé, C.; Gosselin, G. Synthesis and In Vitro Activity Of D- and L-Enantiomers Of 5-(Trifluoromethyl)Uracil Nucleoside Derivatives. Nucleosides Nucleotides Nucleic Acids 2001, 20, 1123–1125. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, D.; Irace, C.; Santamaria, R.; Montesarchio, D. Trifluoromethyl derivatives of canonical nucleosides: Synthesis and bioactivity studies. MedChemComm 2013, 4, 1405–1410. [Google Scholar] [CrossRef]
- Dong, M.; Kirchberger, T.; Huang, X.; Yang, Z.J.; Zhang, L.R.; Guse, A.H.; Zhang, L.H. Trifluoromethylated cyclic-ADP-ribose mimic: Synthesis of 8-trifluoromethyl-N1-[(5″-O-phosphorylethoxy)methyl]-5′-O-phosphorylinosine-5′,5″-cyclic pyrophosphate (8-CF3-cIDPRE) and its calcium release activity in T cells. Org. Biomol. Chem. 2010, 8, 4705–4715. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Yamamoto, K.; Asai, T.; Nakano, M.; Kumadaki, I. Studies on organic fluorine compounds. Part 35. Trifluoromethylation of pyrimidine- and purine-nucleosides with trifluoromethyl–copper complex. J. Chem. Soc. Perkin Trans. 1 1980, 0, 2755–2761. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Dixon, J.A.; O’Hara, F.; Funder, E.D.; Dixon, D.D.; Rodriguez, R.A.; Baxter, R.D.; Herlé, B.; Sach, N.; Collins, M.R.; et al. Practical and innate carbon-hydrogen functionalization of heterocycles. Nature 2012, 492, 95–99. [Google Scholar] [CrossRef]
- Zhou, Q.; Gui, J.; Pan, C.-M.; Albone, E.; Cheng, X.; Suh, E.M.; Grasso, L.; Ishihara, Y.; Baran, P.S. Bioconjugation by Native Chemical Tagging of C–H Bonds. J. Am. Chem. Soc. 2013, 135, 12994–12997. [Google Scholar] [CrossRef] [PubMed]
- Guyon, H.; Chachignon, H.; Cahard, D. CF3SO2X (X = Na, Cl) as reagents for trifluoromethylation, trifluoromethylsulfenyl, sulfinyl and sulfonylation. Part 1: Use of CF3SO2Na. Beilstein J. Org. Chem. 2017, 13, 2764–2799. [Google Scholar] [CrossRef] [PubMed]
- Chrominski, M.; Baranowski, M.R.; Chmielinski, S.; Kowalska, J.; Jemielity, J. Synthesis of Trifluoromethylated Purine Ribonucleotides and Their Evaluation as 19F NMR Probes. J. Org. Chem. 2020, 85, 3440–3453. [Google Scholar] [CrossRef] [PubMed]
- Nakajo, M.; Jinguji, M.; Ito, S.; Tani, A.; Hirahara, M.; Yoshiura, T. Clinical application of 18F-fluorodeoxyglucose positron emission tomography/computed tomography radiomics-based machine learning analyses in the field of oncology. Jpn. J. Radiol. 2024, 42, 28–55. [Google Scholar] [CrossRef]
- Fleisher, A.S.; Pontecorvo, M.J.; Devous, M.D., Sr.; Lu, M.; Arora, A.K.; Truocchio, S.P.; Aldea, P.; Flitter, M.; Locascio, T.; Devine, M.; et al. Positron Emission Tomography Imaging With [18F]flortaucipir and Postmortem Assessment of Alzheimer Disease Neuropathologic Changes. JAMA Neurol. 2020, 77, 829–839. [Google Scholar] [CrossRef]
- Singh, S.B.; Ng, S.J.; Lau, H.C.; Khanal, K.; Bhattarai, S.; Paudyal, P.; Shrestha, B.B.; Naseer, R.; Sandhu, S.; Gokhale, S.; et al. Emerging PET Tracers in Cardiac Molecular Imaging. Cardiol. Ther. 2023, 12, 85–99. [Google Scholar] [CrossRef]
- Ghosh, K.K.; Padmanabhan, P.; Yang, C.-T.; Ng, D.C.E.; Palanivel, M.; Mishra, S.; Halldin, C.; Gulyás, B. Positron emission tomographic imaging in drug discovery. Drug Discov. Today 2022, 27, 280–291. [Google Scholar] [CrossRef]
- Trotter, J.; Pantel, A.R.; Teo, B.K.; Escorcia, F.E.; Li, T.; Pryma, D.A.; Taunk, N.K. Positron Emission Tomography (PET)/Computed Tomography (CT) Imaging in Radiation Therapy Treatment Planning: A Review of PET Imaging Tracers and Methods to Incorporate PET/CT. Adv. Radiat. Oncol. 2023, 8, 101212. [Google Scholar] [CrossRef] [PubMed]
- Wright, J.S.; Sharninghausen, L.S.; Lapsys, A.; Sanford, M.S.; Scott, P.J.H. C–H Labeling with [18F]Fluoride: An Emerging Methodology in Radiochemistry. ACS Cent. Sci. 2024, 10, 1674–1688. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.; Jordan, C.; Gilmour, R. Fluorinated carbohydrates for 18F-positron emission tomography (PET). Chem. Soc. Rev. 2023, 52, 3599–3626. [Google Scholar] [CrossRef] [PubMed]
- van der Born, D.; Pees, A.; Poot, A.J.; Orru, R.V.A.; Windhorst, A.D.; Vugts, D.J. Fluorine-18 labelled building blocks for PET tracer synthesis. Chem. Soc. Rev. 2017, 46, 4709–4773. [Google Scholar] [CrossRef] [PubMed]
- FDA-Approved PET Radiopharmaceuticals. 2024. Available online: http://www.radiopharmaceuticals.info/pet-radiopharmaceuticals.html (accessed on 22 November 2024).
- Fletcher, J.W.; Djulbegovic, B.; Soares, H.P.; Siegel, B.A.; Lowe, V.J.; Lyman, G.H.; Coleman, R.E.; Wahl, R.; Paschold, J.C.; Avril, N.; et al. Recommendations on the Use of 18F-FDG PET in Oncology. J. Nucl. Med. 2008, 49, 480–508. [Google Scholar] [CrossRef] [PubMed]
- Litt, H.K.; Kwon, D.H.; Velazquez, A.I. FDG PET Scans in Cancer Care. JAMA Oncol. 2023, 9, 1304. [Google Scholar] [CrossRef] [PubMed]
- Manabe, O.; Tamaki, N. The future of cardiac disease assessment using 18F-FDG PET/CT. Jpn. J. Radiol. 2021, 39, 511–513. [Google Scholar] [CrossRef] [PubMed]
- Sammartino, A.M.; Falco, R.; Drera, A.; Dondi, F.; Bellini, P.; Bertagna, F.; Vizzardi, E. Vascular inflammation and cardiovascular disease: Review about the role of PET imaging. J. Cardiovasc. Imaging 2023, 39, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Guedj, E.; Varrone, A.; Boellaard, R.; Albert, N.L.; Barthel, H.; van Berckel, B.; Brendel, M.; Cecchin, D.; Ekmekcioglu, O.; Garibotto, V.; et al. EANM procedure guidelines for brain PET imaging using [18F]FDG, version 3. Eur. J. Nucl. Med. Mol. Imaging 2022, 49, 632–651. [Google Scholar] [CrossRef] [PubMed]
- Minoshima, S.; Cross, D.; Thientunyakit, T.; Foster, N.L.; Drzezga, A. 18F-FDG PET Imaging in Neurodegenerative Dementing Disorders: Insights into Subtype Classification, Emerging Disease Categories, and Mixed Dementia with Copathologies. J. Nucl. Med. 2022, 63 (Suppl. S1), 2S–12S. [Google Scholar] [CrossRef]
- Casali, M.; Lauri, C.; Altini, C.; Bertagna, F.; Cassarino, G.; Cistaro, A.; Erba, A.P.; Ferrari, C.; Mainolfi, C.G.; Palucci, A.; et al. State of the art of 18F-FDG PET/CT application in inflammation and infection: A guide for image acquisition and interpretation. Clin. Transl. Imaging 2021, 9, 299–339. [Google Scholar] [CrossRef]
- Pijl, J.P.; Nienhuis, P.H.; Kwee, T.C.; Glaudemans, A.W.J.M.; Slart, R.H.J.A.; Gormsen, L.C. Limitations and Pitfalls of FDG-PET/CT in Infection and Inflammation. Nucl. Med. Semin. 2021, 51, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Lovinfosse, P.; Rousseau, C.; Pierga, J.-Y.; Bouchet, F.; Cochet, A.; Alberini, J.-L.; Girault, S.; Vera, P.; Olivier, P.; Uwer, L.; et al. Dual time point [18F]FLT-PET for differentiating proliferating tissues vs non-proliferating tissues. EJNMMI Res. 2019, 9, 109. [Google Scholar] [CrossRef] [PubMed]
- Alwadani, B.; Dall’Angelo, S.; Fleming, I.N. Clinical value of 3′-deoxy-3′-[18F]fluorothymidine-positron emission tomography for diagnosis, staging and assessing therapy response in lung cancer. Insights Imaging 2021, 12, 90. [Google Scholar] [CrossRef] [PubMed]
- Nishii, R.; Volgin, A.Y.; Mawlawi, O.; Mukhopadhyay, U.; Pal, A.; Bornmann, W.; Gelovani, J.G.; Alauddin, M.M. Evaluation of 2′-deoxy-2′-[18F]fluoro-5-methyl-1-beta-L: -arabinofuranosyluracil ([18F]-L: -FMAU) as a PET imaging agent for cellular proliferation: Comparison with [18F]-D: -FMAU and [18F]FLT. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 990–998. [Google Scholar] [CrossRef]
- Mangner, T.J.; Klecker, R.W.; Anderson, L.; Shields, A.F. Synthesis of 2′-deoxy-2′-[18F]fluoro-β-D-arabinofuranosyl nucleosides, [18F]FAU, [18F]FMAU, [18F]FBAU and [18F]FIAU, as potential PET agents for imaging cellular proliferation: Synthesis of [18F]labelled FAU, FMAU, FBAU, FIAU. Nucl. Med. Biol. 2003, 30, 215–224. [Google Scholar] [CrossRef]
- Chen, K.; Yap, L.-p.; Hughes, L.; Park, R.; Wang, X.; Conti, P. [18F]-FMAU PET assessment of erlotinib therapy in non-small cell lung cancer. J. Nucl. Med. 2012, 53 (Suppl. S1), 454. [Google Scholar]
- Alauddin, M.M.; Conti, P.S.; Fissekis, J.D. Synthesis of [18F]-labeled 2′-deoxy-2′-fluoro-5-methyl-1-β-D-arabinofuranosyluracil ([18F]-FMAU). J. Label. Compd. Radiopharm. 2002, 45, 583–590. [Google Scholar] [CrossRef]
- Robertson, J.; Barr, R.; Shulman, L.N.; Forte, G.B.; Magrini, N. Essential medicines for cancer: WHO recommendations and national priorities. Bull. World Health Organ. 2016, 94, 735–742. [Google Scholar] [CrossRef] [PubMed]
- Society, A.C. Colorectal Cancer Facts & Figures. 2023. Available online: https://www.cancer.org/research/cancer-facts-statistics/colorectal-cancer-facts-figures.html (accessed on 11 October 2023).
- Society, A.C. Colorectal Cancer Is A Major Public Health Problem. 2024. Available online: https://nccrt.org/our-impact/data-and-progress/ (accessed on 20 November 2024).
- Vodenkova, S.; Buchler, T.; Cervena, K.; Veskrnova, V.; Vodicka, P.; Vymetalkova, V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: Past, present and future. Pharmacol. Ther. 2020, 206, 107447. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Gmeiner, W.H. Recent Advances in Our Knowledge of mCRC Tumor Biology and Genetics: A Focus on Targeted Therapy Development. OncoTargets Ther. 2021, 14, 2121–2130. [Google Scholar] [CrossRef]
- Punt, C.J.A.; Koopman, M.; Vermeulen, L. From tumour heterogeneity to advances in precision treatment of colorectal cancer. Nat. Rev. Clin. Oncol. 2017, 14, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Wohlhueter, R.M.; McIvor, R.S.; Plagemann, P.G. Facilitated transport of uracil and 5-fluorouracil, and permeation of orotic acid into cultured mammalian cells. J. Cell. Physiol. 1980, 104, 309–319. [Google Scholar] [CrossRef]
- Yoshioka, A.; Tanaka, S.; Hiraoka, O.; Koyama, Y.; Hirota, Y.; Ayusawa, D.; Seno, T.; Garrett, C.; Wataya, Y. Deoxyribonucleoside triphosphate imbalance. 5-Fluorodeoxyuridine-induced DNA double strand breaks in mouse FM3A cells and the mechanism of cell death. J. Biol. Chem. 1987, 262, 8235–8241. [Google Scholar] [CrossRef]
- Santi, D.V.; Hardy, L.W. Catalytic mechanism and inhibition of tRNA (uracil-5-)methyltransferase: Evidence for covalent catalysis. Biochemistry 1987, 26, 8599–8606. [Google Scholar] [CrossRef]
- Randerath, K.; Tseng, W.C.; Harris, J.S.; Lu, L.J. Specific effects of 5-fluoropyrimidines and 5-azapyrimidines on modification of the 5 position of pyrimidines, in particular the synthesis of 5-methyluracil and 5-methylcytosine in nucleic acids. Recent Results Cancer Res. 1983, 84, 283–297. [Google Scholar]
- Chalabi-Dchar, M.; Fenouil, T.; Machon, C.; Vincent, A.; Catez, F.; Marcel, V.; Mertani, H.C.; Saurin, J.C.; Bouvet, P.; Guitton, J.; et al. A novel view on an old drug, 5-fluorouracil: An unexpected RNA modifier with intriguing impact on cancer cell fate. NAR Cancer 2021, 3, zcab032. [Google Scholar] [CrossRef]
- Udofot, O.; Affram, K.; Israel, B.; Agyare, E. Cytotoxicity of 5-fluorouracil-loaded pH-sensitive liposomal nanoparticles in colorectal cancer cell lines. Integr. Cancer Sci. Ther. 2015, 2, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Heggie, G.D.; Sommadossi, J.P.; Cross, D.S.; Huster, W.J.; Diasio, R.B. Clinical pharmacokinetics of 5-fluorouracil and its metabolites in plasma, urine, and bile. Cancer Res. 1987, 47, 2203–2206. [Google Scholar]
- Ward, S.E.; Kaltenthaler, E.; Cowan, J.; Marples, M.; Orr, B.; Seymour, M.T. The clinical and economic benefits of capecitabine and tegafur with uracil in metastatic colorectal cancer. Br. J. Cancer 2006, 95, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Ai, Y.; Obianom, O.N.; Kuser, M.; Li, Y.; Shu, Y.; Xue, F. Enhanced Tumor Selectivity of 5-Fluorouracil Using a Reactive Oxygen Species-Activated Prodrug Approach. ACS Med Chem Lett 2019, 10, 127–131. [Google Scholar] [CrossRef]
- Ikeda, K.; Yoshisue, K.; Matsushima, E.; Nagayama, S.; Kobayashi, K.; Tyson, C.A.; Chiba, K.; Kawaguchi, Y. Bioactivation of tegafur to 5-fluorouracil is catalyzed by cytochrome P-450 2A6 in human liver microsomes in vitro. Clin. Cancer Res. 2000, 6, 4409–4415. [Google Scholar]
- El Sayed, Y.M.; Sadée, W. Metabolic activation of R,S-1-(tetrahydro-2-furanyl)-5-fluorouracil (ftorafur) to 5-fluorouracil by soluble enzymes. Cancer Res. 1983, 43, 4039–4044. [Google Scholar]
- Capecitabine 2023. Available online: https://clinicaltrials.gov/search?cond=Capecitabine (accessed on 8 October 2023).
- Tegafur. 2023. Available online: https://clinicaltrials.gov/search?cond=Tegafur (accessed on 8 October 2023).
- Montgomery, J.A.; Hewson, K. Nucleosides of 2-fluoroadenine. J. Med. Chem. 1969, 12, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, V.; Plunkett, W. Cellular and clinical pharmacology of fludarabine. Clin. Pharmacokinet. 2002, 41, 93–103. [Google Scholar] [CrossRef]
- Lukenbill, J.; Kalaycio, M. Fludarabine: A review of the clear benefits and potential harms. Leuk. Res. 2013, 37, 986–994. [Google Scholar] [CrossRef]
- Montgomery, J.A.; Clayton, S.D.; Shortnacv, A.T. An improved procedure for the preparation of 9-β-D-Arabitiofuranosyl-2-ñuoroadenine. J. Heterocycl. Chem. 1979, 16, 157–160. [Google Scholar] [CrossRef]
- Blumbergs, P.; Khan, M.S.; Kalamas, R.L. Process for the Preparation of 2-Amino-9-(2,3,5-Tri-O-benzyl-beta-D-arabiofuranosyl) Adenine and Novel Intermediate. U.S. Patent 5,110,919, 5 May 1992. [Google Scholar]
- Shen, C.; Liu, J.; Ouyang, W.; Ding, H.; Bai, J.; Xiao, Q. Practical Synthesis of Fludarabine and Nelarabine. Synthesis 2020, 52, 417–423. [Google Scholar] [CrossRef]
- Ding, H.; Li, C.; Zhou, Y.; Hong, S.; Zhang, N.; Xiao, Q. Stereoselective synthesis of 2′-modified nucleosides by using ortho-alkynyl benzoate as a gold(i)-catalyzed removable neighboring participation group. RSC Adv. 2017, 7, 1814–1817. [Google Scholar] [CrossRef]
- Ricci, F.; Tedeschi, A.; Morra, E.; Montillo, M. Fludarabine in the treatment of chronic lymphocytic leukemia: A review. Ther. Clin. Risk Manag. 2009, 5, 187–207. [Google Scholar]
- Lowe, K.L.; Mackall, C.L.; Norry, E.; Amado, R.; Jakobsen, B.K.; Binder, G. Fludarabine and neurotoxicity in engineered T-cell therapy. Gene Ther. 2018, 25, 176–191. [Google Scholar] [CrossRef]
- Meng, H.; Yang, C.; Ni, W.; Ding, W.; Yang, X.; Qian, W. Antitumor activity of fludarabine against human multiple myeloma in vitro and in vivo. Eur. J. Haematol. 2007, 79, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Wen, C.; Li, Z.; Lin, S.; Gao, S.; Ding, H.; Zou, P.; Xing, Z.; Yu, Y. Fludarabine Inhibits Infection of Zika virus, SFTS Phlebovirus, and Enterovirus A71. Viruses 2021, 13, 774. [Google Scholar] [CrossRef]
- Faderl, S.; Gandhi, V.; Keating, M.J.; Jeha, S.; Plunkett, W.; Kantarjian, H.M. The role of clofarabine in hematologic and solid malignancies—Development of a next-generation nucleoside analog. Cancer 2005, 103, 1985–1995. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, V.; Kantarjian, H.; Faderl, S.; Bonate, P.; Du, M.; Ayres, M.; Rios, M.B.; Keating, M.J.; Plunkett, W. Pharmacokinetics and pharmacodynamics of plasma clofarabine and cellular clofarabine triphosphate in patients with acute leukemias. Clin. Cancer Res. 2003, 9, 6335–6342. [Google Scholar] [PubMed]
- Kantarjian, H.M.; Jeha, S.; Gandhi, V.; Wess, M.; Faderl, S. Clofarabine: Past, present, and future. Leuk. Lymphoma 2007, 48, 1922–1930. [Google Scholar] [CrossRef]
- Montgomery, J.A.; Shortnacy-Fowler, A.T.; Clayton, S.D.; Riordan, J.M.; Secrist III, J.A. Synthesis and biological activity of 2′-fluoro-2-halo derivatives of 9-. beta.-D-arabinofuranosyladenine. J. Med. Chem. 1992, 35, 397–401. [Google Scholar] [CrossRef]
- Bauta, W.E.; Schulmeier, B.E.; Burke, B.; Puente, J.F.; Cantrell, W.R.; Lovett, D.; Goebel, J.; Anderson, B.; Ionescu, D.; Guo, R. A New Process for Antineoplastic Agent Clofarabine. Org. Process Res. Dev. 2004, 8, 889–896. [Google Scholar] [CrossRef]
- Tann, C.H.; Brodfuehrer, P.R.; Brundidge, S.P.; Sapino Jr, C.; Howell, H.G. Fluorocarbohydrates in synthesis. An efficient synthesis of 1-(2-deoxy-2-fluoro-beta-D-arabinofuranosyl)-5-iodouracil (beta.-FIAU) and 1-(2-deoxy-2-fluoro-beta-D-arabinofuranosyl) thymine (beta.-FMAU). J. Org. Chem. 1985, 50, 3644–3647. [Google Scholar] [CrossRef]
- (112) Howell, H.G.; Brodfuehrer, P.R.; Brundidge, S.P.; Benigni, D.A.; Sapino, C., Jr. Antiviral nucleosides. A stereospecific, total synthesis of 2′-fluoro-2′-deoxy-.beta.-D-arabinofuranosyl nucleosides. J. Org. Chem. 1988, 53, 85–88. [Google Scholar] [CrossRef]
- Anderson, B.G.; Bauta, W.E.; Cantrell, J.W.R.; Engles, T.; Lovett, D.P. Isolation, Synthesis, and Characterization of Impurities and Degradants from the Clofarabine Process. Org. Process Res. Dev. 2008, 12, 1229–1237. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Drug Approval Package: Clofarabine Injection; U.S. Food and Drug Administration: Silver Spring, MD, USA, 2004. [Google Scholar]
- Takahashi, T.; Kanazawa, J.; Akinaga, S.; Tamaoki, T.; Okabe, M. Antitumor activity of 2-chloro-9-(2-deoxy-2-fluoro-beta-D-arabinofuranosyl) adenine, a novel deoxyadenosine analog, against human colon tumor xenografts by oral administration. Cancer Chemother. Pharmacol. 1999, 43, 233–240. [Google Scholar] [CrossRef]
- Zhenchuk, A.; Lotfi, K.; Juliusson, G.; Albertioni, F. Mechanisms of anti-cancer action and pharmacology of clofarabine. Biochem. Pharmacol. 2009, 78, 1351–1359. [Google Scholar] [CrossRef]
- Hertel, L.W.; Kroin, J.S.; Misner, J.W.; Tustin, J.M. Synthesis of 2-deoxy-2,2-difluoro-D-ribose and 2-deoxy-2,2′-difluoro-D-ribofuranosyl nucleosides. J. Org. Chem. 1988, 53, 2406–2409. [Google Scholar] [CrossRef]
- de Sousa Cavalcante, L.; Monteiro, G. Gemcitabine: Metabolism and molecular mechanisms of action, sensitivity and chemoresistance in pancreatic cancer. Eur. J. Pharmacol. 2014, 741, 8–16. [Google Scholar] [CrossRef]
- Oh, D.-Y.; Lee, K.-H.; Lee, D.-W.; Yoon, J.; Kim, T.-Y.; Bang, J.-H.; Nam, A.-R.; Oh, K.-S.; Kim, J.-M.; Lee, Y.; et al. Gemcitabine and cisplatin plus durvalumab with or without tremelimumab in chemotherapy-naive patients with advanced biliary tract cancer: An open-label, single-centre, phase 2 study. Lancet Gastroenterol. Hepatol. 2022, 7, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Ioka, T.; Kanai, M.; Kobayashi, S.; Sakai, D.; Eguchi, H.; Baba, H.; Seo, S.; Taketomi, A.; Takayama, T.; Yamaue, H.; et al. Randomized phase III study of gemcitabine, cisplatin plus S-1 versus gemcitabine, cisplatin for advanced biliary tract cancer (KHBO1401- MITSUBA). J. Hepato-Biliary-Pancreat. Sci. 2023, 30, 102–110. [Google Scholar] [CrossRef]
- Beutel, A.K.; Halbrook, C.J. Barriers and opportunities for gemcitabine in pancreatic cancer therapy. Am. J. Physiol. Cell Physiol. 2023, 324, C540–C552. [Google Scholar] [CrossRef]
- Nishimoto, A. Effective combinations of anti-cancer and targeted drugs for pancreatic cancer treatment. World J. Gastroenterol. 2022, 28, 3637–3643. [Google Scholar] [CrossRef] [PubMed]
- Ansari, D.; Tingstedt, B.; Andersson, R. Pancreatic cancer—Cost for overtreatment with gemcitabine. Acta Oncol. 2013, 52, 1146–1151. [Google Scholar] [CrossRef]
- Derissen, E.J.B.; Huitema, A.D.R.; Rosing, H.; Schellens, J.H.M.; Beijnen, J.H. Intracellular pharmacokinetics of gemcitabine, its deaminated metabolite 2′,2′-difluorodeoxyuridine and their nucleotides. Br. J. Clin. Pharmacol. 2018, 84, 1279–1289. [Google Scholar] [CrossRef]
- Derissen, E.J.B.; Beijnen, J.H. Intracellular Pharmacokinetics of Pyrimidine Analogues used in Oncology and the Correlation with Drug Action. Clin. Pharmacokinet. 2020, 59, 1521–1550. [Google Scholar] [CrossRef]
- Ruiz van Haperen, V.W.; Veerman, G.; Vermorken, J.B.; Peters, G.J. 2′,2′-Difluoro-deoxycytidine (gemcitabine) incorporation into RNA and DNA of tumour cell lines. Biochem. Pharmacol. 1993, 46, 762–766. [Google Scholar] [CrossRef]
- Chang, Y.-K.; Lee, J.; Park, G.-S.; Lee, M.; Park, C.H.; Kim, H.K.; Lee, G.; Lee, B.-Y.; Baek, J.Y.; Kim, K.S. An efficient large-scale synthesis of gemcitabine employing a crystalline 2,2-difluoro-α-ribofuranosyl bromide. Tetrahedron 2010, 66, 5687–5691. [Google Scholar] [CrossRef]
- Hui, Y.F.; Reitz, J. Gemcitabine: A cytidine analogue active against solid tumors. Am. J. Health-Syst. Pharm. 1997, 54, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Hernández, P.; Olivera, P.; Dueñas-Gonzalez, A.; Pérez-Pastenes, M.A.; Zárate, A.; Maldonado, V.; Meléndez-Zajgla, J. Gemcitabine activity in cervical cancer cell lines. Cancer Chemother. Pharmacol. 2001, 48, 488–492. [Google Scholar] [CrossRef]
- Noble, S.; Goa, K.L. Gemcitabine. A review of its pharmacology and clinical potential in non-small cell lung cancer and pancreatic cancer. Drugs 1997, 54, 447–472. [Google Scholar] [CrossRef]
- Caplin, M.E. Novel Alternatives to Chemotherapy in Advanced Disease: Gastrin Antibodies. In Pancreatic Disease; Springer: London, UK, 2004; pp. 93–98. [Google Scholar]
- Dyall, J.; Coleman, C.M.; Hart, B.J.; Venkataraman, T.; Holbrook, M.R.; Kindrachuk, J.; Johnson, R.F.; Olinger, G.G., Jr.; Jahrling, P.B.; Laidlaw, M.; et al. Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob. Agents Chemother. 2014, 58, 4885–4893. [Google Scholar] [CrossRef]
- Jang, Y.; Shin, J.S.; Lee, M.K.; Jung, E.; An, T.; Kim, U.I.; Kim, K.; Kim, M. Comparison of Antiviral Activity of Gemcitabine with 2′-Fluoro-2′-Deoxycytidine and Combination Therapy with Remdesivir against SARS-CoV-2. Int. J. Mol. Sci. 2021, 22, 1581. [Google Scholar] [CrossRef]
- Kuivanen, S.; Bespalov, M.M.; Nandania, J.; Ianevski, A.; Velagapudi, V.; De Brabander, J.K.; Kainov, D.E.; Vapalahti, O. Obatoclax, saliphenylhalamide and gemcitabine inhibit Zika virus infection in vitro and differentially affect cellular signaling, transcription and metabolism. Antivir. Res. 2017, 139, 117–128. [Google Scholar] [CrossRef]
- Beran, R.K.F.; Sharma, R.; Corsa, A.C.; Tian, Y.; Golde, J.; Lundgaard, G.; Delaney, W.E.; Zhong, W.D.; Greenstein, A.E. Cellular Growth Kinetics Distinguish a Cyclophilin Inhibitor from an HSP90 Inhibitor as a Selective Inhibitor of Hepatitis C Virus. PLoS ONE 2012, 7, e30286. [Google Scholar] [CrossRef]
- Clouser, C.L.; Holtz, C.M.; Mullett, M.; Crankshaw, D.L.; Briggs, J.E.; O’Sullivan, M.G.; Patterson, S.E.; Mansky, L.M. Activity of a novel combined antiretroviral therapy of gemcitabine and decitabine in a mouse model for HIV-1. Antimicrob. Agents Chemother. 2012, 56, 1942–1948. [Google Scholar] [CrossRef]
- Denisova, O.V.; Kakkola, L.; Feng, L.; Stenman, J.; Nagaraj, A.; Lampe, J.; Yadav, B.; Aittokallio, T.; Kaukinen, P.; Ahola, T.; et al. Obatoclax, saliphenylhalamide, and gemcitabine inhibit influenza a virus infection. J. Biol. Chem. 2012, 287, 35324–35332. [Google Scholar] [CrossRef]
- Kim, M.P.; Gallick, G.E. Gemcitabine Resistance in Pancreatic Cancer: Picking the Key Players. Clin. Cancer Res. 2008, 14, 1284–1285. [Google Scholar] [CrossRef]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Pöttler, M.; Lan, B.; Grützmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 4504. [Google Scholar] [CrossRef]
- Shimma, N.; Umeda, I.; Arasaki, M.; Murasaki, C.; Masubuchi, K.; Kohchi, Y.; Miwa, M.; Ura, M.; Sawada, N.; Tahara, H.; et al. The design and synthesis of a new tumor-selective fluoropyrimidine carbamate, capecitabine. Bioorg. Med. Chem. 2000, 8, 1697–1706. [Google Scholar] [CrossRef]
- Sofia, M.J.; Bao, D.; Chang, W.; Du, J.; Nagarathnam, D.; Rachakonda, S.; Reddy, P.G.; Ross, B.S.; Wang, P.; Zhang, H.-R.; et al. Discovery of a β-d-2′-Deoxy-2′-α-fluoro-2′-β-C-methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis C Virus. J. Med. Chem. 2010, 53, 7202–7218. [Google Scholar] [CrossRef]
- Singh, U.S.; Konreddy, A.K.; Kothapalli, Y.; Liu, D.; Lloyd, M.G.; Annavarapu, V.; White, C.A.; Bartlett, M.G.; Moffat, J.F.; Chu, C.K. Prodrug Strategies for the Development of β-l-5-((E)-2-Bromovinyl)-1-((2S,4S)-2-(hydroxymethyl)-1,3-(dioxolane-4-yl))uracil (l-BHDU) against Varicella Zoster Virus (VZV). J. Med. Chem. 2023, 66, 7038–7053. [Google Scholar] [CrossRef] [PubMed]
- Mehellou, Y.; Balzarini, J.; McGuigan, C. Aryloxy Phosphoramidate Triesters: A Technology for Delivering Monophosphorylated Nucleosides and Sugars into Cells. ChemMedChem 2009, 4, 1779–1791. [Google Scholar] [CrossRef]
- Song, X.; Lorenzi, P.L.; Landowski, C.P.; Vig, B.S.; Hilfinger, J.M.; Amidon, G.L. Amino Acid Ester Prodrugs of the Anticancer Agent Gemcitabine: Synthesis, Bioconversion, Metabolic Bioevasion, and hPEPT1-Mediated Transport. Mol. Pharm. 2005, 2, 157–167. [Google Scholar] [CrossRef]
- Koolen, S.L.W.; Witteveen, P.O.; Jansen, R.S.; Langenberg, M.H.G.; Kronemeijer, R.H.; Nol, A.; Garcia-Ribas, I.; Callies, S.; Benhadji, K.A.; Slapak, C.A.; et al. Phase I Study of Oral Gemcitabine Prodrug (LY2334737) Alone and in Combination with Erlotinib in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2011, 17, 6071–6082. [Google Scholar] [CrossRef] [PubMed]
- Pratt, S.E.; Durland-Busbice, S.; Shepard, R.L.; Heinz-Taheny, K.; Iversen, P.W.; Dantzig, A.H. Human Carboxylesterase-2 Hydrolyzes the Prodrug of Gemcitabine (LY2334737) and Confers Prodrug Sensitivity to Cancer Cells. Clin. Cancer Res. 2013, 19, 1159–1168. [Google Scholar] [CrossRef]
- Bender, D.M.; Bao, J.; Dantzig, A.H.; Diseroad, W.D.; Law, K.L.; Magnus, N.A.; Peterson, J.A.; Perkins, E.J.; Pu, Y.J.; Reutzel-Edens, S.M.; et al. Synthesis, Crystallization, and Biological Evaluation of an Orally Active Prodrug of Gemcitabine. J. Med. Chem. 2009, 52, 6958–6961. [Google Scholar] [CrossRef]
- Alexander, P.; Kucera, G.; Pardee, T.S. Improving nucleoside analogs via lipid conjugation: Is fatter any better? Crit. Rev. Oncol. Hematol. 2016, 100, 46–56. [Google Scholar] [CrossRef]
- Galmarini, C.M.; Myhren, F.; Sandvold, M.L. CP-4055 and CP-4126 are active in ara-C and gemcitabine-resistant lymphoma cell lines. Br. J. Haematol. 2009, 144, 273–275. [Google Scholar] [CrossRef]
- Bergman, A.M.; Adema, A.D.; Balzarini, J.; Bruheim, S.; Fichtner, I.; Noordhuis, P.; Fodstad, O.; Myhren, F.; Sandvold, M.L.; Hendriks, H.R.; et al. Antiproliferative activity, mechanism of action and oral antitumor activity of CP-4126, a fatty acid derivative of gemcitabine, in in vitro and in vivo tumor models. Investig. New Drugs 2011, 29, 456–466. [Google Scholar] [CrossRef]
- Slusarczyk, M.; Lopez, M.H.; Balzarini, J.; Mason, M.; Jiang, W.G.; Blagden, S.; Thompson, E.; Ghazaly, E.; McGuigan, C. Application of ProTide technology to gemcitabine: A successful approach to overcome the key cancer resistance mechanisms leads to a new agent (NUC-1031) in clinical development. J. Med. Chem. 2014, 57, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- Rauchwerger, D.R.; Firby, P.S.; Hedley, D.W.; Moore, M.J. Equilibrative-sensitive nucleoside transporter and its role in gemcitabine sensitivity. Cancer Res. 2000, 60, 6075–6079. [Google Scholar] [PubMed]
- Bouffard, D.Y.; Laliberté, J.; Momparler, R.L. Kinetic studies on 2′,2′-difluorodeoxycytidine (Gemcitabine) with purified human deoxycytidine kinase and cytidine deaminase. Biochem. Pharmacol. 1993, 45, 1857–1861. [Google Scholar] [CrossRef] [PubMed]
- Abbruzzese, J.L.; Grunewald, R.; Weeks, E.A.; Gravel, D.; Adams, T.; Nowak, B.; Mineishi, S.; Tarassoff, P.; Satterlee, W.; Raber, M.N.; et al. A phase I clinical, plasma, and cellular pharmacology study of gemcitabine. J. Clin. Oncol. 1991, 9, 491–498. [Google Scholar] [CrossRef]
- Saiki, Y.; Hirota, S.; Horii, A. Attempts to remodel the pathways of gemcitabine metabolism: Recent approaches to overcoming tumours with acquired chemoresistance. Cancer Drug Resist. 2020, 3, 819–831. [Google Scholar] [CrossRef]
- NUC-1031. 2024. Available online: https://clinicaltrials.gov/search?term=NUC-1031 (accessed on 16 November 2024).
- Helwick, C. NUC-1031/Cisplatin Fails to Improve Outcomes in Advanced Biliary Tract Cancer. 2023. Available online: https://ascopost.com/issues/october-10-2023/nuc-1031cisplatin-fails-to-improve-outcomes-in-advanced-biliary-tract-cancer/ (accessed on 16 November 2024).
- Zhang, L.; Qi, K.; Xu, J.; Xing, Y.; Wang, X.; Tong, L.; He, Z.; Xu, W.; Li, X.; Jiang, Y. Design, Synthesis, and Anti-Cancer Evaluation of Novel Cyclic Phosphate Prodrug of Gemcitabine. J. Med. Chem. 2023, 66, 4150–4166. [Google Scholar] [CrossRef]
- Tsume, Y.; Hilfinger, J.M.; Amidon, G.L. Enhanced cancer cell growth inhibition by dipeptide prodrugs of floxuridine: Increased transporter affinity and metabolic stability. Mol. Pharm. 2008, 5, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Power, D.G.; Kemeny, N.E. The role of floxuridine in metastatic liver disease. Mol. Cancer Ther. 2009, 8, 1015–1025. [Google Scholar] [CrossRef]
- Floxuridine. 2023. Available online: https://www.clinicaltrials.gov/ct2/results?cond=&term=Floxuridine&cntry=&state=&city=&dist= (accessed on 31 March 2023).
- Murakami, Y.; Kazuno, H.; Emura, T.; Tsujimoto, H.; Suzuki, N.; Fukushima, M. Different mechanisms of acquired resistance to fluorinated pyrimidines in human colorectal cancer cells. Int. J. Oncol. 2000, 17, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Noordhuis, P.; Holwerda, U.; Van der Wilt, C.L.; Van Groeningen, C.J.; Smid, K.; Meijer, S.; Pinedo, H.M.; Peters, G.J. 5-Fluorouracil incorporation into RNA and DNA in relation to thymidylate synthase inhibition of human colorectal cancers. Ann. Oncol. 2004, 15, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Hoffer, M.; Duschinsky, R.; Fox, J.J.; Yung, N. Simple Syntheses of Pyrimidine-2′-Deoxy-Ribonucleosides1. J. Am. Chem. Soc. 1959, 81, 4112–4113. [Google Scholar] [CrossRef]
- Robins, M.J.; MacCoss, M.; Naik, S.R.; Ramani, G. Nucleic acid related compounds. 21. Direct fluorination of uracil and cytosine bases and nucleosides using trifluoromethyl hypofluorite. Mechanism, stereochemistry, and synthetic applications. J. Am. Chem. Soc. 1976, 98, 7381–7389. [Google Scholar] [CrossRef] [PubMed]
- Hajime, A. Stereoselective Synthesis of Anomers of 5-Substituted 2′-Deoxyuridines. Bull. Chem. Soc. Jpn. 1987, 60, 2073–2077. [Google Scholar]
- Wang, Z.X.; Duan, W.; Wiebe, L.I.; Balzarini, J.; De Clercq, E.; Knaus, E.E. Synthesis of 1-(2-deoxy-beta-D-ribofuranosyl)-2,4-difluoro-5-substituted-benzene thymidine mimics, some related alpha-anomers, and their evaluation as antiviral and anticancer agents. Nucleosides Nucleotides Nucleic Acids 2001, 20, 11–40. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Drug Approval Package: Floxuridine for Injection USP, 500 mg/vial; U.S. Food and Drug Administration: Silver Spring, MD, USA, 2001. [Google Scholar]
- Hocek, M.; Votruba, I. Covalent analogues of DNA base-pairs and triplets. Part 2: Synthesis and cytostatic activity of bis(purin-6-yl)acetylenes,-diacetylenes and related compounds. Bioorg. Med. Chem. Lett. 2002, 12, 1055–1058. [Google Scholar] [CrossRef] [PubMed]
- CLINIC, M. Capecitabine (Oral Route). Available online: https://www.mayoclinic.org/drugs-supplements/capecitabine-oral-route/proper-use/drg-20062501 (accessed on 9 April 2023).
- Miwa, M.; Ura, M.; Nishida, M.; Sawada, N.; Ishikawa, T.; Mori, K.; Shimma, N.; Umeda, I.; Ishitsuka, H. Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. Eur. J. Cancer 1998, 34, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Capecitabine. Available online: https://www.clinicaltrials.gov/ct2/results?cond=&term=Capecitabine&cntry=&state=&city=&dist= (accessed on 9 April 2023).
- Mekala, N.; Moturu, M.V.R.K.; Dammalapati, R.V.L.N.; Parimi, A.R. Safe and Alternate Process for the Reductions of Methanesulfonates: Application in the Synthesis of 1,2,3-Triacetyl-5-deoxy-d-ribofuranoside. Org. Process Res. Dev. 2016, 20, 609–614. [Google Scholar] [CrossRef]
- Gelmon, K.; Chan, A.; Harbeck, N. The role of capecitabine in first-line treatment for patients with metastatic breast cancer. Oncologist 2006, 11 (Suppl. S1), 42–51. [Google Scholar] [CrossRef] [PubMed]
- Bensalem, A.; Bouzid, K. Docetaxel plus capecitabine in the treatment of previous anthracycline-treated patients with metastatic breast cancer. J. Clin. Oncol. 2009, 27 (Suppl. S15), e12014. [Google Scholar] [CrossRef]
- Adjei, A.A. A review of the pharmacology and clinical activity of new chemotherapy agents for the treatment of colorectal cancer. Brit. J. Clin. Pharmacol. 1999, 48, 265–277. [Google Scholar] [CrossRef]
- Ishitsuka, H. Capecitabine: Preclinical pharmacology studies. Investig. New Drugs 2000, 18, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Koukourakis, G.V.; Kouloulias, V.; Koukourakis, M.J.; Zacharias, G.A.; Zabatis, H.; Kouvaris, J. Efficacy of the oral fluorouracil pro-drug capecitabine in cancer treatment: A review. Molecules 2008, 13, 1897–1922. [Google Scholar] [CrossRef]
- Ciccolini, J.; Fina, F.; Bezulier, K.; Giacometti, S.; Roussel, M.; Evrard, A.; Cuq, P.; Romain, S.; Martin, P.M.; Aubert, C. Transmission of apoptosis in human colorectal tumor cells exposed to capecitabine, Xeloda, is mediated via Fas. Mol. Cancer Ther. 2002, 1, 923–927. [Google Scholar] [PubMed]
- Kato, H.; Ichinose, Y.; Ohta, M.; Hata, E.; Tsubota, N.; Tada, H.; Watanabe, Y.; Wada, H.; Tsuboi, M.; Hamajima, N.; et al. A Randomized Trial of Adjuvant Chemotherapy with Uracil–Tegafur for Adenocarcinoma of the Lung. New Engl. J. Med. 2004, 350, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Kinouchi, M.; Ishida, K.; Fujibuchi, W.; Naitoh, T.; Ogawa, H.; Ando, T.; Yazaki, N.; Watanabe, K.; Haneda, S.; et al. 5-fu metabolism in cancer and orally-administrable 5-fu drugs. Cancers 2010, 2, 1717–1730. [Google Scholar] [CrossRef]
- Tegafur, Gimeracil, and Oteracil. 2024. Available online: https://clinicaltrials.gov/search?term=Tegafur,%20gimeracil,%20and%20oteracil&page=1 (accessed on 24 October 2024).
- Blum, M.; Suzuki, A.; Ajani, J.A. A Comprehensive Review of S-1 in the Treatment of Advanced Gastric Adenocarcinoma. Future Oncol. 2011, 7, 715–726. [Google Scholar] [CrossRef]
- Komatsu, T.; Yamazaki, H.; Shimada, N.; Nakajima, M.; Yokoi, T. Roles of cytochromes P450 1A2, 2A6, and 2C8 in 5-fluorouracil formation from tegafur, an anticancer prodrug, in human liver microsomes. Drug Metab. Dispos. 2000, 28, 1457–1463. [Google Scholar] [CrossRef]
- Takiuchi, H.; Ajani, J.A. Uracil-tegafur in gastric carcinoma: A comprehensive review. J. Clin. Oncol. 1998, 16, 2877–2885. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute, NIH. Tegafur-Uracil. Available online: https://www.cancer.gov/publications/dictionaries/cancer-drug/def/tegafur-uracil (accessed on 10 November 2024).
- Hoff, P.M.; Pazdur, R. UFT Plus Oral Leucovorin: A New Oral Treatment for Colorectal Cancer. Oncologist 1998, 3, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Zasada, A.; Mironiuk-Puchalska, E.; Koszytkowska-Stawińska, M. Synthesis of Tegafur by the Alkylation of 5-Fluorouracil under the Lewis Acid and Metal Salt-Free Conditions. Org. Process Res. Dev. 2017, 21, 885–889. [Google Scholar] [CrossRef]
- Cubero, D.I.; Cruz, F.M.; Santi, P.; Silva, I.D.; Del Giglio, A. Tegafur-uracil is a safe alternative for the treatment of colorectal cancer in patients with partial dihydropyrimidine dehydrogenase deficiency: A proof of principle. Ther. Adv. Med. Oncol. 2012, 4, 167–172. [Google Scholar] [CrossRef]
- Engel, D.; Nudelman, A.; Tarasenko, N.; Levovich, I.; Makarovsky, I.; Sochotnikov, S.; Tarasenko, I.; Rephaeli, A. Novel prodrugs of tegafur that display improved anticancer activity and antiangiogenic properties. J. Med. Chem. 2008, 51, 314–323. [Google Scholar] [CrossRef]
- Balboni, B.; El Hassouni, B.; Honeywell, R.J.; Sarkisjan, D.; Giovannetti, E.; Poore, J.; Heaton, C.; Peterson, C.; Benaim, E.; Lee, Y.B.; et al. RX-3117 (fluorocyclopentenyl cytosine): A novel specific antimetabolite for selective cancer treatment. Expert Opin. Investig. Drugs 2019, 28, 311–322. [Google Scholar] [CrossRef]
- Sarkisjan, D.; Julsing, J.R.; El Hassouni, B.; Honeywell, R.J.; Kathmann, I.; Matherly, L.H.; Lee, Y.B.; Kim, D.J.; Peters, G.J. RX-3117 (Fluorocyclopentenyl-Cytosine)-Mediated Down-Regulation of DNA Methyltransferase 1 Leads to Protein Expression of Tumor-Suppressor Genes and Increased Functionality of the Proton-Coupled Folate Carrier. Int. J. Mol. Sci. 2020, 21, 2717. [Google Scholar] [CrossRef] [PubMed]
- Peters, G.J.; Smid, K.; Vecchi, L.; Kathmann, I.; Sarkisjan, D.; Honeywell, R.J.; Losekoot, N.; Ohne, O.; Orbach, A.; Blaugrund, E.; et al. Metabolism, mechanism of action and sensitivity profile of fluorocyclopentenylcytosine (RX-3117; TV-1360). Investig. New Drugs 2013, 31, 1444–1457. [Google Scholar] [CrossRef] [PubMed]
- RX-3117. 2023. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=RX-3117&cntry=&state=&city=&dist= (accessed on 12 March 2023).
- RX-3117. 2024. Available online: https://www.clinicaltrials.gov/search?term=RX-3117 (accessed on 11 November 2024).
- Choi, W.J.; Chung, H.J.; Chandra, G.; Alexander, V.; Zhao, L.X.; Lee, H.W.; Nayak, A.; Majik, M.S.; Kim, H.O.; Kim, J.H.; et al. Fluorocyclopentenyl-cytosine with Broad Spectrum and Potent Antitumor Activity. J. Med. Chem. 2012, 55, 4521–4525. [Google Scholar] [CrossRef]
- Blagden, S.; Spiliopoulou, P.; Spiers, L.; Gnanaranjan, C.; Qi, C.; Woodcock, V.K.; Moschandreas, J.; Tyrrell, H.E.J.; Griffiths, L.; Butcher, C.; et al. A phase I first-in-human, dose-escalation and expansion study to evaluate the safety and tolerability of NUC-3373 in patients with locally advanced, unresectable or metastatic solid malignancies. Ann. Oncol. 2018, 29, viii145. [Google Scholar] [CrossRef]
- Vande Voorde, J.; Liekens, S.; McGuigan, C.; Murziani, P.G.; Slusarczyk, M.; Balzarini, J. The cytostatic activity of NUC-3073, a phosphoramidate prodrug of 5-fluoro-2′-deoxyuridine, is independent of activation by thymidine kinase and insensitive to degradation by phosphorolytic enzymes. Biochem. Pharmacol. 2011, 82, 441–452. [Google Scholar] [CrossRef] [PubMed]
- NUC-3373. 2023. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=NUC+3373&cntry=&state=&city=&dist= (accessed on 5 March 2023).
- Hammond, W.A.; Swaika, A.; Mody, K. Pharmacologic resistance in colorectal cancer: A review. Ther. Adv. Med. Oncol. 2016, 8, 57–84. [Google Scholar] [CrossRef] [PubMed]
- Slusarczyk, M.; Serpi, M.; Ghazaly, E.; Kariuki, B.M.; McGuigan, C.; Pepper, C. Single Diastereomers of the Clinical Anticancer ProTide Agents NUC-1031 and NUC-3373 Preferentially Target Cancer Stem Cells In Vitro. J. Med. Chem. 2021, 64, 8179–8193. [Google Scholar] [CrossRef]
- Bré, J.; Dickson, A.L.; Read, O.J.; Zhang, Y.; McKissock, F.G.; Mullen, P.; Tang, P.; Zickuhr, G.M.; Czekster, C.M.; Harrison, D.J. The novel anti-cancer fluoropyrimidine NUC-3373 is a potent inhibitor of thymidylate synthase and an effective DNA-damaging agent. Cancer Chemother. Pharmacol. 2023, 91, 401–412. [Google Scholar] [CrossRef] [PubMed]
- McGuigan, C.; Murziani, P.; Slusarczyk, M.; Gonczy, B.; Vande Voorde, J.; Liekens, S.; Balzarini, J. Phosphoramidate ProTides of the Anticancer Agent FUDR Successfully Deliver the Preformed Bioactive Monophosphate in Cells and Confer Advantage over the Parent Nucleoside. J. Med. Chem. 2011, 54, 7247–7258. [Google Scholar] [CrossRef] [PubMed]
- Ghazaly, E.; Woodcock, V.K.; Spilipoulou, P.; Spiers, L.; Moschandreas, J.; Griffiths, L.; Gnanaranjan, C.; Harrison, D.J.; Evans, T.R.J.; Blagden, S.P. Interim pharmacokinetic (PK) and pharmacodynamic (PD) data from the first-in-human study of NUC-3373, a pyrimidine nucleotide analogue, in patients with advanced solid tumors. Ann. Oncol. 2017, 28, v128. [Google Scholar] [CrossRef]
- Evans, T.R.J.; Blagden, S.P.; Graham, J.S.; Ciombor, K.K.; De Gramont, A.; Tabernero, J.; Berlin, J. NuTide:302: A phase Ib study to assess the safety, pharmacokinetics and clinical activity of the ProTide NUC-3373 when combined with standard agents used in colorectal cancer. J. Clin. Oncol. 2019, 37, TPS719. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor or Virus | Cell Lines | IC50 | CC50/tox | Ref |
|---|---|---|---|---|
| Multiple Myeloma (MM) | RPMI 8226 | 1.54 µg/mL | - | [104] |
| MM.1S | 13.48 µg/mL | - | ||
| MM.1R | 33.79 µg/mL | - | ||
| ZIKV | Vero Cells | 0.13 +/− 0.04 µM | 3.10 µM | [105] |
| BHK-21 Cells | 0.41 +/− 0.04 µM | 3.61 µM | ||
| SFTSV | Vero Cells | 0.83 +/− 0.03 µM | 3.10 µM | [105] |
| BHK-21 Cells | 0.27 +/− 0.001 µM | 3.61 µM | ||
| EV-A71 | Vero Cells | 0.04 +/− 0.001 µM | 3.10 µM | [105] |
| Compound | Cell Lines | Antiproliferative Activity IC50 (µM) |
|---|---|---|
| Clofarabine (52) | HCT116 | 0.12 |
| HT-29 | 0.77 | |
| DLD-1 | 0.07 | |
| WiDr | 0.67 |
| Compound | Cervical Cancer Cell Lines | IC50 (µM) | IC30 (µM) | Ref. |
|---|---|---|---|---|
| Gemcitabine | SiHa Cells | 203 ± 12.3 µM | 0.1 ± 0.05 µM | [129] |
| CaLo Cells | 0.89 ± 0.10 µM | 0.2 ± 0.06 µM | ||
| InBl Cells | 0.63 ± 0.07 µM | 0.07 ± 0.01 µM | ||
| HeLa Cells | 0.32 ± 0.02 µM | 0.05 ± 0.01 µM | ||
| C33A Cells | 0.27 ± 0.08 µM | 0.04 ± 0.01 µM | ||
| CasKi Cells | 0.11 ± 0.04 µM | 0.02 ± 0.09 µM |
| Compound | Virus | Cell Line | EC50 | CC50 | Ref |
|---|---|---|---|---|---|
| Gemcitabine (53) | MERS-CoV | Vero E6 Cells | 1.2 µM | >10 µM | [132] |
| SARS-CoV | Vero E6 Cells | 4.9 µM | >10 µM | [132] | |
| ZIKA | RPE Cells | 0.01 µM | >10 µM | [134] | |
| HCV | Huh-7 Cells | 12 nM | >44,444 nM | [135] | |
| HIV | U373-MAGI-CXCR4CEM | 16.3 nM | ND | [136] | |
| IAV (Influenza A) | RPE Cells | 0.068 ± 0.005 µM | ND | [137] | |
| HSV-1 | RPE Cells | >4 logs at 3 µM | ND | [137] | |
| SARS-CoV-2 | Vero Cells | 1.2 ± 1.1 µM | >300 µM | [133] |
| Compound | Cell Lines | IC50 (µM) |
|---|---|---|
| Floxuridine (FUDR, 54) | L1210 | <0.02 ± 0.002 |
| L929 | >25 | |
| HeLa S3 | >25 | |
| CCRF-CEM | 0.05 ± 0.004 |
| Compound | Human Colorectal and Hepatoma Cells | IC50 (µM) | Ref. |
|---|---|---|---|
| Capecitabine (55) | LS174T WT | 890 ± 48 µM | [180] |
| LS174T-c2 | 330 ± 4 µM | ||
| LS174T WT + HepG2 | 630 ± 14 µM | ||
| LS174T-c2 + HepG2 | 89 ± 6 µM |
| Compound | Cells | IC50 | Ref |
|---|---|---|---|
| Tegafur (56) | HT-29 | 201 µM | [191] |
| BxPC-3 | 172 µM | [191] | |
| SK-ES | 174 µM | [191] |
| Compound | Cancer Cell Line | IC50 | Ref |
|---|---|---|---|
| RX-3117 (57) | NCIH226 (lung) | 0.25 μM | [197] |
| HCT116 (colon) | 0.19 μM | ||
| U251 (brain) | 0.83 μM | ||
| K562 (Leukemia) | 0.82 μM | ||
| PC-3 (prostate) | 0.63 μM | ||
| HepG2 (liver) | 0.79 μM | ||
| UMRC2 (kidney) | 0.83 μM |
| Compound | Tumor Cells Lines | IC50 (µM) |
|---|---|---|
| NUC-3373 (58) | L1210/W | 0.011 ± 0.007 |
| L1210/TK− | 0.045 ± 0.027 | |
| CEM/0 | 0.068 ± 0.035 | |
| CEM/TK− | 0.31 ± 0.06 | |
| HeLa | 0.065 ± 0.013 | |
| HeLa/TK− | 2.5 ± 1.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kothapalli, Y.; Lesperance, T.A.; Jones, R.A.; Chu, C.K.; Singh, U.S. Chemical Space of Fluorinated Nucleosides/Nucleotides in Biomedical Research and Anticancer Drug Discovery. Chemistry 2025, 7, 7. https://doi.org/10.3390/chemistry7010007
Kothapalli Y, Lesperance TA, Jones RA, Chu CK, Singh US. Chemical Space of Fluorinated Nucleosides/Nucleotides in Biomedical Research and Anticancer Drug Discovery. Chemistry. 2025; 7(1):7. https://doi.org/10.3390/chemistry7010007
Chicago/Turabian StyleKothapalli, Yugandhar, Tucker A. Lesperance, Ransom A. Jones, Chung K. Chu, and Uma S. Singh. 2025. "Chemical Space of Fluorinated Nucleosides/Nucleotides in Biomedical Research and Anticancer Drug Discovery" Chemistry 7, no. 1: 7. https://doi.org/10.3390/chemistry7010007
APA StyleKothapalli, Y., Lesperance, T. A., Jones, R. A., Chu, C. K., & Singh, U. S. (2025). Chemical Space of Fluorinated Nucleosides/Nucleotides in Biomedical Research and Anticancer Drug Discovery. Chemistry, 7(1), 7. https://doi.org/10.3390/chemistry7010007