
Crystalline Diradical Dianions and Radical Anions of Indenofluorenediones

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
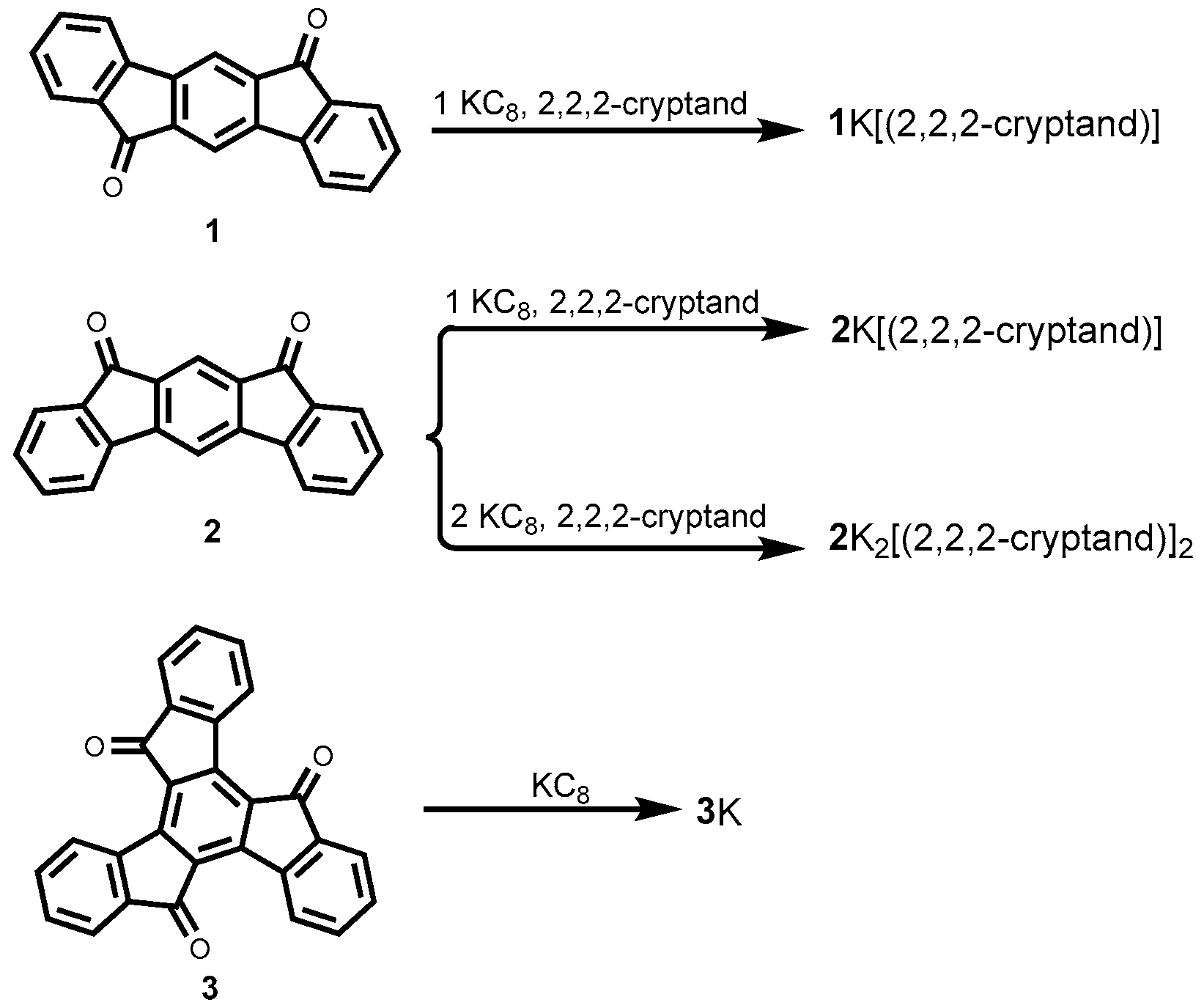
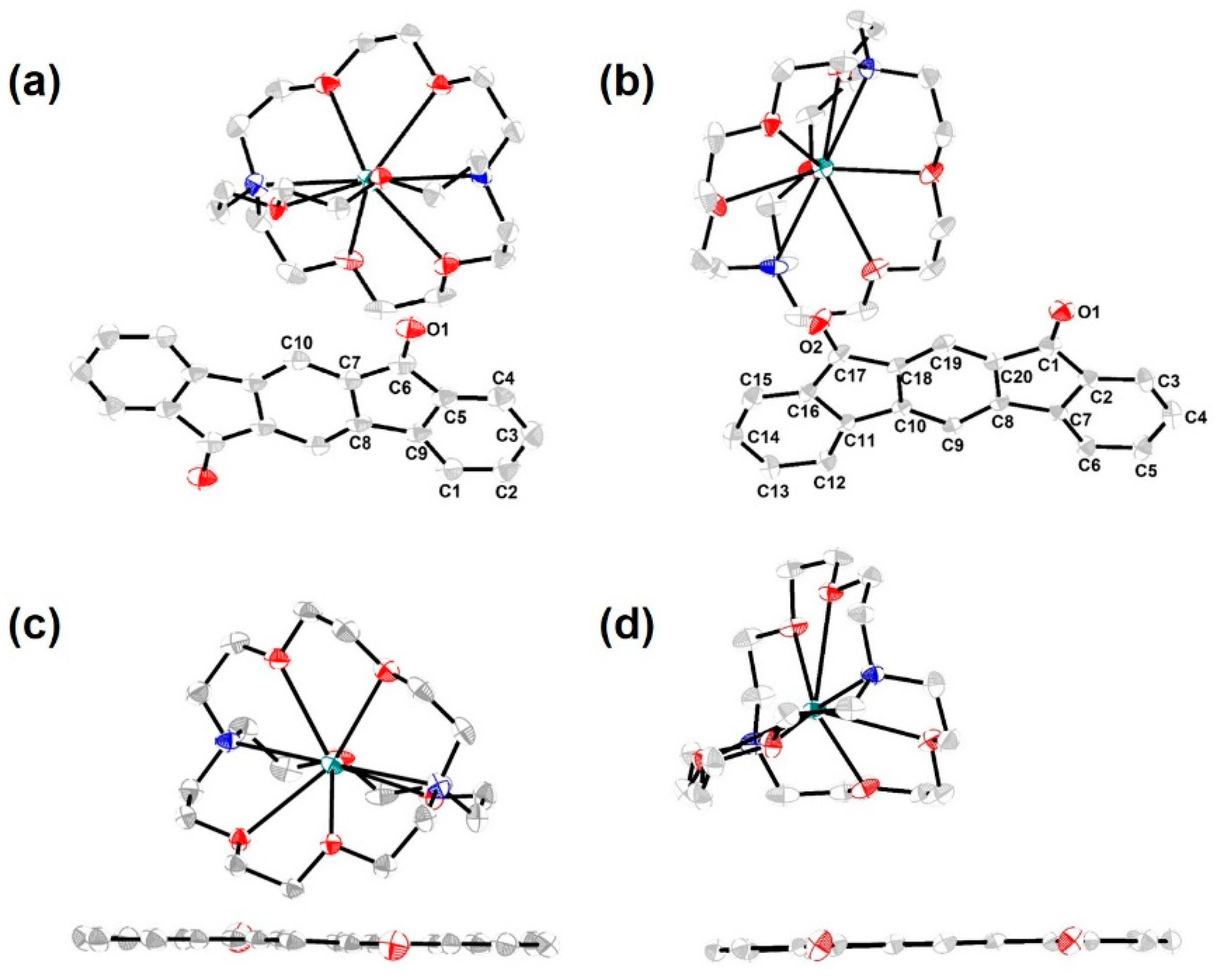
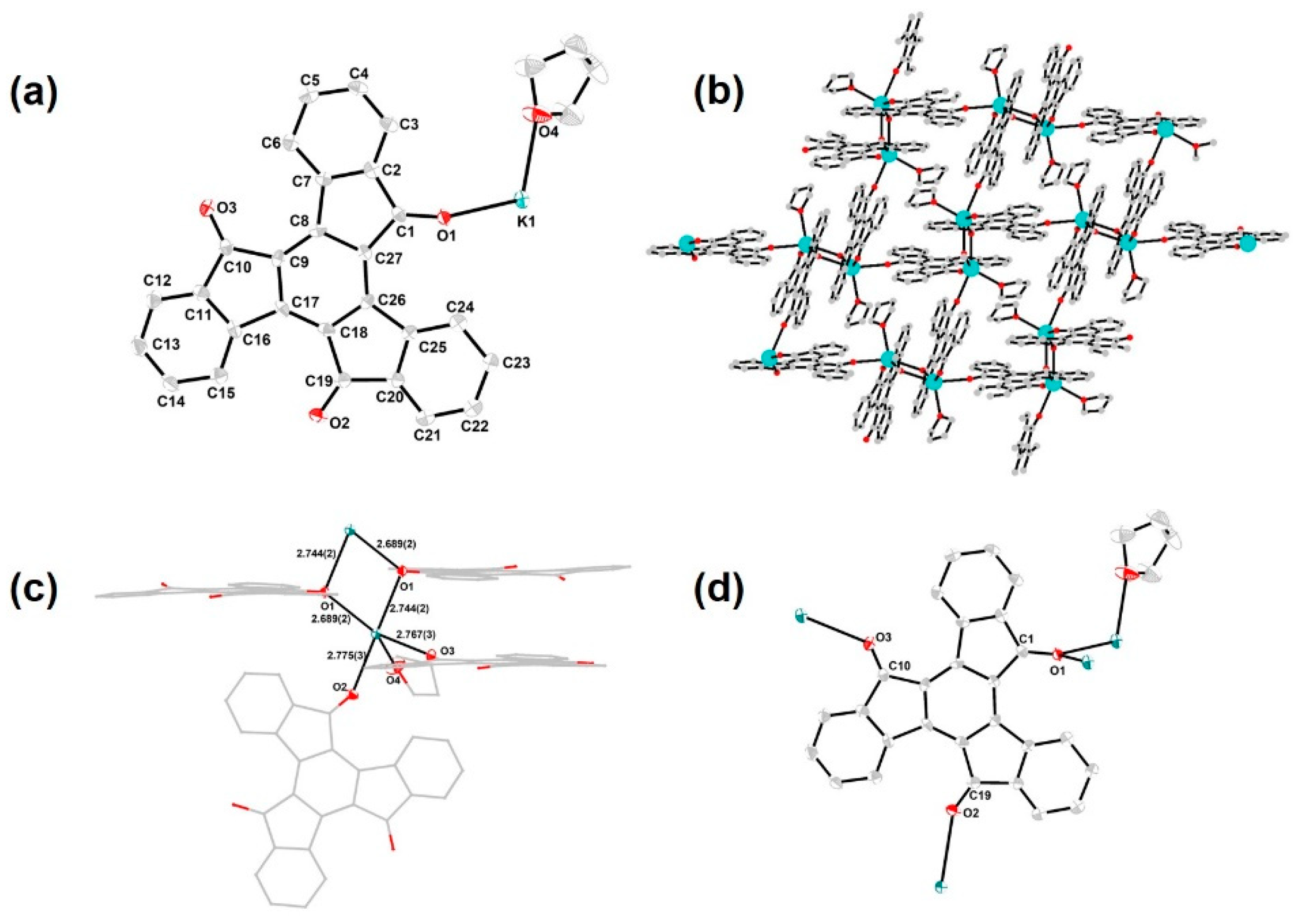
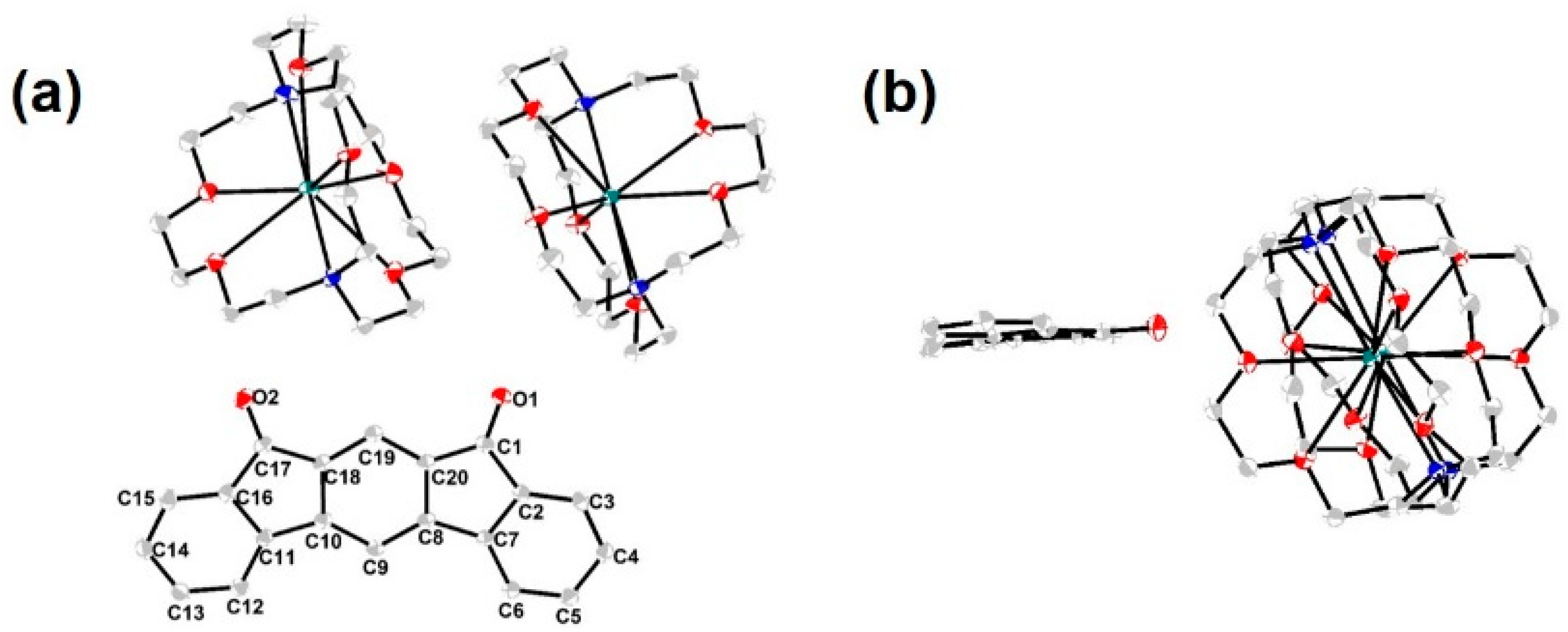
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gámez-Valenzuela, S.; Echeverri, M.; Gómez-Lor, B.; Martínez, J.I.; Carmen Ruiz Delgado, M. In silico design of 2D polymers containing truxene-based platforms: Insights into their structural and electronic properties. J. Mater. Chem. C 2020, 8, 15416–15425. [Google Scholar] [CrossRef]
- Samorí, P.; Palermo, V. Flexible Carbon-Based Electronics; Wiley-VCH: Weinheim, Germany, 2018. [Google Scholar]
- Klauk, H. Organic Electronics: Materials, Manufacturing, and Applications; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Klauk, H. Organic Electronics II: More Materials and Applications; Wiley-VCH: Weinheim, Germany, 2012. [Google Scholar]
- Quinn, J.T.E.; Zhu, J.; Li, X.; Wang, J.; Li, Y. Recent progress in the development of n-type organic semiconductors for organic field effect transistors. J. Mater. Chem. C 2017, 5, 8654–8681. [Google Scholar] [CrossRef]
- Anthony, J.E.; Facchetti, A.; Heeney, M.; Marder, S.R.; Zhan, X. n-Type organic semiconductors in organic electronics. Adv. Mater. 2010, 22, 3876–3892. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Kumaki, D.; Nishida, J.; Tokito, S.; Yamashita, Y. High Performance n-type field-effect transistors based on indenofluorenedione and diindenopyrazinedione derivatives. Chem. Mater. 2008, 20, 2615–2617. [Google Scholar] [CrossRef]
- Can, A.; Facchetti, A.; Usta, H. Indenofluorenes for organic optoelectronics: The dance of fused five- and six-membered rings enabling structural versatility. J. Mater. Chem. C 2022, 10, 8496–8535. [Google Scholar] [CrossRef]
- Jung, B.J.; Tremblay, N.J.; Yeh, M.-L.; Katz, H.E. Molecular design and synthetic approaches to electron-transporting organic transistor semiconductors. Chem. Mater. 2011, 23, 568–582. [Google Scholar] [CrossRef]
- Homnick, P.J.; Lahti, P.M. Modular electron donor group tuning of frontier energy levels in diarylaminofluorenone push–pull molecules. Phys. Chem. Chem. Phys. 2012, 14, 11961–11968. [Google Scholar] [CrossRef]
- Gong, X.; Iyer, P.K.; Moses, D.; Bazan, G.C.; Heeger, A.J.; Xiao, S.S. Stabilized blue emission from polyfluorene-based light-emitting diodes: Elimination of fluorenone defects. Adv. Funct. Mater. 2003, 13, 325–330. [Google Scholar] [CrossRef]
- Uckert, F.; Setayesh, S.; Müllen, K. A precursor route to 2,7-poly(9-fluorenone). Macromolecules 1999, 32, 4519–4524. [Google Scholar] [CrossRef]
- Al-Betar, A.R.F.; Pickup, P.G. Electrochemical n-doping of polyfluorenone films. Synth. Met. 2019, 254, 128–133. [Google Scholar] [CrossRef]
- Jiao, Y.; Zhang, L.; Lin, R. Heat resistant fluorenone-based polyimines as novel light-emitting polymers. J. Wuhan Univ. Technol. (Mater. Sci.) 2017, 32, 469–472. [Google Scholar] [CrossRef]
- Venkatesan, R.; Somanathan, N.; Rajeswari, N. Structure and properties of functionalized polyfluorenone containing hetero aromatic side chains. Chin. J. Polym. Sci. 2014, 32, 667–674. [Google Scholar] [CrossRef]
- Zhang, S.; Nie, G.; Han, X.; Xu, J.; Li, M.; Cai, T. Electrosyntheses of high quality free-standing poly(9-fluorenone) films in boron trifluoride diethyl etherate. Electrochim. Acta 2006, 51, 5738–5745. [Google Scholar] [CrossRef]
- Cihaner, A.; Tirke, S.; Önal, A.M. Electrochemical polymerization of 9-fluorenone. J. Electroanal. Chem. 2004, 568, 151–156. [Google Scholar] [CrossRef]
- Oldridge, L.; Kastler, M.; Mullen, K. Synthesis of a soluble poly(fluorenone). Chem. Commun. 2006, 8, 885–887. [Google Scholar] [CrossRef]
- Lee, J.; Kim, B.; Park, Y.; Kim, S.; Park, J. Fluorine effects in new indenofluorenedione derivatives for electron transporting layer in OLED devices. J. Nanosci. Nanotechnol. 2014, 14, 6431–6434. [Google Scholar] [CrossRef]
- Qashou, S.I.; Rashad, M.; Mahmoud, A.Z.; Darwish, A.A.A. The promotion of Indeno [1, 2-b] flourene-6, 12 dione thin film to be changed into stable aromatic compound under the effect of annealing treatment. Vacuum 2019, 162, 199–207. [Google Scholar] [CrossRef]
- Fan, Z.P.; Li, X.Y.; Sun, B.; Sun, C.L.; Shi, Z.F.; Shao, X.F.; Zhang, H.L. Constructing two-dimensional crossed molecular packing through branching chain engineering of amino-indenofluorene derivatives. J. Mater. Chem. C 2022, 10, 8666–8673. [Google Scholar] [CrossRef]
- Usta, H.; Facchetti, A.; Marks, T.J. Synthesis and characterization of electron-deficient and highly soluble (Bis)Indenofluorene building blocks for n-type semiconducting polymers. Org. Lett. 2008, 10, 1385–1388. [Google Scholar] [CrossRef]
- Miyata, Y.; Minari, T.; Nemoto, T.; Isoda, S.; Komatsu, K. Synthesis of fluorinated anti-fluorenacenedione and the structural, electronic, and field-effect properties. Org. Biomol. Chem. 2007, 5, 2592–2598. [Google Scholar] [CrossRef]
- Romain, M.; Chevrier, M.; Bebiche, S.; Mohammed-Brahim, T.; Rault-Berthelot, J.; Jacques, E.; Poriel, C. The structure–property relationship study of electron-deficient dihydroindeno[2,1-b]fluorene derivatives for n-type organic field effect transistors. J. Mater. Chem. C 2015, 3, 5742–5753. [Google Scholar] [CrossRef]
- Usta, H.; Facchetti, A.; Marks, T.J. Air-stable, solution-processable n-channel and ambipolar semiconductors for thin-film transistors based on the Indenofluorenebis(dicyanovinylene) Core. J. Am. Chem. Soc. 2008, 130, 8580–8581. [Google Scholar] [CrossRef] [PubMed]
- Usta, H.; Risko, C.; Wang, Z.; Huang, H.; Deliomeroglu, M.K.; Zhukhovitskiy, A.; Facchetti, A.; Marks, T.J. Design, Synthesis, and characterization of ladder-type molecules and polymers. Air-stable, solution-processable n-channel and ambipolar semiconductors for thin-film transistors via experiment and theory. J. Am. Chem. Soc. 2009, 131, 5586–5608. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhang, Y.; Ng, M.-K. Derivatives of 4,9-dihydro-s-indaceno[1,2-b:5,6-b‘]dithiophene-4,9-dione: synthesis and properties. J. Org. Chem. 2007, 72, 6364–6371. [Google Scholar] [CrossRef]
- Gomez-Esteban, S.; Benito-Hernandez, A.; Termine, R.; Hennrich, G.; Navarrete, J.T.L.; Delgado, M.C.R.; Golemme, A.; Gómez-Lor, B. High-mobility self-assembling truxenone-based n-type organic semiconductors. Chem. Eur. J. 2018, 24, 3576–3583. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, C.B.; Voroshazi, E.; Holliday, S.; Cnops, K.; Cheyns, D.; Mcculloch, I. Electron-deficient truxenone derivatives and their use in organic photovoltaics. J. Mater. Chem. A 2014, 2, 12348–12354. [Google Scholar] [CrossRef]
- Isoda, K.; Yasuda, T.; Kato, T. Truxene-based columnar liquid crystals: Self-assembled structures and electro-active properties. Chem. Asian J. 2009, 4, 1619–1625. [Google Scholar] [CrossRef]
- Yang, X.; Hu, Y.; Dunlap, N.; Wang, X.; Huang, S.; Su, Z.; Sharma, S.; Jin, Y.; Huang, F.; Wang, X.; et al. A truxenone-based covalent organic framework as an all-solid-state lithium-ion battery cathode with high capacity. Angew. Chem. Int. Ed. 2020, 132, 20565–20569. [Google Scholar] [CrossRef]
- Holliday, S. Truxenone Based Acceptors. In Synthesis and Characterisation of Non-Fullerene Electron Acceptors for Organic Photovoltaics; Springer: Cham, Switzerland, 2018. [Google Scholar] [CrossRef]
- Liu, M.; Liu, J.; Li, J.; Zhao, Z.; Zhou, K.; Li, Y.; He, P.; Wu, J.; Bao, Z.; Yang, Q.; et al. Blending aryl ketone in covalent organic frameworks to promote photoinduced electron transfer. J. Am. Chem. Soc. 2023, 145, 9198–9206. [Google Scholar] [CrossRef]
- Kumar, S.; Koo, Y.H.; Higashino, T.; Matsuda, W.; Ghosh, S.; Tsutsui, Y.; Suda, M.; Imahori, H.; Suzuki, K.; Kaji, H.; et al. Truxenone triimide: Two-dimensional molecular arrangements of triangular molecules for air stable n-type semiconductors. Adv. Electron. Mater. 2022, 8, 2101390. [Google Scholar] [CrossRef]
- Chen, C.; Ruan, H.P.; Feng, Z.; Fang, Y.; Zhao, Y.; Su, Y.; Wang, X. Crystalline diradical dianions of azaacenes. Angew. Chem. Int. Ed. 2020, 59, 11794–11799. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.T.; Tang, S.X.; Su, Y.T.; Wang, X.P. Recent advances in stable main group element radicals: Preparation and characterization. Chem. Soc. Rev. 2022, 51, 5930–5973. [Google Scholar] [CrossRef] [PubMed]
- Mutoh, K.; Kobayashi, Y.; Nakashima, T. A hexaarylbiimidazole-terarylene hybrid: Visible-to-NIR-II absorption via sequential photochromic reactions. Angew. Chem. Int. Ed. 2024, 63, e202410115. [Google Scholar]
- Kopp, S.M.; Redman, A.J.; Rončević, I.; Schröder, L.; Bogani, L.; Anderson, H.L.; Timmel, C.R. Charge and spin transfer dynamics in a weakly coupled porphyrin dimer. J. Am. Chem. Soc. 2024, 146, 21476–21489. [Google Scholar] [CrossRef]
- Behrendt, A.; Screttas, C.G.; Bethell, D.; Schiemann, O.; Steele, B.R. Magnetic and electrochemical investigations on anions derived from oligoketones containing fluorenone and benzophenone units. An approach to the design of stable multiradical organic materials. J. Chem. Soc. Perkin Trans. 1998, 2, 2039–2045. [Google Scholar] [CrossRef]
- Cǎndida, M.; Shohoji, B.L.; Luisa, M.; Franco, T.M.B.; Celina, M.; Lazana, R.; Sato, K.; Takui, T.; Itoh, K. ESR Study of High spin states derived from tribenzoylenebenzene. Mol. Cryst. Liq. Cryst. 1997, 305, 353–365. [Google Scholar]
- So, H.; Kim, J.H.; Lee, J.H.; Hwang, H.; An, D.K.; Lee, K.M. Planarity of terphenyl rings possessing o-carborane cages: Turning on intramolecular-charge-transfer-based emission. Chem. Commun. 2019, 55, 14518–14521. [Google Scholar] [CrossRef]
- Wei, Y.; Zheng, X.; Lin, D.; Yuan, H.; Yin, Z.; Yang, L.; Yu, Y.; Wang, S.; Xie, L.H.; Huang, W. Superelectrophilic-Initiated C–H functionalization at the β-position of thiophenes: A one-pot synthesis of trans-stereospecific saddle-shaped cyclic compounds. J. Org. Chem. 2019, 84, 10701–10709. [Google Scholar] [CrossRef]
- Rochford, L.A.; Ramadan, A.J.; Holliday, S.; Jones, T.S.; Nielsen, C.B. The effect of fluorination on the surface structure of truxenones. RSC Adv. 2016, 6, 67315–67318. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of density functionals by combining the method of constraint satisfaction with parametrization for thermochemistry, thermochemical kinetics, and noncovalent interactions. J. Chem. Theory Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef]
- Li, Y.; Toscano, M.; Mazzone, G.; Russo, N. Antioxidant properties and free radical scavenging mechanisms of cyclocurcumin. New J. Chem. 2018, 42, 12698–12705. [Google Scholar] [CrossRef]
- Dong, X.; Sun, Q.; Feng, Z.; Ruan, H.; Tang, S.; Liu, M.; Zhao, Y.; Su, Y.; Wang, X. Room-temperature reversible σ-dimerization of a phenalenyl radical. Chin. J. Chem. 2022, 40, 1655–1661. [Google Scholar] [CrossRef]
- Dimitrić Marković, J.M.; Marković, Z.S.; Pašti, I.A.; Brdarić, T.P.; Popović-Bijelića, A.; Mojović, M. A joint application of spectroscopic, electrochemical and theoretical approaches in evaluation of the radical scavenging activity of 3-OH flavones and their iron complexes towards different radical species. Dalton Trans. 2012, 41, 7295–7303. [Google Scholar] [CrossRef] [PubMed]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Kanetomo, T.; Naoi, Y.; Enomoto, M. Gadolinium-triradical complex with ground S = 10 state: Synthesis, structural characterization and magnetic studies. Eur. J. Inorg. Chem. 2021, 2021, 1130–1136. [Google Scholar] [CrossRef]
- Shimizu, D.; Osuka, A. A Benzene-1,3,5-triaminyl radical fused with ZnII-Porphyrins: Remarkable stability and a high-spin quartet ground state. Angew. Chem. Int. Ed. 2018, 57, 3733–3736. [Google Scholar] [CrossRef]
- Saito, T.; Nishihara, S.; Kataoka, Y.; Nakanishi, Y.; Matsui, T.; Kitagawa, Y.; Kawakami, T.; Okumura, M.; Yamaguchi, K. Transition state optimization based on approximate spin-projection (AP) method. Chem. Phys. Lett. 2009, 483, 168–171. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Okumura, M.; Mori, W.; Maki, J.; Takada, K.; Noro, T.; Tanaka, K. Comparison between spin restricted and unrestricted Post-Hartree—Fock calculations of effective exchange integrals in Ising and Heisenberg models. Chem. Phys. Lett. 1993, 210, 201–210. [Google Scholar] [CrossRef]
- Kertesz, M. Pancake Bonding: An unusual Pi-stacking interaction. Chem. Eur. J. 2019, 25, 400–416. [Google Scholar] [CrossRef]
- Cui, H.; Wang, L.; Ruan, H.; Liu, M.; Feng, Z.; Wang, J.; Zhao, Y.; Wang, X. Controlling the unpaired electron by electrostatic attraction in the solid state. Chem. Commun. 2021, 57, 13345–13348. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dong, X.; Wang, T.; Zhao, Y.; Sun, Q.; Tang, S.; Zhao, Y.; Wang, X. Crystalline Diradical Dianions and Radical Anions of Indenofluorenediones. Chemistry 2025, 7, 27. https://doi.org/10.3390/chemistry7010027
Dong X, Wang T, Zhao Y, Sun Q, Tang S, Zhao Y, Wang X. Crystalline Diradical Dianions and Radical Anions of Indenofluorenediones. Chemistry. 2025; 7(1):27. https://doi.org/10.3390/chemistry7010027
Chicago/Turabian StyleDong, Xue, Tao Wang, Yu Zhao, Quanchun Sun, Shuxuan Tang, Yue Zhao, and Xinping Wang. 2025. "Crystalline Diradical Dianions and Radical Anions of Indenofluorenediones" Chemistry 7, no. 1: 27. https://doi.org/10.3390/chemistry7010027
APA StyleDong, X., Wang, T., Zhao, Y., Sun, Q., Tang, S., Zhao, Y., & Wang, X. (2025). Crystalline Diradical Dianions and Radical Anions of Indenofluorenediones. Chemistry, 7(1), 27. https://doi.org/10.3390/chemistry7010027