Abstract
Sesamin, a tetrahydrofuran lignan, has gained significant attention over the past few decades due to its versatile medicinal activities. However, until now, the research on sesamin analogues has not been explored extensively. In this study, a series of new N-aryl-azasesamins were synthesized for the first time using sesamin as a raw material. The mechanism of the key breakage of the ethereal bond of the tetrahydrofuran ring in sesamin has been studied. The configuration of C6 in N-aryl-azasesamins was confirmed through NMR and X-ray single crystal refraction analyses. The results showed that the configuration of N-aryl-azasesamins was opposite to sesamin in C6. Subsequently, the N-aryl-azasesamins were evaluated for their antifungal and antitumor activities via micro-broth dilution and MTT assays. It was observed that none of the N-aryl-azasesamins exhibited inhibitory activity against the growth of C. albicans and C. neoformans at a concentration of 100 μg/mL. Most analogues showed no activity against HepG2 cells. However, 21c and 21k demonstrated antitumor activity after 24 h of incubation with IC50 values of 6.49 μM and 4.73 μM, respectively. These results suggest that some N-aryl-azasesamins exhibit significantly enhanced antitumor activity compared with sesamin.
1. Introduction
Natural products and their analogues play a vital role in the discovery and development of therapeutic agents [1]. Based on the structure–activity relationship of the core skeletons of natural products, new therapeutic compounds with improved biological efficacies and reduced toxicities can be synthesized [2]. Sesamin 1 (Figure 1) is a lipid-soluble or poorly water-soluble tetrahydrofuran lignan (2.5 μg/mL) [3] which was first isolated from Sesamum indicum L. in 1894 [4]. The Sesamum indicum L. is a major source of sesamin [5]. However, 30 other medicinal plants in some specific genera also contain minor amounts of sesamin [6]. Sesamin has been a research hotspot for scientists for decades due to its a wide range of biological activities, such as in anti-inflammation [7,8], neurodegenerative diseases [9], eye problems [10], cardiovascular diseases [11,12,13], lung diseases [14], anticancer effects [15,16,17,18,19], and so on. Sesamin has been widely studied in both preclinical and clinical trials for several diseases, including ischemic brain stroke [20], depression [21], Parkinson’s disease [22], osteoarthritis [23], diabetic retinopathy [10], acute hepatic injury [24], etc. Interestingly, the study by Wadhwa et al. showed that sesamin is an interesting scaffold for developing novel therapeutic agents against fungal infections due to its low toxicity, easy availability, and significant antifungal activity [25]. However, its poor water solubility has limited its widespread application.
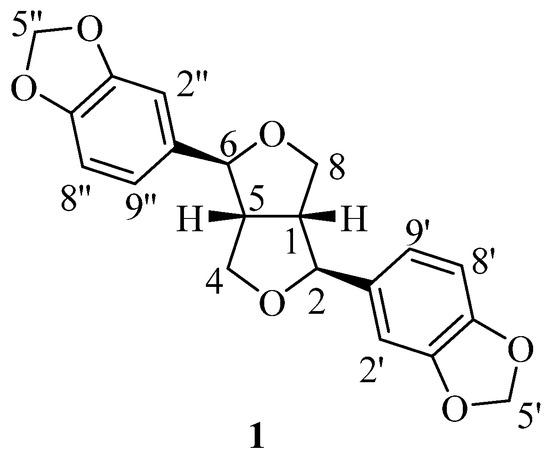
Figure 1.
Structure of sesamin.
For many years, nitrogen-containing heterocycles have captivated scientists due to their diverse structure and significant biological roles [26]. The pyrrolidine ring, also known as tetrahydropyrrole, is a key five-membered heterocyclic compound featuring one nitrogen atom. It is the core structure of a multitude of biologically and pharmacologically active molecules [27,28]. Compounds bearing pyrrolidine scaffolds are extensively used as intermediates in drug research and development studies for the development of new drug candidates [29,30]. Some pyrrolidine derivatives are known to be employed as pharmacophore groups, with some having antifungal [31], antiviral [32], antitumor [33], anti-inflammatory [34], anticonvulsant [35] activities, etc. Notable drugs incorporating a pyrrolidine ring include clemastine, procyclidine, clindamycin, enalapril, bepridil, ethosuximide, etc. In the aza-lignan research aspect, the Barker group synthesized a series of aza-analogues of fragransin A2 and galbelgin. Their findings indicated that these aza-derivatives exhibited greater anticancer activity compared with their corresponding tetrahydrofuran counterparts [36].
During the screening of antibacterial activity in our research group, it was found that sesamin exhibited an inhibitory effect on both Gram-positive bacteria (MRSA) and Gram-negative bacteria (E. coli). However, sesamin was observed to be precipitated during the antibacterial experiments due to its poor solubility, making it impossible to determine its minimum inhibitory concentration in microscopic assays. Therefore, analogues were initially designed to transform the tetrahydrofuran ring of sesamin into a tetrahydropyrrole ring, given the extensive use of the tetrahydropyrrole scaffold in drug research and development. These products may improve solubility and bioactivities or facilitate the synthesis of water-soluble quaternary ammonium salts. Consequently, in the present study, the synthesis of azasesamin was carried out and the antifungal and antitumor activities of azasesamins were evaluated.
2. Results and Discussion
2.1. Synthesis
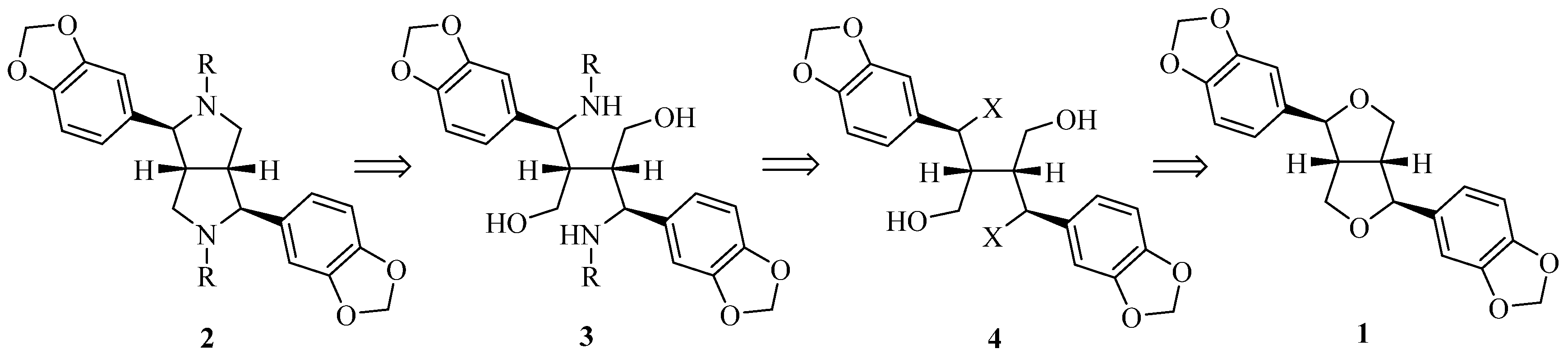
Retrosynthetically (Figure 2), azasesamin 2 can be synthesized from 3 by an intramolecular Mitsunobu reaction. Compound 3, in turn, can be obtained from 4 by an intermolecular SN2 reaction with amine. The ethereal bond of the tetrahydrofuran ring in sesamin 1 is broken by hydrogen halide acid to generate 4. First, hydrobromic acid or hydrochloric acid was selected to open the tetrahydrofuran ring of 1 to form benzyl halide 4. When the progress of the reaction was monitored with thin layer chromatography (TLC), an obvious new spot was observed, indicating the formation of a new product. A molecular ion peak in LC-MS corresponding to benzyl halide 4 was identified. However, the new spot disappeared after workup using saturated NaHCO3 solution. Thus, it can be inferred that benzyl halide 4 may be unstable.
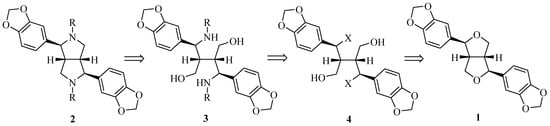
Figure 2.
Retrosynthetic analysis of azasesamin.
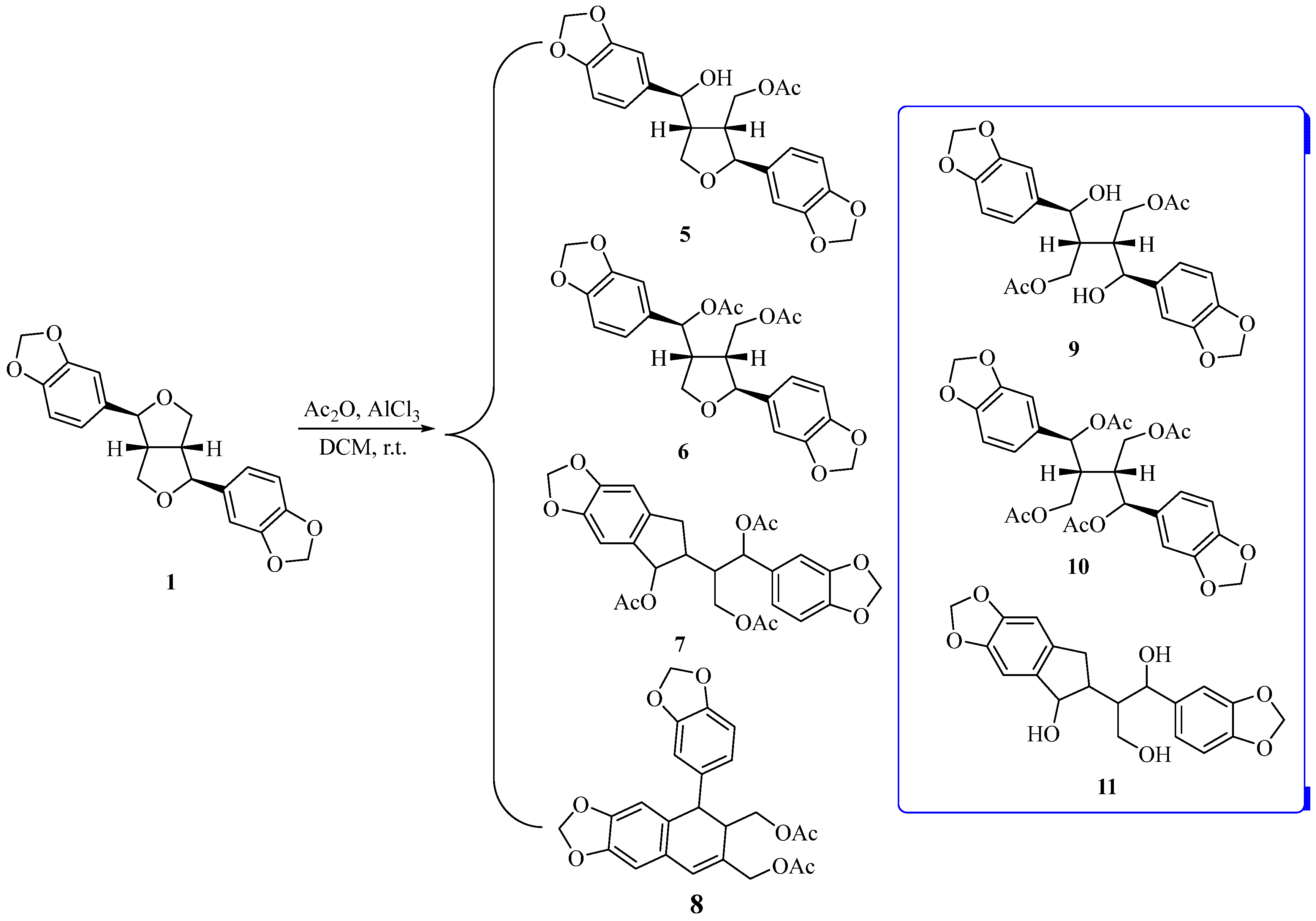
A new strategy was required for the breakage of the ethereal bond of 1, since benzyl halide 4 could not be obtained. Mincione et al. reported that aliphatic ethers react with Ti(NO3)3 in acetic anhydride via cleavage of the ethereal bond and formation of the corresponding acetyloxy derivative [37]. Therefore, a Lewis acid was selected for cleavage of the ethereal bond in 1 (Scheme 1). We found that Ti(NO3)3, FeCl3, or AlCl3 can open the tetrahydrofuran ring of 1. After separation using silica gel column chromatography, four main products 5–8 were obtained by the reaction of sesamin 1 with Ti(NO3)3, FeCl3, or AlCl3. However, the desired products with two opened furan rings (9 and 10) were not observed. Although pure compound 7 was not isolated via silica gel column chromatography, a molecular ion peak in LC-MS corresponding to compound 7 was identified, and a distinct CH3 peak of acetyl group was detected in the 1H NMR. After hydrolyzing compound 7 with K2CO3 of methanol/water solution and purifying it via silica gel column chromatography, compound 11 was obtained. The structure of compound 7 was then further verified. After screening reaction conditions such as temperature, solvents, etc., it was found that using AlCl3 as the Lewis acid, CH2Cl2 as the solvent, and a reaction temperature of 0 °C, the overall yield of compounds 5, 6 were highest (28%). With the optimized condition, the yields of compounds 5–8 were 11%, 17%, 8%, and 35%, respectively. Therefore, these conditions were chosen for opening the tetrahydrofuran ring of 1.
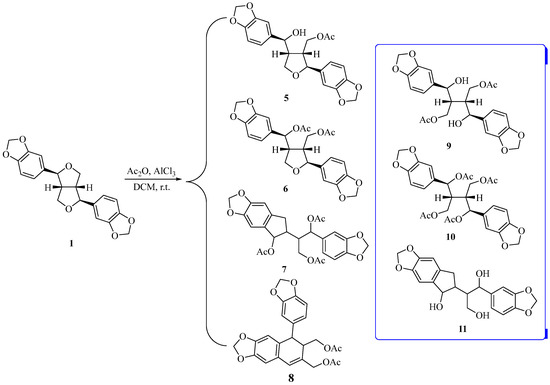
Scheme 1.
Cleavage of the ethereal bond of the tetrahydrofuran ring in 1.
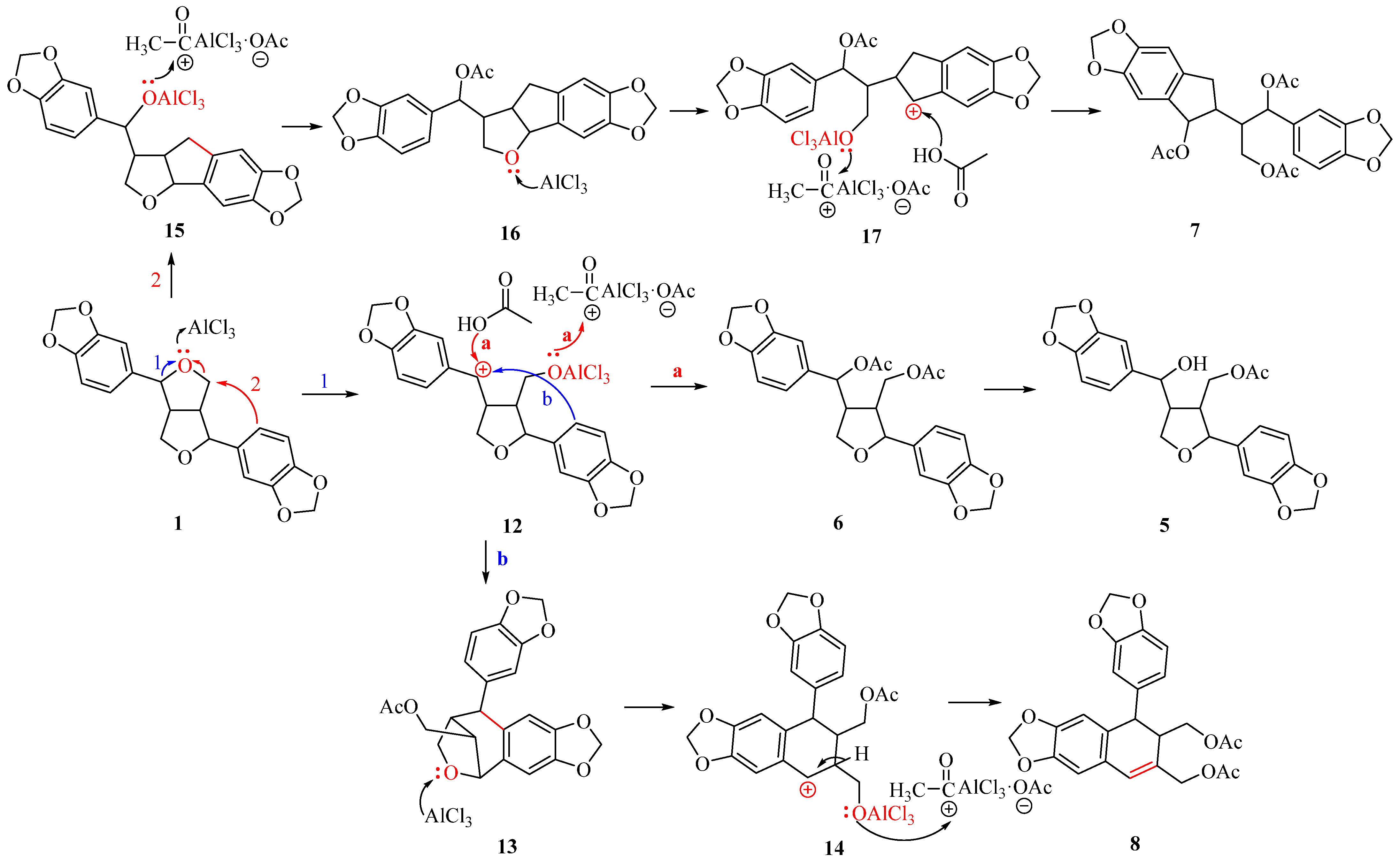
Based on the products obtained from the cleavage of the ethereal bond reaction, we deduced that 1 undergoes the following reaction mechanism under the action of AlCl3 (Scheme 2). One of the ethereal bonds of the tetrahydrofuran ring in 1 cleavage produces carbon cation 12 (route 1). When 12 is captured by acetic acid, and the oxygen anion captures the acetyl cation present in the system, product 6 is obtained. The benzyl hydroxyacetate ester in compound 6 undergoes selective cleavage to yield product 5. When 12 undergoes a Friedel–Crafts reaction at the C9’ position, it forms intermediate 13. Another ether bond continues to cleavage under the influence of AlCl3, generating cation 14, which then undergoes an elimination reaction, resulting in acetylation of the oxygen anion to give product 8. When the C9’ aromatic ring of 1 attacks C8, the C8-O bond breaks under the action of AlCl3, leading to the formation of cation 15 (route 2). This cation then undergoes acetylation to yield compound 16. Another ether bond continues to open under the action of AlCl3, yielding cation 17. Similarly, the carbon cation is captured by acetic acid, and the oxygen anion captures the acetyl cation to produce product 7.
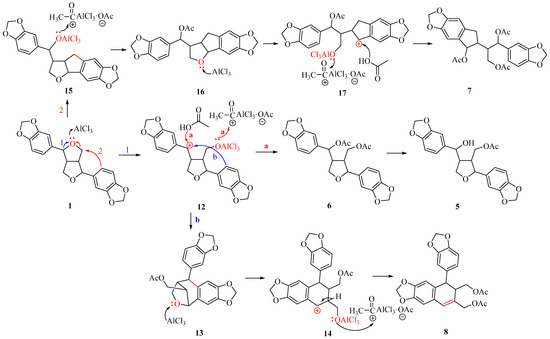
Scheme 2.
Mechanism for the synthesis of compounds 5–8.
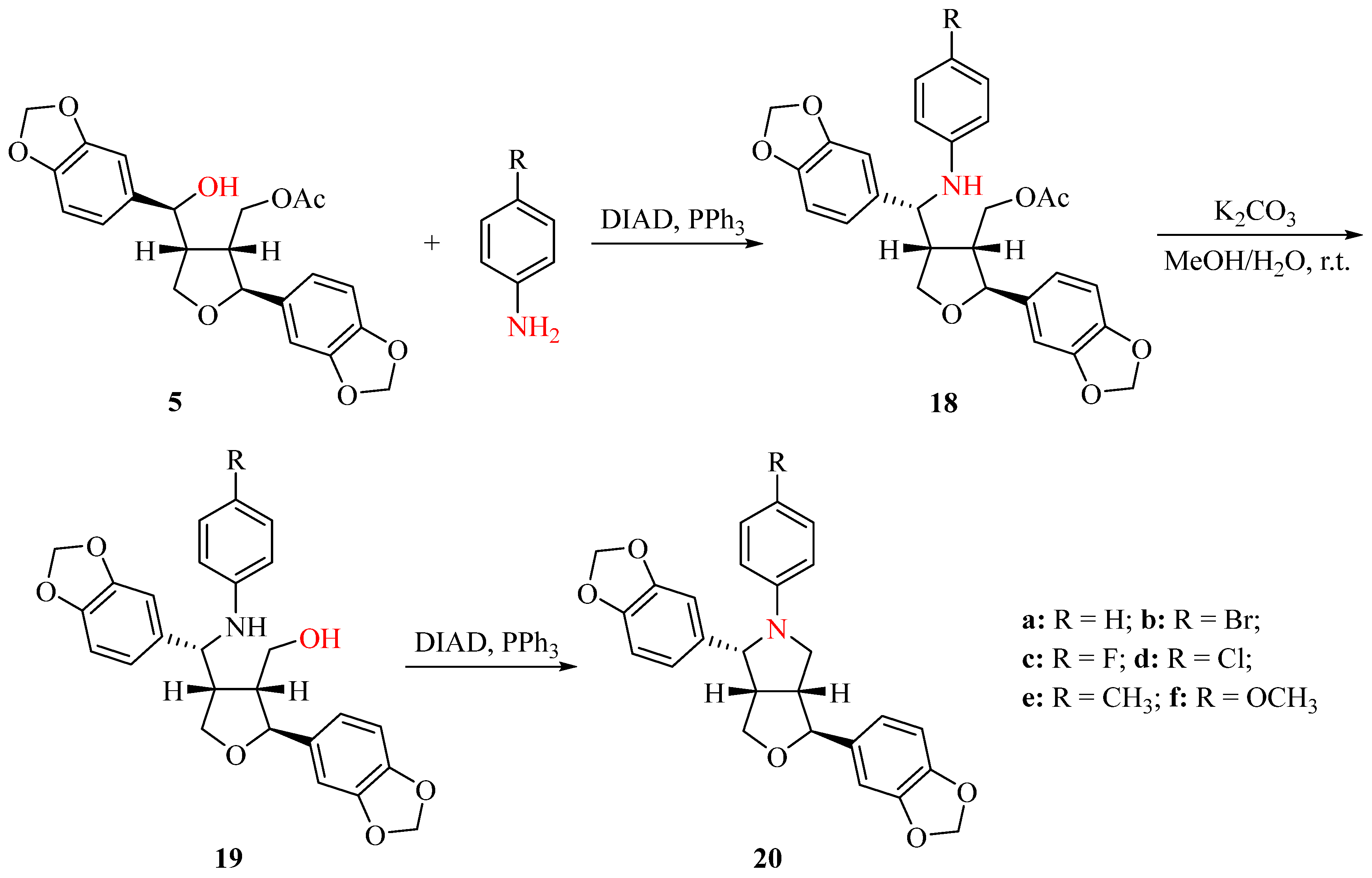
Due to the inability to obtain compounds 9 and 10 under acidic conditions from 1, we used compound 5 as the starting material to synthesize N-aryl-azasesamins 20 (Scheme 3). First, compound 5 was coupled with an aromatic amine via the intermolecular Mitsunobu reaction in the presence of DIAD and PPh3 in DCM to give amine 18 (yield: 68.9–91.7%). The acetyl ester of compound 18 was then hydrolyzed under K2CO3 to obtain compound 19 (yield: 60.1–94.5%). Finally, an intramolecular Mitsunobu reaction under the same condition as the first step yielded N-aryl-azasesamins 20a–20f (yield: 80.1–92.1%). The yield of the intermolecular Mitsunobu reaction was higher than that of the intramolecular Mitsunobu reaction, and the yield of some acetyl ester hydrolysis was low, possibly due to the poor solubility of the product. All products can be obtained through silica gel column chromatography.
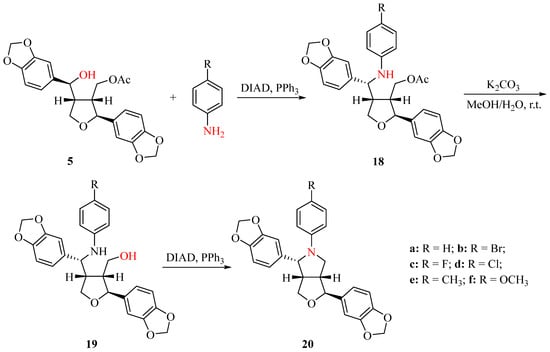
Scheme 3.
Synthesis of N-aryl-azasesamins 20.
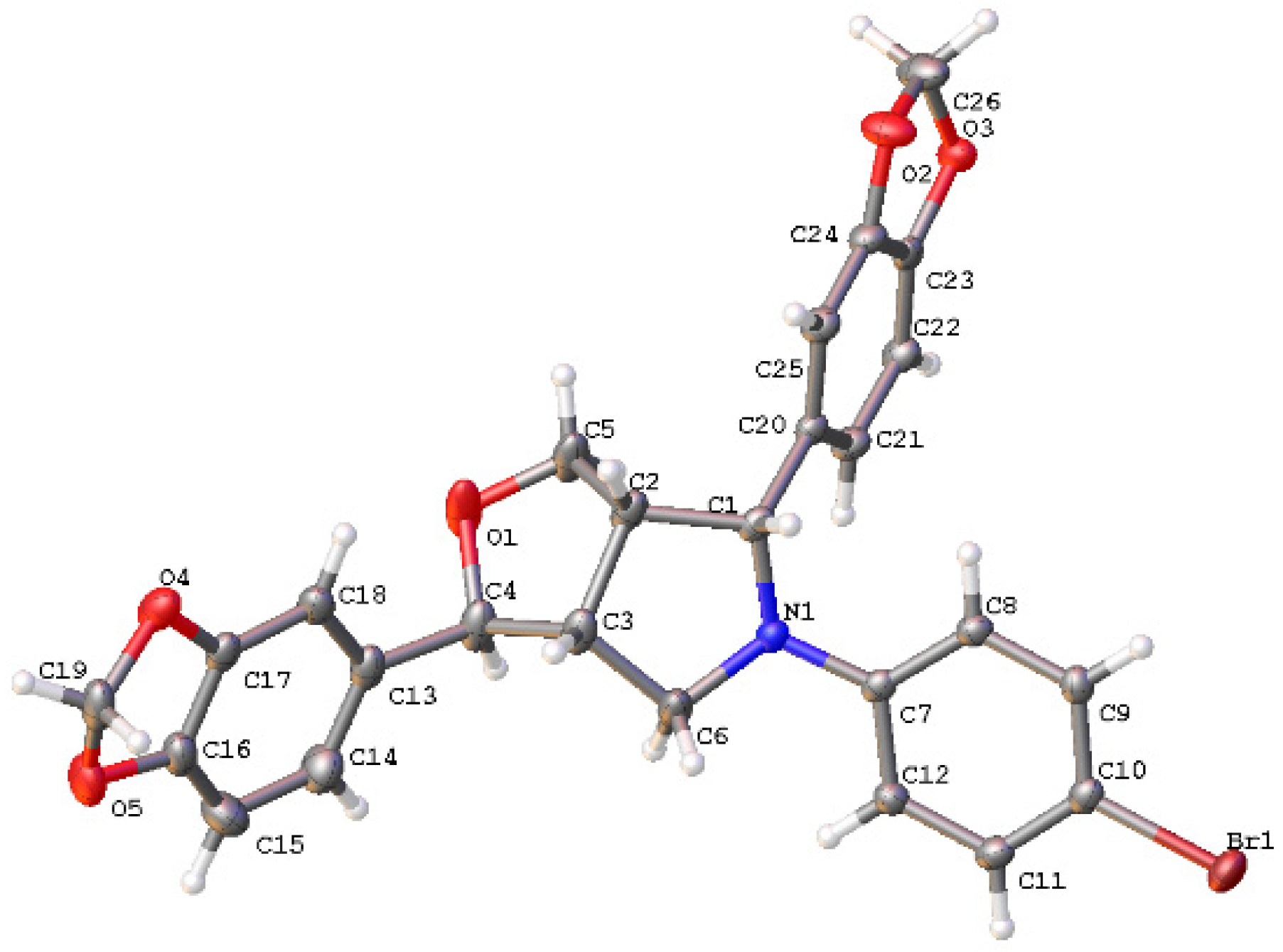
In order to determine the configuration of 20 in C6, compound 20b was analyzed by means of 1D, 2D NMR spectroscopic and X-ray analysis. Based on the analyses using 1H NMR, 13C NMR (Figures S35 and S36), H-H COSY, HSQC, and HMBC (Figures S67–S69), it was determined that δ 4.73 (d, J = 5.76 Hz, 1H) corresponds to H6, while δ 2.96–2.86 (m, 1H) corresponds to H1. The correlation between H6 and H1 was observed in the NOESY (Figure S70), suggesting that H6 and H1 might be in a cis direction according to the stable conformation of tetrahydrofuran. To further confirm the configuration of C6, single crystals of compound 20b were obtained, and X-ray analysis confirmed that H1 and H6 are cis (Figure 3, CCDC: 2168972). The crystallographic data and structural refinement parameters for compound 20b were summarized in Table S1. The configurations of C1, C2, C5, and C6 in 1 are R, S, R, and S, respectively [4]. Therefore, we concluded that the configuration of the compound 20b in C6 is R. The configuration of C6 in compounds 20b is opposite to that of C6 in 1. Compounds 20a–20f were synthesized using the same method, so it can infer that the configuration of C6 in compound 20 is R. However, since compound 18 was synthesized from compound 5 via the Mitsunobu reaction, it can be inferred that H1 and H6 in compound 5 are trans based on the Mitsunobu reaction mechanism. This suggests that acetic acid attacks the carbon cation at C6 from the same direction as H5, resulting in product 5 with the same configuration as 1.
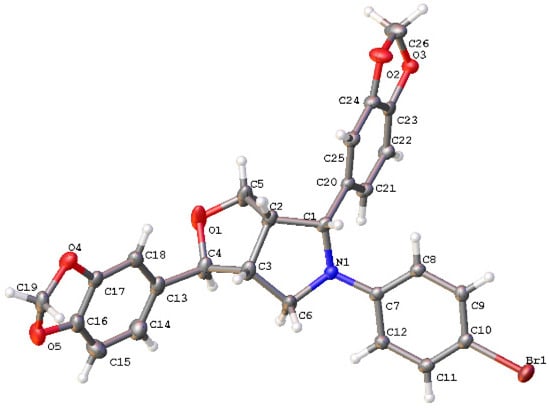
Figure 3.
Crystallographic structural diagram of compound 20b.
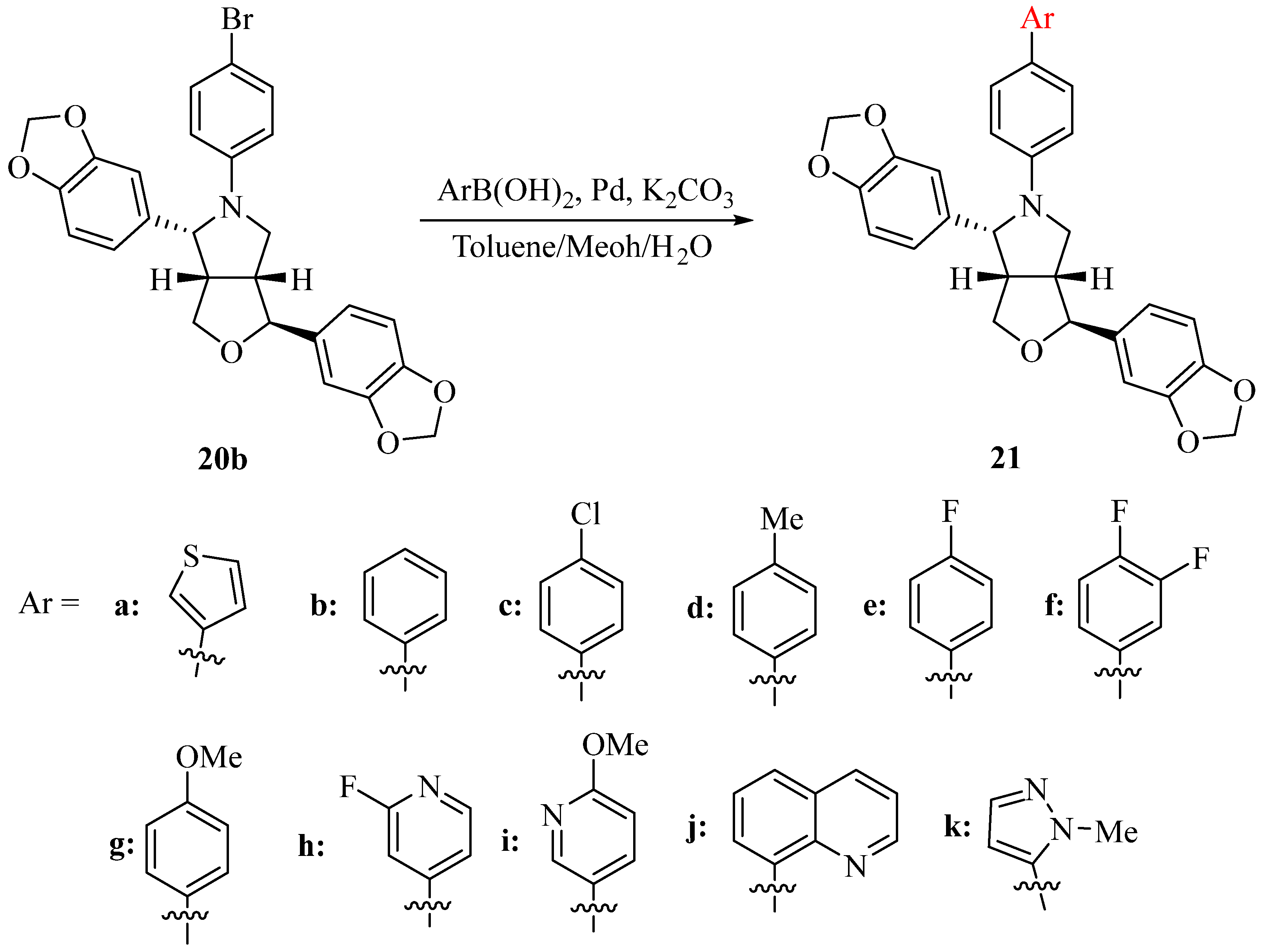
In order to further increase the structural diversity of the N-aryl-azasesamin, another 11 N-aryl-azasesamin was synthesized by Suzuki reaction between 20b and aryl boric acids (Scheme 4). The yield of the Suzuki reaction ranged from 60.6% (compound 21i) to 91.5% (compound 21j). Heterocycles can further introduce into N-aryl-azasesamin. However, when aliphatic boric acid was used, the target product was not obtained. Instead, the debrominated product (compound 20a) was formed. This may be due to interference during the transmetalation step based on the mechanism of the Suzuki reaction.
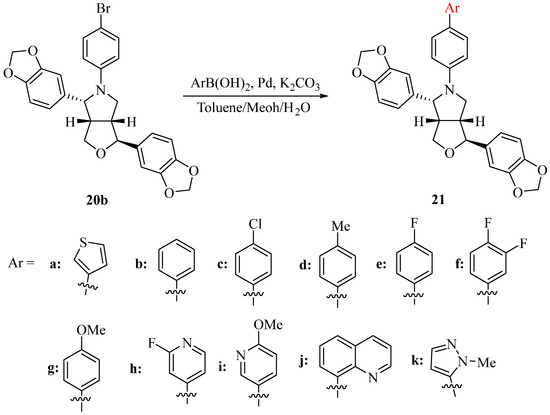
Scheme 4.
Synthesis of N-aryl-azasesamin 21.
2.2. Bioactivity Evaluation
The N-aryl-azasesamins were evaluated for their activities against C. albicans and C. neoformans. Compared with the positive control fluconazole (the inhibition rates were 99.98% and 101.05% at a concentration of 10 μg/mL, respectively), the inhibition rates of N-aryl-azasesamins for the two fungi were less than 50% at a concentration of 100 μg/mL. The results indicate that these compounds have no antifungal activity, which may be attributed to the difference in C6 configuration between these N-aryl-azasesamins and natural sesamin, as natural sesamin possesses antibacterial activity. Therefore, azasesamins with configurations identical to natural sesamin will be synthesized to investigate the configuration–activity relationship.
The cytotoxic activity of the N-aryl-azasesamins were evaluated against HepG2 cells, and the IC50 values are shown in Table 1. Sesamin exhibited no activity against HepG2 cells. Among N-aryl-azasesamins 21a–21k, only 21C and 21K demonstrated moderate anticancer activity with IC50 values of 6.49 μM and 4.73 μM, respectively, compared with the positive control paclitaxel (IC50 = 3.10 μM). The results indicate that N-aryl-azasesamins 21C and 21K significantly enhance their antitumor activity compared with sesamin. Therefore, additional azasesamins containing chlorine atoms and pyrazole heterocycles can be synthesized. The anticancer activities of the N-aryl-azasesamins will be evaluated across various cancer cell lines to better understand their structure–activity relationship.

Table 1.
Cytotoxic activity of N-aryl-azasesamins in HepG2 cells a.
3. Materials and Methods
3.1. General Chemical Procedures
1H and 13C NMR spectra were recorded using a Bruker Avance-III 400 MHz spectrometer with the indicated solvent. Chemical shifts (δ) were expressed in ppm with reference to the solvent signals (CDCl3; 1H: 7.26 ppm; 13C: 77.16 ppm) and coupling constants (J) were indicated in Hz. All NMR experiments were conducted using standard pulse sequences provided by the manufacturer. Column chromatography (CC) was carried out on silica gel (200−400 mesh, Qingdao Marine Chemical Factory, Qingdao, China). Thin-layer chromatography was conducted on Whatman glass-backed plates coated with 0.25 mm layers of silica gel 60. The crystal structure was measured with a Bruker D8 Venture single crystal diffractometer. The diffraction points of the crystals were measured by the GaKα ray with graphite monochromatic radiation (λ = 1.34139 Å). The HRESIMS were performed on an Agilent Technologies 6540 time-of-flight mass spectrometer (Agilent Technologies, Inc., Santa Clara, CA, USA).
C. albicans was inoculated in YM liquid medium and C. neoformans was inoculated in Sabouraud dextrose broth medium. All strains were inoculated in the corresponding liquid medium and stored at −80 °C until further use. Prior to each experiment, the strains were re-inoculated on solid agar culture plates and passaged at least once to revive the strains. HepG2 cell line was obtained from Shanghai Cell Bank. All reagents or solvents were purchased from commercial suppliers and were of reagent or HPLC grade and used without further purification.
3.2. Experimental Section
(1) Cleavage of the ethereal bond.
To a solution of 1 (1.1 g, 3.2 mmol) in CH2Cl2 (20 mL), acetic anhydride (5 mL) was added. The reaction mixture was cooled down to 0 °C, followed by the addition of anhydrous AlCl3 (1.2 g, 9.0 mmol) in an ice bath. Then, hydrochloric acid (20 mL, 2 mol/L) was added to quench the reaction, and the mixture was extracted with EtOAc (2 × 50 mL). The combined organic layers were washed with brine (30 mL) and dried over anhydrous Na2SO4. The solvent was removed under vacuo and the residue was purified via flash column chromatography (petroleum ether/EtOAc = 2:1) to obtain the products 5–8.
Compound 5: colorless solid, yield: 11.0%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 6.84–6.70 (m, 6H), 5.94 (d, J = 4.60 Hz, 4H), 4.83 (d, J = 5.76 Hz, 1H), 4.80 (d, J = 5.04 Hz, 1H), 4.33 (dd, J = 11.12, 6.36 Hz, 1H), 4.23–4.12 (m, 2H), 4.12–4.03 (m, 1H), 2.86–2.75 (m, 1H), 2.39–2.28 (m, 1H), 2.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm): 171.0, 148.2, 147.9, 147.4, 147.1, 137.2, 136.5, 119.7, 119.3, 108.5, 108.2, 106.6, 106.3, 101.3, 101.1, 83.7, 72.6, 69.8, 62.8, 48.7, 47.0, 21.0.
Compound 6: yellow transparent oil, yield: 17.0%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 6.85 (dd, J = 7.96, 1.84 Hz, 1H), 6.83–6.73 (m, 5H), 5.95 (s, 2H), 5.94 (d, J = 1.40 Hz, 2H), 5.84 (d, J = 9.04 Hz, 1H), 4.85 (d, J = 4.28 Hz, 1H), 4.26 (t, J = 7.68 Hz, 1H), 4.18–4.04 (m, 2H), 3.94 (t, J = 9.00 Hz, 1H), 3.09–2.96 (m, 1H), 2.29–2.20 (m, 1H), 2.03 (s, 3H), 2.02 (s, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm): 170.9, 169.9, 148.1, 148.0, 147.9, 147.1, 136.4, 132.6, 121.4, 118.9, 108.5, 108.2, 107.5, 106.1, 101.4, 101.2, 83.8, 74.2, 70.7, 62.5, 47.9, 45.1, 21.3, 21.0.
Compound 8: colorless transparent oil, yield: 35.0%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 6.68–6.62 (m, 2H), 6.58 (s, 1H), 6.51–6.44 (m, 3H), 5.91 (dd, J = 5.32, 1.16 Hz, 2H), 5.86 (dd, J = 5.60, 1.20 Hz, 2H), 4.61 (d, J = 13.04 Hz, 1H), 4.52 (d, J = 12.88 Hz, 1H), 4.18 (dd, J = 10.92, 5.04 Hz, 1H), 4.00 (s, 1H), 3.84 (dd, J = 10.88, 9.00 Hz, 1H), 2.77–2.69 (m, 1H), 2.06 (s, 3H), 1.93 (s, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm): 171.0, 170.7, 147.6, 147.5, 146.8, 146.1, 137.3, 129.6, 129.0, 128.0, 126.4, 120.6, 110.2, 108.1, 108.1, 107.4, 101.2, 100.9, 66.5, 64.4, 45.2, 43.2, 21.0, 20.8.
(2) Synthesis of compound 11.
Compound 7 (249.1 mg, 0.5 mmol) was dissolved in CH3OH (15 mL) and H2O (5 mL), followed by the addition of K2CO3 (276.4 mg, 2.0 mmol). The mixture was stirred at room temperature overnight. The solvent was then removed under reduced pressure, and the residue was purified directly using silica gel column chromatography (petroleum ether/EtOAc = 1:1), yielding 149.6 mg of the product 11.
Compound 11: white solid, yield: 80.4%. 1H NMR (400 MHz, DMSO-d6, J in Hz) δ (ppm): 7.01 (s, 1H), 6.83 (d, J = 7.80 Hz, 1H), 6.63–6.55 (m, 2H), 6.09 (s, 1H), 5.97 (s, 2H), 5.89 (d, J = 11.68 Hz, 2H), 5.21 (d, J = 7.24 Hz, 1H), 4.57 (t, J = 8.52 Hz, 1H), 4.53–4.41 (m, 2H), 3.96 (d, J = 9.80 Hz, 1H), 3.86–3.77 (m, 1H), 3.71–3.62 (m, 1H), 3.53–3.44 (m, 1H), 3.20–3.11 (m, 1H), 1.90–1.79 (m, 1H), 1.65–1.53 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 147.3, 145.6, 145.5, 145.4, 139.9, 135.2, 132.7, 122.2, 109.0, 108.4, 107.9, 106.2, 100.8, 100.5, 67.4, 59.4, 58.7, 45.9, 44.8, 43.4. HRMS m/z 395.1110 [M + Na]+ (calcd for C20H20O7Na, 395.1101).
(3) General procedure for the synthesis of compounds 18a–18f.
To a stirred solution of compound 5 (324.7 mg, 0.8 mmol) in CH2Cl2 (25 mL), arylamine (2.4 mmol) and PPh3 (411.3 mg, 1.6 mmol) were added successively. The reaction mixture was cooled down to 0 °C and DIAD (320 μL, 1.6 mmol) was added with a syringe. At the end of the reaction, the mixture was diluted with 20 mL of H2O, then extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with brine (40 mL) and dried over anhydrous Na2SO4. The solvent was removed under vacuo and the residue was then purified using flash column chromatography to obtain the products 18.
Compound 18a: colorless transparent oil, yield: 90.4%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 7.11 (t, J = 7.48 Hz, 2H), 6.83–6.72 (m, 6H), 6.67 (t, J = 7.20 Hz, 1H), 6.53 (d, J = 7.84 Hz, 2H), 5.95 (s, 2H), 5.92 (dd, J = 6.20, 1.32 Hz, 2H), 4.91 (d, J = 7.12 Hz, 1H), 4.61 (d, J = 4.52 Hz, 1H), 4.42–4.24 (m, 2H), 4.19–4.09 (m, 2H), 2.84–2.74 (m, 1H), 2.61–2.53 (m, 1H), 2.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm): 170.9, 148.3, 148.1, 147.2, 146.9, 146.3, 137.1, 136.2, 129.4, 120.0, 119.1, 117.8, 113.7, 113.5, 108.6, 108.3, 107.0, 106.1, 101.2, 83.4, 70.4, 62.4, 55.8, 49.8, 48.4, 21.0.
Compound 18b: colorless transparent oil, yield: 82.9%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 7.18 (d, J = 8.68 Hz, 2H), 6.82–6.73 (m, 6H), 6.40 (d, J = 8.68 Hz, 2H), 5.95 (s, 2H), 5.93 (d, J = 4.60 Hz, 2H), 4.88 (d, J = 7.28 Hz, 1H), 4.55 (d, J = 4.40 Hz, 1H), 4.40–4.24 (m, 2H), 4.17–4.06 (m, 2H), 2.81–2.71 (m, 1H), 2.64–2.53 (m, 1H), 2.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm): 170.8, 148.3, 148.1, 147.2, 147.0, 145.4, 136.9, 135.6, 132.2, 132.1, 119.9, 119.1, 115.1, 109.4, 108.7, 108.3, 106.9, 106.0, 101.3, 101.2, 83.3, 70.2, 62.3, 55.8, 49.8, 48.3, 20.9.
Compound 18c: colorless transparent oil, yield: 68.9%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 6.84–6.72 (m, 8H), 6.45 (s, 2H), 5.95 (s, 2H), 5.93 (dd, J = 4.28, 1.12 Hz, 2H), 4.90 (d, J = 7.00 Hz, 1H), 4.51 (d, J = 4.56 Hz, 1H), 4.40–4.23 (m, 2H), 4.14 (d, J = 4.20 Hz, 2H), 2.84–2.70 (m, 1H), 2.60–2.52 (m, 1H), 2.01 (s, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm): 170.9, 148.3, 148.1, 147.2, 147.0, 137.0, 120.0, 119.1, 116.0, 115.8, 114.4, 108.63, 108.3, 106.9, 106.1, 101.3, 101.2, 83.4, 70.4, 62.4, 56.5, 49.7, 48.3, 21.0.
Compound 18d: colorless transparent oil, yield: 81.6%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 7.05 (d, J = 8.76 Hz, 2H), 6.82–6.71 (m, 6H), 6.43 (d, J = 8.76 Hz, 2H), 5.96 (s, 2H), 5.93 (dd, J = 5.24, 1.16 Hz, 2H), 4.88 (d, J = 7.32 Hz, 1H), 4.85 (br s, 1H), 4.55 (d, J = 4.20 Hz, 1H), 4.40–4.24 (m, 2H), 4.19–4.06 (m, 2H), 2.80–2.72 (m, 1H), 2.65–2.55 (m, 1H), 2.01 (s, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm): 170.8, 148.4, 148.1, 147.2, 147.0, 144.9, 136.9, 135.7, 129.3, 122.4, 119.9, 119.1, 114.6, 108.7, 108.4, 106.9, 106.0, 101.3, 101.2, 83.4, 70.3, 62.3, 55.9, 49.8, 48.4, 21.0.
Compound 18e: colorless transparent oil, yield: 91.7%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 6.92 (d, J = 8.20 Hz, 2H), 6.82–6.71 (m, 6H), 6.46 (d, J = 6.12 Hz, 2H), 5.95 (s, 2H), 5.92 (dd, J = 6.00, 1.12 Hz, 2H), 4.91 (d, J = 6.88 Hz, 1H), 4.56 (d, J = 4.44 Hz, 1H), 4.41–4.22 (m, 2H), 4.14 (d, J = 4.08 Hz, 2H), 2.88–2.73 (m, 1H), 3.60–2.48 (m, 1H), 2.20 (s, 3H), 2.01 (s, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm): 170.8, 148.2, 148.1, 147.2, 146.8, 137.1, 129.9, 120.0, 119.1, 113.7, 108.6, 108.3, 107.0, 106.1, 101.2, 83.5, 70.5, 62.5, 49.7, 48.2, 29.8, 21.0, 20.5.
Compound 18f: colorless transparent oil, yield: 82.3%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 6.81–6.68 (m, 8H), 6.56–6.42 (m, 2H), 5.95 (s, 2H), 5.92 (d, J = 3.12 Hz, 2H), 4.92 (d, J = 6.52 Hz, 1H), 4.49 (brs, 1H), 4.38–4.22 (m, 2H), 4.20–4.08 (m, 2H), 3.70 (s, 3H), 2.90–2.70 (m, 1H), 2.58–2.42 (m, 1H), 2.01 (s, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm): 170.9, 148.2, 148.1, 147.1, 137.1, 119.0, 115.0, 108.6, 108.3, 106.1, 101.2, 101.2, 83.5, 70.7, 62.5, 55.8, 49.6, 21.0.
(4) General procedure for the synthesis of compounds 19a–19f.
To a solution of compound 18 (0.7 mmol) in methanol (30 mL), K2CO3 (107.8 mg, 0.8 mmol), and H2O (5 mL) were added. The reaction mixture was then stirred at room temperature. At the end of the reaction, CH2Cl2 (100 mL) was added, and then the organic layers were washed with H2O (30 mL) and brine (40 mL) and dried over anhydrous Na2SO4. The solvent was removed under vacuo and the residue was purified using flash column chromatography to obtain the product 19.
Compound 19a: white solid, yield: 70.8%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 7.11 (t, J = 8.56 Hz, 2H), 6.84–6.70 (m, 6H), 6.67 (t, J = 7.52 Hz, 1H), 6.55 (d, J = 7.48 Hz, 2H), 5.95 (s, 2H), 5.91 (dd, J = 6.40, 1.44 Hz, 2H), 4.90 (d, J = 7.20 Hz, 1H), 4.75 (d, J = 4.28 Hz, 1H), 4.20–4.03 (m, 2H), 4.00–3.80 (m, 2H), 2.85–2.75 (m, 1H), 2.51–2.40 (m, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 148.2, 148.1, 147.1, 146.7, 146.4, 137.6, 136.5, 129.4, 119.9, 118.9, 117.8, 113.7, 108.5, 108.3, 107.1, 106.0, 101.7, 83.1, 70.2, 60.8, 55.7, 53.0, 48.4.
Compound 19b: white solid, yield: 60.1%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 7.18 (d, J = 8.68 Hz, 2H), 6.82–6.71 (m, 6H), 6.41 (d, J = 8.72 Hz, 2H), 5.95 (s, 2H), 5.93 (d, J = 4.80 Hz, 2H), 4.85 (d, J = 7.28 Hz, 1H), 4.69 (d, J = 3.92 Hz, 1H), 4.16 (dd, J = 9.52, 3.00 Hz, 1H), 4.06 (dd, J = 9.40, 6.16 Hz, 1H), 3.93–3.84 (m, 2H), 2.84–2.74 (m, 1H), 2.53–2.46 (m, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 148.3, 148.1, 147.1, 146.9, 137.4, 132.1, 119.9, 118.9, 115.3, 108.6, 108.4, 107.0, 106.0, 101.3, 101.2, 83.1, 70.2, 60.9, 53.0, 48.4, 31.7.
Compound 19c: white solid, yield: 80.1%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 6.85–6.71 (m, 8H), 6.51–6.44 (m, 2H), 5.95 (s, 2H), 5.92 (dd, J = 4.64, 1.44 Hz, 2H), 4.91 (d, J = 7.08 Hz, 1H), 4.66 (d, J = 4.36 Hz, 1H), 4.16 (dd, J = 9.52, 4.04 Hz, 1H), 4.09 (dd, J = 9.44, 6.32 Hz, 1H), 3.96–3.84 (m, 2H), 2.85–2.75 (m, 1H), 2.51–2.40 (m, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 155.0, 148.3, 148.1, 147.1, 146.8, 142.5, 137.5, 136.1, 120.0, 118.9, 116.0, 115.7, 114.8, 108.6, 108.3, 107.1, 106.0, 101.2, 101.2, 83.1, 70.2, 60.8, 56.7, 52.9, 48.4.
Compound 19d: white solid, yield: 86.1%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 7.09–7.01 (m, 2H), 6.83–6.70 (m, 6H), 6.47 (d, J = 8.80 Hz, 2H), 5.95 (s, 2H), 5.93 (dd, J = 5.36, 1.40 Hz, 2H), 4.86 (d, J = 7.20 Hz, 1H), 4.69 (d, J = 4.04 Hz, 1H), 4.16 (dd, J = 9.56, 3.56 Hz, 1H), 4.07 (dd, J = 9.52, 6.20 Hz, 1H), 3.93–3.83 (m, 2H), 2.85–2.75 (m, 1H), 2.54–2.43 (m, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 148.3, 148.1, 147.1, 146.9, 145.0, 137.4, 136.0, 129.2, 122.5, 119.9, 118.9, 114.8, 108.6, 108.3, 107.0, 106.0, 101.3, 101.2, 83.1, 70.2, 60.9, 56.0, 53.0, 48.4.
Compound 19e: white solid, yield: 94.5%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 6.93 (d, J = 8.20 Hz, 2H), 6.84–6.70 (m, 6H), 6.48 (d, J = 8.32 Hz, 2H), 5.98–5.85 (m, 4H), 4.93 (t, J = 7.16 Hz, 1H), 4.72 (t, J = 3.68 Hz, 1H), 4.15 (dd, J = 9.40, 3.72 Hz, 1H), 4.08 (dd, J = 9.32, 6.32 Hz, 1H), 3.95–3.80 (m, 2H), 2.87–2.74 (m, 1H), 2.50–2.38 (m, 1H), 2.20 (t, J = 11.72 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm): 148.2, 148.1, 147.0, 146.7, 144.0, 137.6, 136.7, 129.9, 127.1, 119.9, 118.9, 113.9, 108.5, 108.3, 107.1, 106.0, 101.1, 83.1, 70.2, 60.7, 56.0, 53.0, 48.5, 20.5.
Compound 19f: white solid, yield: 73.7%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 6.83–6.69 (m, 8H), 6.51 (d, J = 8.88 Hz, 2H), 5.94 (s, 2H), 5.91 (dd, J = 5.12, 1.40 Hz, 2H), 4.95 (d, J = 7.00 Hz, 1H), 4.65 (d, J = 4.52 Hz, 1H), 4.17–4.04 (m, 2H), 3.93–3.84 (m, 2H), 3.70 (s, 3H), 2.81–2.73 (m, 1H), 2.45–2.37 (m, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 152.4, 148.2, 148.0, 147.0, 146.7, 140.3, 137.6, 136.6, 119.9, 118.9, 115.3, 115.0, 108.5, 108.3, 107.1, 106.0, 101.1, 83.0, 70.2, 60.6, 56.8, 55.8, 52.9, 48.5.
(5) General procedure for the synthesis of N-aryl-azasesamins 20.
To a stirred solution of compound 19 (0.4 mmol) in CH2Cl2 (15 mL), PPh3 (204.9 mg, 0.8 mmol) was added. The reaction mixture was cooled down to 0 °C and then DIAD (155 μL, 0.8 mmol) was added. At the end of the reaction, the mixture was diluted with H2O (20 mL) and extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with brine (40 mL) and dried over anhydrous Na2SO4. The solvent was removed under vacuo to obtain residue, which was then purified using flash column chromatography to obtain product 20.
Compound 20a: white solid, yield: 90.1%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 7.15 (t, J = 7.72 Hz, 2H), 6.88–6.71 (m, 7H), 6.60 (d, J = 8.24 Hz, 2H), 5.95 (s, 2H), 5.94 (d, J = 3.28 Hz, 2H), 4.75 (d, J = 5.84 Hz, 1H), 4.70 (d, J = 8.28 Hz, 1H), 3.91 (dd, J = 9.72, 3.48 Hz, 1H), 3.82 (t, J = 8.52 Hz, 1H), 3.63–3.53 (m, 2H), 3.46–3.36 (m, 1H), 2.93–2.85 (m, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 148.3, 148.1, 147.2, 146.6, 136.4, 134.1, 128.9, 120.0, 119.3, 118.0, 115.5, 108.5, 108.3, 107.4, 106.4, 101.2, 101.1, 86.8, 70.4, 65.4, 55.1, 51.3, 50.2. HRMS m/z 430.1648 [M + H]+ (calcd for C26H24NO5, 430.1649).
Compound 20b: white solid, yield: 92.1%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 7.21 (d, J = 8.96 Hz, 2H), 6.87–6.66 (m, 6H), 6.45 (d, J = 9.00 Hz, 2H), 5.95 (s, 2H), 5.95 (d, J = 1.80 Hz, 2H), 4.73 (d, J = 5.76 Hz, 1H), 4.67 (d, J = 8.28 Hz, 1H), 3.90–3.78 (m, 2H), 3.60–3.51 (m, 2H), 3.47–3.36 (m, 1H), 2.96–2.86 (m, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 148.2, 148.1, 147.2, 147.1, 146.8, 136.2, 133.4, 131.6, 119.9, 119.3, 117.0, 110.3, 108.6, 108.3, 107.3, 106.4, 101.2, 86.7, 70.2, 65.5, 55.0, 51.3, 50.2. HRMS m/z 508.0778 [M + H]+ (calcd for C26H23BrNO5, 508.0754).
Compound 20c: white solid, yield: 91.3%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 6.88–6.73 (m, 8H), 6.58 (dd, J = 8.96, 4.40 Hz, 2H), 5.95 (s, 2H), 5.94 (d, J = 6.56 Hz, 2H), 4.73 (d, J = 6.12 Hz, 1H), 4.60 (d, J = 8.16 Hz, 1H), 3.88 (dd, J = 9.76, 3.12 Hz, 1H), 3.80 (t, J = 8.44 Hz, 1H), 3.56 (t, J = 6.92 Hz, 1H), 3.50–3.35 (m, 2H), 2.92–2.82 (m, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 155.3, 148.1, 147.2, 146.7, 144.7, 136.1, 133.4, 120.1, 119.4, 117.1, 117.1, 115.5, 115.3, 108.6, 108.3, 107.5, 106.4, 101.2, 87.0, 70.3, 66.0, 56.2, 51.1, 50.3. HRMS m/z 448.1555 [M + H]+ (calcd for C26H23FNO5, 448.1555).
Compound 20d: white solid, yield: 90.3%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 7.08 (d, J = 8.96 Hz, 2H), 6.88–6.68 (m, 6H), 6.53 (d, J = 8.96 Hz, 2H), 5.95 (s, 2H), 5.95 (d, J = 1.72 Hz, 2H), 4.75 (d, J = 5.80 Hz, 1H), 4.67 (d, J = 8.28 Hz, 1H), 3.88 (dd, J = 9.88, 3.68 Hz, 1H), 3.82 (dd, J = 9.32, 7.80 Hz, 1H), 3.61–3.51 (m, 2H), 3.47–3.36 (m, 1H), 2.96–2.86 (m, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 148.2, 148.1, 147.2, 146.8, 146.5, 136.1, 133.3, 128.8, 123.3, 120.0, 119.3, 116.7, 108.6, 108.3, 107.3, 106.4, 101.2, 101.2, 86.7, 70.2, 65.7, 55.3, 51.2, 50.2. HRMS m/z 464.1261 [M + H]+ (calcd for C26H23ClNO5, 464.1259).
Compound 20e: white solid, yield: 86.8%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 6.95 (d, J = 8.36 Hz, 2H), 6.88–6.73 (m, 6H), 6.54 (d, J = 8.44 Hz, 2H), 5.95 (s, 2H), 5.93 (d, J = 3.36 Hz, 2H), 4.72 (d, J = 6.08 Hz, 1H), 4.62 (d, J = 8.20 Hz, 1H), 3.90 (dd, J = 9.80, 3.20 Hz, 1H), 3.79 (t, J = 9.16 Hz, 1H), 3.55 (dd, J = 9.20, 6.84 Hz, 1H), 3.48 (dd, J = 9.72, 8.04 Hz, 1H), 3.44–3.33 (m, 1H), 2.91–2.80 (m, 1H), 2.21 (s, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm): 148.1, 148.0, 147.2, 146.6, 146.2, 136.3, 134.1, 129.4, 120.0, 119.4, 116.1, 108.5, 108.3, 107.5, 106.5, 101.2, 101.1, 87.0, 70.4, 65.6, 55.7, 51.2, 50.2, 20.5. HRMS m/z 444.1806 [M + H]+ (calcd for C27H26NO5, 444.1805).
Compound 20f: white solid, yield: 80.1%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 6.91–6.70 (m, 10H), 5.95 (s, 2H), 5.93 (d, J = 1.56 Hz, 2H), 4.78 (d, J = 3.36 Hz, 1H), 4.57 (d, J = 7.96 Hz, 1H), 3.91 (d, J = 8.84 Hz, 1H), 3.79 (t, J = 8.68 Hz, 1H), 3.72 (s, 3H), 3.66–3.49 (m, 1H), 3.39 (t, J = 9.32 Hz, 2H), 2.89–2.80 (m, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 148.1, 148.0, 147.2, 146.6, 136.2, 120.2, 119.5, 117.8, 114.4, 108.5, 108.3, 107.7, 106.5, 101.2, 101.1, 87.2, 70.5, 65.9, 55.7, 53.6, 51.1, 50.3. HRMS m/z 460.1761 [M + H]+ (calcd for C27H26NO6, 460.1755).
(6) General procedure for the synthesis of N-aryl-azasesamins 21.
Compound 20 (0.2 mmol), arylboric acid (0.2 mmol), K2CO3 (49.7 mg, 0.4 mmol), Pd (PPh3)4 (20.8 mg, 0.02 mmol), and toluene (20 mL) were added to a 50 mL round-bottom flask. The reaction was heated to 80 °C under nitrogen and reacted overnight. At the end of the reaction, toluene was removed by use of a rotary evaporator, and the concentrate was diluted with H2O (20 mL) and extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with brine (20 mL) and dried over anhydrous Na2SO4. The solvent was removed under vacuo to obtain residue, which was then purified using flash column chromatography to obtain the product 21.
Compound 21a: white solid, yield: 75.0%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 7.39 (d, J = 8.72 Hz, 2H), 7.34–7.27 (m, 2H), 7.25 (t, J = 1.40Hz, 1H), 6.89–6.71 (m, 6H), 6.62 (d, J = 8.72 Hz, 2H), 5.96 (s, 2H), 5.95 (d, J = 3.08 Hz, 2H), 4.76 (t, J = 6.20 Hz, 2H), 3.94 (dd, J = 9.92, 3.72 Hz, 1H), 3.83 (dd, J = 9.28, 7.80 Hz, 1H), 3.67–3.55 (m, 2H), 3.48–3.38 (m, 1H), 2.97–2.87 (m, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 148.1, 148.1, 147.4, 147.2, 146.7, 142.5, 136.3, 134.0, 127.0, 126.2, 125.9, 120.0, 119.3, 118.1, 115.7, 108.6, 108.3, 107.4, 106.4, 101.2, 86.7, 70.3, 65.6, 55.0, 51.3, 50.2. HRMS m/z 512.1533 [M + H]+ (calcd for C30H26NO5S, 512.1526).
Compound 21b: white solid, yield: 85.3%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 7.50 (dd, J = 8.44, 1.32 Hz, 2H), 7.42 (d, J = 8.72 Hz, 2H), 7.37 (t, J = 7.48 Hz, 2H), 7.28–7.22 (m, 1H), 6.89–6.73 (m, 8H), 5.96 (s, 2H), 5.95 (t, J = 2.16 Hz, 2H), 4.85 (d, J = 5.84 Hz, 1H), 4.78 (d, J = 8.28 Hz, 1H), 3.99 (dd, J = 10.08, 3.64 Hz, 1H), 3.84 (dd, J = 9.36, 7.84 Hz, 1H), 3.71–3.61 (m, 2H), 3.50–3.41 (m, 1H), 2.99–2.90 (m, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 148.2, 148.1, 147.2, 146.9, 141.0, 136.2, 128.8, 127.6, 126.6, 126.5, 120.2, 119.4, 116.2, 108.6, 108.3, 107.5, 106.4, 101.2, 86.6, 70.2, 66.3, 55.5, 51.2, 50.2. HRMS m/z 506.1957 [M + H]+ (calcd for C32H28NO5, 506.1962).
Compound 21c: white solid, yield: 81.3%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 7.44–7.31 (m, 6H), 6.88–6.76 (m, 6H), 6.66 (d, J = 8.72 Hz, 2H), 5.96 (s, 2H), 5.95 (d, J = 2.32 Hz, 2H), 4.78 (t, J = 6.12 Hz, 2H), 3.95 (dd, J = 10.00, 3.84 Hz, 1H), 3.84 (dd, J = 9.32, 7.80 Hz, 1H), 3.70–3.56 (m, 2H), 3.49–3.39 (m, 1H), 2.98–2.89 (m, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 148.2, 148.1, 147.6, 147.2, 146.8, 139.6, 136.2, 133.7, 132.2, 129.4, 128.9, 128.9, 127.7, 127.4, 120.0, 119.3, 115.7, 108.7, 108.6, 108.3, 107.3, 106.4, 101.2, 86.6, 70.3, 65.6, 55.0, 51.3, 50.2. HRMS m/z 540.1581 [M + H]+ (calcd for C32H27ClNO5, 540.1572).
Compound 21d: white solid, yield: 88.7%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 7.40 (d, J = 6.00 Hz, 2H), 7.38 (d, J = 6.48 Hz, 2H), 7.18 (d, J = 8.00 Hz, 2H), 6.88–6.76 (m, 6H), 6.66 (d, J = 8.12 Hz, 2H), 5.96 (s, 2H), 5.95 (d, J = 2.44 Hz, 2H), 4.76 (dd, J = 9.92, 5.84 Hz, 2H), 3.95 (dd, J = 9.96, 3.64 Hz, 1H), 3.84 (dd, J = 9.32, 7.84 Hz, 1H), 3.67–3.56 (m, 2H), 3.48–3.39 (m, 1H), 2.97–2.87 (m, 1H), 2.36 (s, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm): 148.1, 148.1, 147.4, 147.2, 146.7, 138.3, 136.3, 136.0, 134.0, 130.8, 129.5, 127.3, 126.4, 126.3, 120.0, 119.3, 115.7, 108.6, 108.3, 107.4, 106.4, 101.2, 101.2, 86.7, 70.3, 65.6, 55.0, 51.4, 50.2, 21.2. HRMS m/z 520.2126 [M + H]+ (calcd for C33H30NO5, 520.2118).
Compound 21e: white solid, yield: 71.9%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 7.44 (dd, J = 8.76, 5.40 Hz, 2H), 7.34 (d, J = 8.72 Hz, 2H), 7.05 (t, J = 8.72 Hz, 2H), 6.89–6.72 (m, 6H), 6.65 (d, J = 8.72 Hz, 2H), 5.96 (s, 2H), 5.95 (d, J = 2.56 Hz, 2H), 4.76 (t, J = 5.56 Hz, 2H), 3.95 (dd, J = 9.92, 3.80 Hz, 1H), 3.84 (dd, J = 9.32, 7.80 Hz, 1H), 3.68–3.55 (m, 2H), 3.48–3.40 (m, 1H), 2.96–2.90 (m, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 160.8, 148.2, 148.1, 147.5, 147.2, 146.8, 137.3, 136.3, 133.9, 129.8, 128.0, 127.9, 127.4, 120.0, 119.3, 115.7, 115.7, 115.5, 108.6, 108.3, 107.4, 106.4, 101.2, 86.7, 70.3, 65.5, 55.0, 51.4, 50.2. HRMS m/z 524.1863 [M + H]+ (calcd for C32H27FNO5, 524.1868).
Compound 21f: white solid, yield: 66.5%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 7.32 (d, J = 8.52 Hz, 2H), 7.29–7.24 (m, 1H), 7.21–7.09 (m, 2H), 6.88–6.73 (m, 6H), 6.66 (d, J = 8.60 Hz, 2H), 5.95 (s, 2H), 5.94 (d, J = 2.92 Hz, 2H), 4.78 (dd, J = 7.88, 5.92 Hz, 2H), 3.95 (dd, J = 9.96, 3.80 Hz, 1H), 3.83 (t, J = 8.92 Hz, 1H), 3.70–3.57 (m, 2H), 3.50–3.38 (m, 1H), 3.00–2.89 (m, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 148.2, 148.1, 147.6, 147.2, 146.8, 138.3, 136.2, 133.5, 128.8, 127.4, 122.2, 122.1, 122.1, 120.0, 119.3, 117.5, 117.4, 115.8, 115.2, 115.0, 108.6, 108.3, 107.3, 106.4, 101.2, 86.6, 70.2, 65.7, 55.0, 51.3, 50.2. HRMS m/z 542.1777 [M + H]+ (calcd for C32H26F2NO5, 542.1774).
Compound 21g: white solid, yield: 74.5%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 7.43 (d, J = 8.68 Hz, 2H), 7.36 (d, J = 8.68 Hz, 2H), 6.94–6.77 (m, 8H), 6.67 (d, J = 8.68 Hz, 2H), 5.96 (s, 2H), 5.95 (d, J = 2.60 Hz, 2H), 4.76 (dd, J = 14.20, 5.84 Hz, 2H), 3.95 (dd, J = 9.92, 3.56 Hz, 1H), 3.88–3.83 (m, 1H), 3.82 (s, 3H), 3.65–3.56 (m, 2H), 3.49–3.37 (m, 1H), 2.97–2.86 (m, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 158.5, 148.1, 148.1, 147.2, 147.2, 146.7, 136.3, 134.0, 133.9, 130.6, 127.5, 127.5, 127.4, 127.1, 120.0, 119.3, 115.8, 114.2, 108.6, 108.3, 107.4, 106.4, 101.2, 101.2, 86.8, 70.3, 65.6, 55.4, 55.1, 51.3, 50.2. HRMS m/z 536.2068 [M + H]+ (calcd for C33H30NO6, 536.2068).
Compound 21h: white solid, yield: 63.1%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 8.15 (d, J = 5.36 Hz, 1H), 7.47 (d, J = 8.76 Hz, 2H), 7.30 (d, J = 5.36 Hz, 1H), 7.02 (s, 1H), 6.87–6.70 (m, 6H), 6.65 (d, J = 8.80 Hz, 2H), 5.96 (s, 2H), 5.95 (d, J = 1.20 Hz, 2H), 4.84 (d, J = 8.36 Hz, 1H), 4.80 (d, J = 5.44 Hz, 1H), 3.94 (dd, J = 10.00, 4.24 Hz, 1H), 3.86 (dd, J = 9.32, 7.80 Hz, 1H), 3.75 (t, J = 8.36 Hz, 1H), 3.62 (dd, J = 9.40, 6.48 Hz, 1H), 3.51–3.43 (m, 1H), 3.03–2.94 (m, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 166.0, 149.1, 148.3, 148.1, 147.8, 147.6, 147.3, 146.9, 136.1, 133.4, 127.6, 125.5, 123.6, 119.9, 119.2, 118.4, 118.4, 115.3, 108.7, 108.3, 107.2, 106.3, 105.3, 101.3, 101.2, 86.4, 70.1, 65.4, 54.4, 51.4, 50.1. HRMS m/z 525.1814 [M + H]+ (calcd for C31H26FN2O5, 525.1820).
Compound 21i: white solid, yield: 60.6%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 8.30 (d, J = 2.20 Hz, 1H), 7.71 (dd, J = 8.56, 2.20 Hz, 1H), 7.32 (d, J = 8.60 Hz, 2H), 6.88–6.75 (m, 7H), 6.66 (d, J = 8.60 Hz, 2H), 5.96 (s, 2H), 5.95–5.89 (m, 2H), 4.77 (t, J = 5.96 Hz, 2H), 3.96 (s, 3H), 3.93 (d, J = 3.60 Hz, 1H), 3.84 (t, J = 8.24 Hz, 1H), 3.69–3.55 (m, 2H), 3.48–3.40 (m, 1H), 2.99–2.90 (m, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 162.9, 148.2, 148.1, 147.6, 147.2, 146.7, 144.0, 137.3, 136.2, 133.8, 130.2, 127.4, 127.1, 124.2, 123.6, 119.9, 119.3, 119.2, 115.8, 110.8, 108.6, 108.3, 107.3, 106.4, 101.2, 86.7, 70.3, 65.5, 54.9, 53.8, 51.3, 50.2. HRMS m/z 537.2020 [M + H]+ (calcd for C32H29N2O6, 537.2020).
Compound 21j: green solid, yield: 91.5%. 1H NMR (400 MHz, DMSO-d6, J in Hz) δ (ppm): 8.86 (dd, J = 3.72, 1.36 Hz, 1H), 8.37 (dd, J = 8.24, 1.28 Hz, 1H), 7.88 (d, J = 7.32 Hz, 1H), 7.67 (d, J = 6.24 Hz, 1H), 7.60 (t, J = 7.68 Hz, 1H), 7.52 (dd, J = 8.24, 4.12 Hz, 1H), 7.46 (d, J = 8.56 Hz, 2H), 6.99–6.79 (m, 6H), 6.65 (d, J = 8.56 Hz, 2H), 6.01 (s, 2H), 5.99–5.94 (m, 2H), 4.83 (d, J = 8.52 Hz, 1H), 4.74 (d, J = 6.16 Hz, 1H), 3.99 (dd, J = 10.04, 3.08 Hz, 1H), 3.77 (t, J = 7.56 Hz, 1H), 3.60 (t, J = 8.28 Hz, 1H), 3.53–3.45 (m, 1H), 3.43–3.39 (m, 1H), 2.94–2.87 (m, 1H). 13C NMR (100 MHz, DMSO-d6) δ (ppm): 149.9, 147.5, 147.4, 147.0, 146.5, 146.0, 145.3, 139.9, 136.9, 136.4, 134.6, 131.0, 129.3, 128.5, 127.8, 126.8, 126.5, 121.2, 120.0, 119.1, 114.1, 108.3, 108.0, 107.3, 106.3, 100.9, 85.5, 69.4, 64.5, 53.8, 51.2, 49.4. HRMS m/z 557.2077 [M + H]+ (calcd for C35H29N2O5, 557.2071).
Compound 21k: white solid, yield: 75.1%. 1H NMR (400 MHz, CDCl3, J in Hz) δ (ppm): 7.50 (d, J = 1.92 Hz, 1H), 7.19 (d, J = 8.72 Hz, 2H), 6.88–6.72 (m, 6H), 6.63 (d, J = 8.72 Hz, 2H), 6.22 (d, J = 1.80 Hz, 1H), 5.96 (s, 2H), 5.95–5.90 (m, 2H), 4.79 (t, J = 4.44 Hz, 2H), 3.94 (dd, J = 9.88, 3.96 Hz, 1H), 3.88 (s, 3H), 3.85 (dd, J = 9.40, 7.80 Hz, 1H), 3.70 (dd, J = 9.80, 8.24 Hz, 1H), 3.60 (dd, J = 9.40, 6.44 Hz, 1H), 3.50–3.42 (m, 1H), 3.00–2.93 (m, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 148.2, 148.1, 147.2, 146.8, 138.0, 136.2, 133.6, 129.7, 129.4, 124.2, 123.6, 119.9, 119.2, 116.0, 115.0, 113.2, 108.7, 108.3, 107.2, 106.3, 105.5, 101.2, 86.5, 70.2, 65.5, 54.6, 51.4, 50.1, 29.8. HRMS m/z 510.2030 [M + H]+ (calcd for C30H28N3O5, 510.2023).
(7) Crystal structure determination.
A crystal with optimum size was placed on a Bruker D8 Venture single crystal diffractometer. Under the optimized conditions of graphite monochromatic GaKα ray (λ = 1.34139 Å) and 213.96 K, the diffraction points were measured in a fixed range of θ. A direct method was used to solve the structure, and the hydrogen and nonhydrogen atoms were hydrogenated by use of isotropic and anisotropic thermal parameters, respectively. The crystal structure was corrected and analyzed by SHELX 2018 (Sheldrick, 2018) and SHELXL 2018 (Sheldrick, 2015). The relevant crystallographic and structural correction data are shown in Table S1.
(8) Bioactivity evaluation.
Antifungal activity evaluation: The broth microdilution method based on the Clinical and Laboratory Standards Institute (CLSI) was used to evaluate the antifungal activity of N-aryl-azasesamins. The compounds were dissolved in DMSO at a stock concentration of 4 mg/mL. Exponentially growing cultures (OD600 = 0.03–0.06) of each strain were prepared from overnight cultures. Cultures were diluted in broth (1:10) and added to a 96-well plate (195 μL/well). C. neoformans was used directly without dilutions. Fluconazole was used as positive control for C. albicans and C. neoformans. Plates were read at 600 nm after 48 h incubation. Inhibition was calculated by subtracting the absorbance of the blank wells, dividing by the average value for the DMSO only wells, and multiplying by 100.
Antitumor activity evaluation: HepG2 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin in an incubator set to 5% CO2, 37 °C, and 100% humidity conditions. The medium was normally exchanged every 2–3 days, and cells were passaged every 6–7 days. During the exponential growth phase, cells were passaged 3 to 4 times before being used for experiments.
For the assays, cells were detached using 0.25% trypsin, and the resulting suspension was seeded into 96-well culture plates at a density of approximately 5000 cells/mL/well. Cells were treated with increasing concentrations of N-aryl-azasesamins solution and cultured for 24 h. About 10 μL of freshly prepared MTT (5 mg/mL) solution was added to each well and incubated for 4 h. Following incubation, the supernatant was removed, and 150 μL of DMSO was added to dissolve the formazan crystals. Optical density (OD) values were measured at 570 nm with a reference wavelength of 450 nm using a microplate reader. The IC50 values of each compound, representing the concentration required to achieve 50% inhibition of cell growth, were calculated.
(9) Data Analysis.
Data were averaged and statistically analyzed using SPSS 17.0 (IBM, Armonk, NY, USA) with one-way ANOVA (Tukey test) and Student’s T-tests to compare mean differences at a significance level of p < 0.05. GraphPad Prism 9 (GraphPad Software, San Diego, CA, USA) was used for IC50 calculation. There were three biological and three technical replicates performed for each sample.
4. Conclusions
In summary, in the present work, 17 N-aryl-azasesamins were successfully synthesized for the first time, through cleavage of the ethereal bond, Mitsunobu and Suzuki reactions, etc. The mechanism for synthesis of compounds 5–8 was deduced, and configurations of the products were confirmed. Based on the bioactivity evaluation of these analogues, none of the analogues showed activity against C. albicans and C. neoformans. However, against HepG2 cells, only analogues 21C and 21K demonstrated moderate anticancer activity. It is concluded that some N-aryl-azasesamins containing chlorine atoms or a substructure of methylpyrazole have significantly improved antitumor activities compared with sesamin. The configuration of C6 in the N-aryl-azasesamins synthesized in this study differs from that of natural sesamin, which may be the primary reason for the poor antibacterial and antitumor activities observed in these analogues. Therefore, azasesamins with configurations identical to natural sesamin, as well as those containing chlorine atoms and pyrazole heterocycles, will be synthesized. To better understand the structure–activity relationship, the anticancer activities of these N-aryl-azasesamins will be evaluated across various cancer cell lines. Furthermore, quaternary ammonium salts of the N-aryl-azasesamins will also be synthesized to improve their solubility.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/chemistry6060079/s1, Table S1: Crystallographic data and structural refinement parameters of compound 20b; Figures S1 and S2: 1H and 13C NMR spectrum of compound 5; Figures S3 and S4: 1H and 13C NMR spectrum of compound 6; Figures S5 and S6: 1H and 13C NMR spectrum of compound 8; Figures S7 and S8: 1H and 13C NMR spectrum of compound 11; Figures S9 and S10: 1H and 13C NMR spectrum of compound 18a; Figures S11 and S12: 1H and 13C NMR spectrum of compound 18b; Figures S13 and S14: 1H and 13C NMR spectrum of compound 18c; Figures S15 and S16: 1H and 13C NMR spectrum of compound 18d; Figures S17 and S18: 1H and 13C NMR spectrum of compound 18e; Figures S19 and S20: 1H and 13C NMR spectrum of compound 18f; Figures S21 and S22: 1H and 13C NMR spectrum of compound 19a; Figures S23 and S24: 1H and 13C NMR spectrum of compound 19b; Figures S25 and S26: 1H and 13C NMR spectrum of compound 19c; Figures S27 and S28: 1H and 13C NMR spectrum of compound 19d; Figures S29 and S30: 1H and 13C NMR spectrum of compound 19e; Figures S31 and S32: 1H and 13C NMR spectrum of compound 19f; Figures S33 and S34: 1H and 13C NMR spectrum of compound 20a; Figures S35 and S36: 1H and 13C NMR spectrum of compound 20b; Figures S37 and S38: 1H and 13C NMR spectrum of compound 20c; Figures S39 and S40: 1H and 13C NMR spectrum of compound 20d; Figures S41 and S42: 1H and 13C NMR spectrum of compound 20e; Figures S43 and S44: 1H and 13C NMR spectrum of compound 20f; Figures S45 and S46: 1H and 13C NMR spectrum of compound 21a; Figures S47 and S48: 1H and 13C NMR spectrum of compound 21b; Figures S49 and S50: 1H and 13C NMR spectrum of compound 21c; Figures S51 and S52: 1H and 13C NMR spectrum of compound 21d; Figures S53 and S54: 1H and 13C NMR spectrum of compound 21e; Figures S55 and S56: 1H and 13C NMR spectrum of compound 21f; Figures S57 and S58: 1H and 13C NMR spectrum of compound 21g; Figures S59 and S60: 1H and 13C NMR spectrum of compound 21h; Figures S61 and S62: 1H and 13C NMR spectrum of compound 21i; Figures S63 and S64: 1H and 13C NMR spectrum of compound 21j; Figures S65 and S66: 1H and 13C NMR spectrum of compound 21k; Figures S67–S70: 2D NMR spectra of compound 20b; Figure S71: HRMS of compound 11; Figures S72–S77: HRMS of compound 20a–20f; Figures S78–S88: HRMS of compound 21a–21k.
Author Contributions
J.W. investigation; X.Q. validation, writing—original draft; Y.S.J. formal analysis, writing—original draft; C.L. formal analysis; X.S. validation; X.X. formal analysis; S.S. validation; Y.G. supervision, conceptualization, funding acquisition, project administration, writing—review and editing; L.L.W. validation, writing—review and editing; H.B. supervision, project administration, conceptualization. All authors have read and agreed to the published version of the manuscript.
Funding
This research work was funded by the National Natural Science Foundation of China (No. 21967005), the Natural Science Foundation of Guangxi (Nos. 2023GXNSFAA026340, AD20238016), and the Research Start-up Foundation of Guangxi Minzu University (No. 2019KJQD07).
Data Availability Statement
Data are contained within the article and supplementary materials.
Acknowledgments
The authors would like to thank Guoqing Chen from The Hong Kong Polytechnic University for the discussion of the anticancer section.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod. 2016, 79, 629–661. [Google Scholar] [CrossRef] [PubMed]
- Dholwani, K.K.; Saluja, A.K.; Gupta, A.R.; Shah, D.R. A review on plant-derived natural products and their analogs with anti-tumor activity. Indian J. Pharmacol. 2008, 40, 49–58. [Google Scholar]
- Sato, H.; Aoki, A.; Tabata, A.; Kadota, K.; Tozuka, Y.; Seto, Y.; Onoue, S. Development of sesamin-loaded solid dispersion with α-glycosylated stevia for improving physicochemical and nutraceutical properties. J. Funct. Foods 2017, 35, 325–331. [Google Scholar]
- Zhang, Y.; Liu, F.; Lin, Y.; Li, L.; Chen, M.; Ni, L.; Rather, M.A. A Comprehensive Review on Distribution, Pharmacological Properties, and Mechanisms of Action of Sesamin. J. Chem. 2022, 2022, 4236525. [Google Scholar]
- Jeng, K.; Hou, R. Sesamin and Sesamolin: Natures Therapeutic Lignans. Curr. Enzym. Inhib. 2005, 1, 11–20. [Google Scholar] [CrossRef]
- Dar, A.A.; Arumugam, N. Lignans of sesame: Purification methods, biological activities and biosynthesis—A review. Bioorg. Chem. 2013, 50, 1–10. [Google Scholar] [PubMed]
- Majdalawieh, A.F.; Yousef, S.M.; Abu-Yousef, I.A.; Nasrallah, G.K. Immunomodulatory and anti-inflammatory effects of sesamin: Mechanisms of action and future directions. Crit. Rev. Food Sci. Nutr. 2021, 62, 5081–5112. [Google Scholar] [PubMed]
- Kitipaspallop, W.; Phuwapraisirisan, P.; Kim, W.K.; Chanchao, C.; Pimtong, W. Sesamin lacks zebrafish embryotoxicity but exhibits evidence of anti-angiogenesis, anti-oxidant and anti-inflammatory activities. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2023, 269, 109637. [Google Scholar]
- Fukunaga, M.; Ohnishi, M.; Shiratsuchi, A.; Kawakami, T.; Takahashi, M.; Motomura, M.; Egusa, K.; Urasaki, T.; Inoue, A. Sesamin increases heme oxygenase-1 protein in RAW 264.7 macrophages through inhibiting its ubiquitination process. Eur. J. Pharmacol. 2014, 741, 214–221. [Google Scholar] [CrossRef]
- Ahmad, S.; ElSherbiny, N.M.; Jamal, M.S.; Alzahrani, F.A.; Haque, R.; Khan, R.; Zaidi, S.K.; AlQahtani, M.H.; Liou, G.I.; Bhatia, K. Anti-inflammatory role of sesamin in STZ induced mice model of diabetic retinopathy. J. Neuroimmunol. 2016, 295–296, 47–53. [Google Scholar] [CrossRef]
- Dalibalta, S.; Majdalawieh, A.F.; Manjikian, H. Health benefits of sesamin on cardiovascular disease and its associated risk factors. Saudi Pharm. J. 2020, 28, 1276–1289. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.T.; Ren, J.Y.; Zhu, S.Q.; Zhang, Z.A.; Guo, Z.H.; An, J.Q.; Yin, B.W.; Ma, Y.X. The Effects of Sesamin Supplementation on Obesity, Blood Pressure, and Lipid Profile: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Front. Endocrinol. 2022, 13, 842152. [Google Scholar]
- Helli, B.; Mowla, K.; Mohammadshahi, M.; Jalali, M.T. Effect of Sesamin Supplementation on Cardiovascular Risk Factors in Women with Rheumatoid Arthritis. J. Am. Coll. Nutr. 2015, 35, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Yashaswini, P.S.; Sadashivaiah, B.; Ramaprasad, T.R.; Singh, S.A. In vivo modulation of LPS induced leukotrienes generation and oxidative stress by sesame lignans. J. Nutr. Biochem. 2017, 41, 151–157. [Google Scholar] [CrossRef]
- Majdalawieh, A.F.; Mansour, Z.R. Sesamol, a major lignan in sesame seeds (Sesamum indicum): Anti-cancer properties and mechanisms of action. Eur. J. Pharmacol. 2019, 855, 75–89. [Google Scholar] [CrossRef]
- Majdalawieh, A.F.; Massri, M.; Nasrallah, G.K. A comprehensive review on the anti-cancer properties and mechanisms of action of sesamin, a lignan in sesame seeds (Sesamum indicum). Eur. J. Pharmacol. 2017, 815, 512–521. [Google Scholar] [CrossRef]
- Chen, Y.; Li, H.; Zhang, W.; Qi, W.; Lu, C.; Huang, H.; Yang, Z.; Liu, B.; Zhang, L. Sesamin suppresses NSCLC cell proliferation and induces apoptosis via Akt/p53 pathway. Toxicol. Appl. Pharmacol. 2020, 387, 114848. [Google Scholar] [CrossRef]
- Kong, X.; Ma, M.Z.; Zhang, Y.; Weng, M.Z.; Gong, W.; Guo, L.Q.; Zhang, J.X.; Wang, G.D.; Su, Q.; Quan, Z.W.; et al. Differentiation therapy: Sesamin as an effective agent in targeting cancer stem-like side population cells of human gallbladder carcinoma. BMC Complement. Altern. Med. 2014, 14, 254. [Google Scholar] [CrossRef]
- Deng, P.; Wang, C.; Chen, L.; Wang, C.; Du, Y.; Yan, X.; Chen, M.; Yang, G.; He, G. Sesamin Induces Cell Cycle Arrest and Apoptosis through the Inhibition of Signal Transducer and Activator of Transcription 3 Signalling in Human Hepatocellular Carcinoma Cell Line HepG2. Biol. Pharm. Bull. 2013, 36, 1540–1548. [Google Scholar] [CrossRef]
- Ahmad, S.; Elsherbiny, N.M.; Haque, R.; Khan, M.B.; Ishrat, T.; Shah, Z.A.; Khan, M.M.; Ali, M.; Jamal, A.; Katare, D.P.; et al. Sesamin attenuates neurotoxicity in mouse model of ischemic brain stroke. Neuro Toxicol. 2014, 45, 100–110. [Google Scholar]
- Zhao, Y.; Wang, Q.; Jia, M.; Fu, S.; Pan, J.; Chu, C.; Liu, X.; Liu, X.; Liu, Z. (+)-Sesamin attenuates chronic unpredictable mild stress-induced depressive-like behaviors and memory deficits via suppression of neuroinflammation. J. Nutr. Biochem. 2019, 64, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Baluchnejadmojarad, T.; Mansouri, M.; Ghalami, J.; Mokhtari, Z.; Roghani, M. Sesamin imparts neuroprotection against intrastriatal 6-hydroxydopamine toxicity by inhibition of astroglial activation, apoptosis, and oxidative stress. Biomed. Pharmacother. 2017, 88, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Phitak, T.; Pothacharoen, P.; Settakorn, J.; Poompimol, W.; Caterson, B.; Kongtawelert, P. Chondroprotective and anti-inflammatory effects of sesamin. Phytochemistry 2012, 80, 77–88. [Google Scholar] [CrossRef]
- Chiang, H.M.; Chang, H.; Yao, P.W.; Chen, Y.S.; Jeng, K.C.; Wang, J.S.; Hou, C.W. Sesamin reduces acute hepatic injury induced by lead coupled with lipopolysaccharide. J. Chin. Med. Assoc. 2014, 77, 227–233. [Google Scholar] [CrossRef]
- Wadhwa, K.; Kaur, H.; Kapoor, N.; Brogi, S. Identification of Sesamin from Sesamum indicum as a Potent Antifungal Agent Using an Integrated in Silico and Biological Screening Platform. Molecules 2023, 28, 4658. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A.K.; Singh, H.; Vijayan, V.; Kumar, D.; Naik, J.; Thareja, S.; Yadav, J.P.; Pathak, P.; Grishina, M.; et al. Nitrogen Containing Heterocycles as Anticancer Agents: A Medicinal Chemistry Perspective. Pharmaceuticals 2023, 16, 299. [Google Scholar] [CrossRef] [PubMed]
- Bhat, C.; Tilve, S.G. Recent advances in the synthesis of naturally occurring pyrrolidines, pyrrolizidines and indolizidine alkaloids using proline as a unique chiral synthon. RSC Adv. 2014, 4, 5405–5452. [Google Scholar] [CrossRef]
- Li Petri, G.; Raimondi, M.V.; Spanò, V.; Holl, R.; Barraja, P.; Montalbano, A. Pyrrolidine in Drug Discovery: A Versatile Scaffold for Novel Biologically Active Compounds. Top. Curr. Chem. 2021, 379, 34–79. [Google Scholar] [CrossRef]
- Jeelan Basha, N.; Basavarajaiah, S.M.; Shyamsunder, K. Therapeutic potential of pyrrole and pyrrolidine analogs: An update. Mol. Divers. 2022, 26, 2915–2937. [Google Scholar] [CrossRef]
- Bhat, A.A.; Singh, I.; Tandon, N.; Tandon, R. Structure activity relationship (SAR) and anticancer activity of pyrrolidine derivatives: Recent developments and future prospects (A review). Eur. J. Med. Chem. 2023, 246, 114954–114998. [Google Scholar] [CrossRef]
- Łukowska-Chojnacka, E.; Kowalkowska, A.; Gizińska, M.; Koronkiewicz, M.; Staniszewska, M. Synthesis of tetrazole derivatives bearing pyrrolidine scaffold and evaluation of their antifungal activity against Candida albicans. Eur. J. Med. Chem. 2019, 164, 106–120. [Google Scholar] [CrossRef] [PubMed]
- Moni, L.; Banfi, L.; Basso, A.; Carcone, L.; Rasparini, M.; Riva, R. Ugi and Passerini Reactions of Biocatalytically Derived Chiral Aldehydes: Application to the Synthesis of Bicyclic Pyrrolidines and of Antiviral Agent Telaprevir. J. Org. Chem. 2015, 80, 3411–3428. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, X.; Lin, Y.; Wang, Y.; Wu, S.; Xia, K.; Xu, C.; Ma, H.; Zheng, J.; Luo, L.; et al. Design, synthesis, and evaluation of pyrrolidine based CXCR4 antagonists with in vivo anti-tumor metastatic activity. Eur. J. Med. Chem. 2020, 205, 112537–112550. [Google Scholar] [CrossRef] [PubMed]
- Jan, M.S.; Ahmad, S.; Hussain, F.; Ahmad, A.; Mahmood, F.; Rashid, U.; Abid, O.u.R.; Ullah, F.; Ayaz, M.; Sadiq, A. Design, synthesis, in-vitro, in-vivo and in-silico studies of pyrrolidine-2,5-dione derivatives as multitarget anti-inflammatory agents. Eur. J. Med. Chem. 2020, 186, 111863–111878. [Google Scholar] [CrossRef]
- Góra, M.; Czopek, A.; Rapacz, A.; Dziubina, A.; Głuch-Lutwin, M.; Mordyl, B.; Obniska, J. Synthesis, Anticonvulsant and Antinociceptive Activity of New Hybrid Compounds: Derivatives of 3-(3-Methylthiophen-2-yl)-pyrrolidine-2,5-dione. Int. J. Mol. Sci. 2020, 21, 5750. [Google Scholar] [CrossRef]
- Jung, E.-K.; Dittrich, N.; Pilkington, L.I.; Rye, C.E.; Leung, E.; Barker, D. Synthesis of aza-derivatives of tetrahydrofuran lignan natural products. Tetrahedron 2015, 71, 9439–9456. [Google Scholar] [CrossRef]
- Mincione, E.; Lanciano, F. Thallium nitrate as a reagent for the conversion of epoxides into α-hydroxynitrate esters and for the cleavage of aliphatic ethers. Tetrahedron Lett. 1980, 21, 1149–1150. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).