
Hydrogenation Studies of Iridium Pyridine Diimine Complexes with O- and S-Donor Ligands (Hydroxido, Methoxido and Thiolato) #


Abstract

1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
- (a)
- NMR spectroscopy
- (b)
- X-ray crystallography
2.2.1. Theoretical Methods
DFT Calculations
Local Coupled Cluster Calculations
3. Hydrogenation Reactions
3.1. Comparison of the Sulphido, Hydroxido, and Methoxido Systems
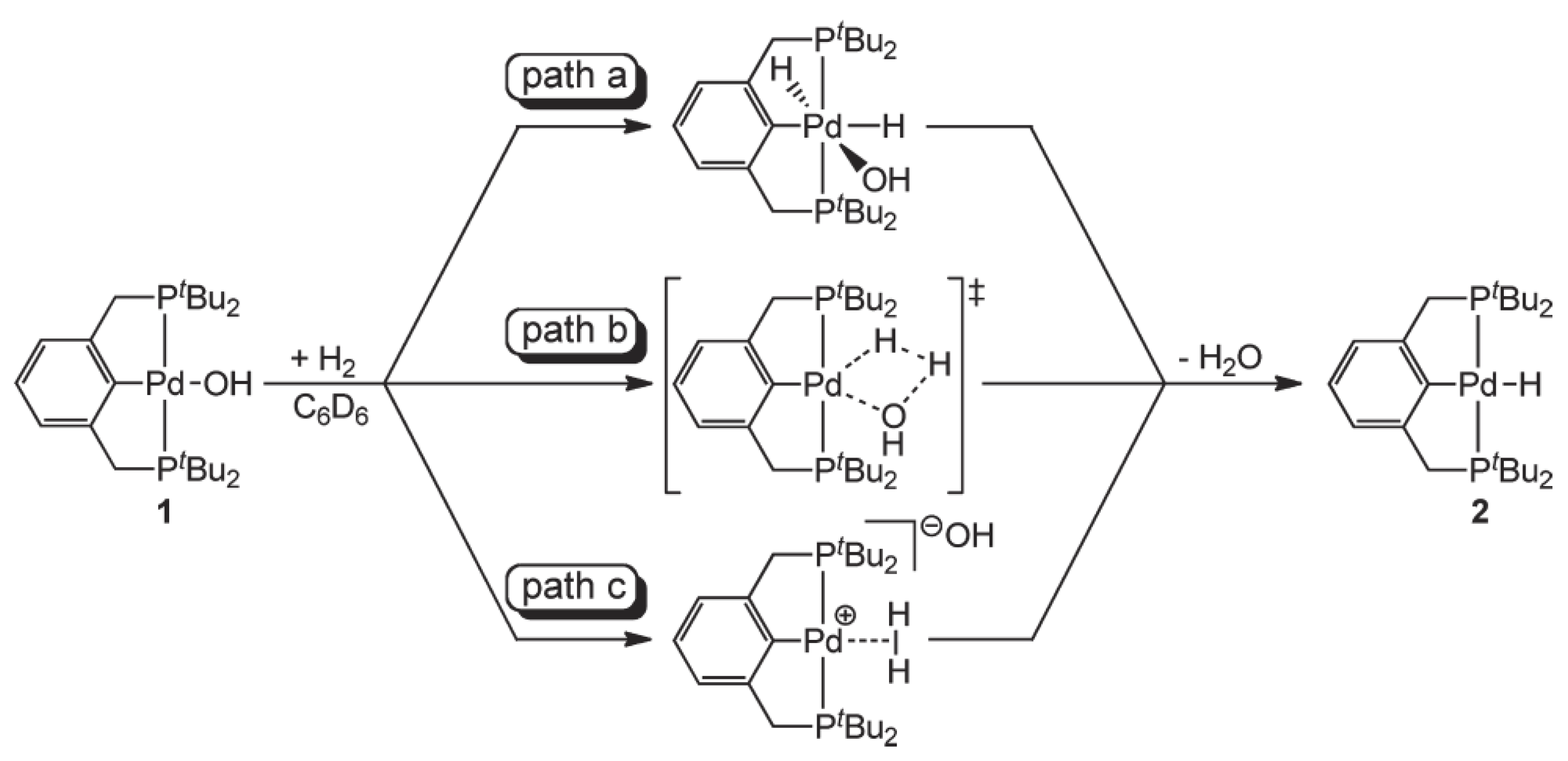
3.1.1. O-Donors
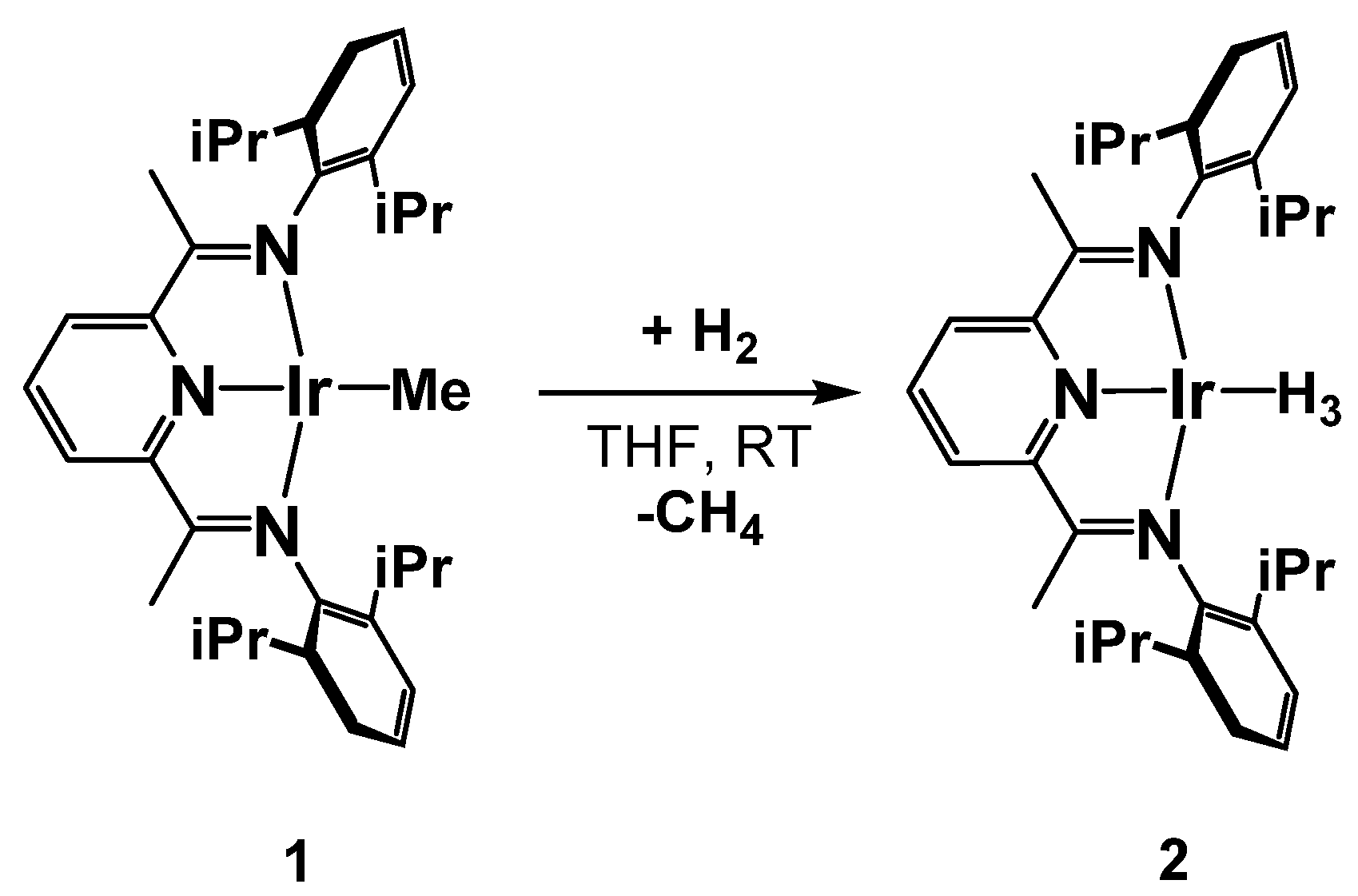
3.1.2. S-Donors
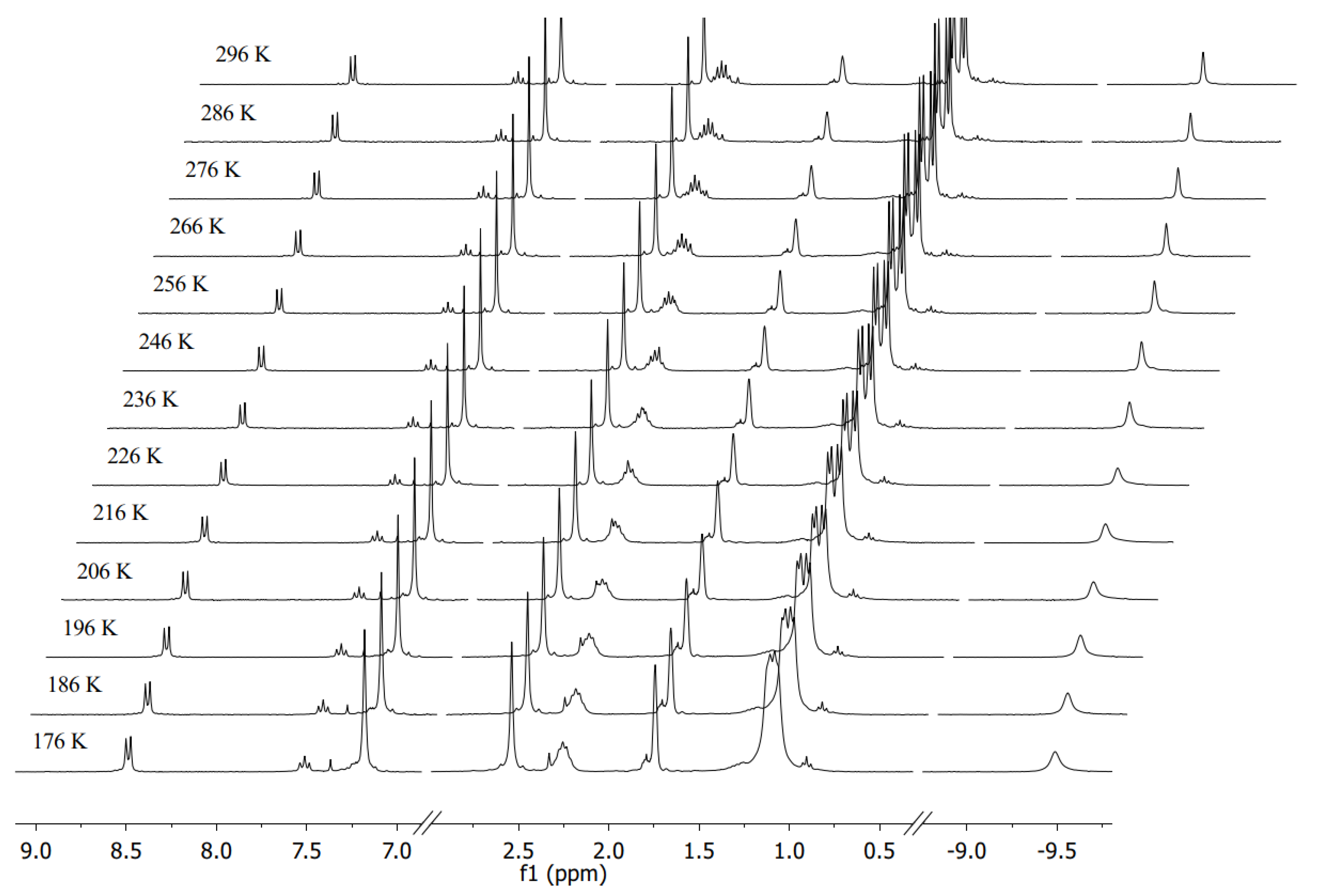
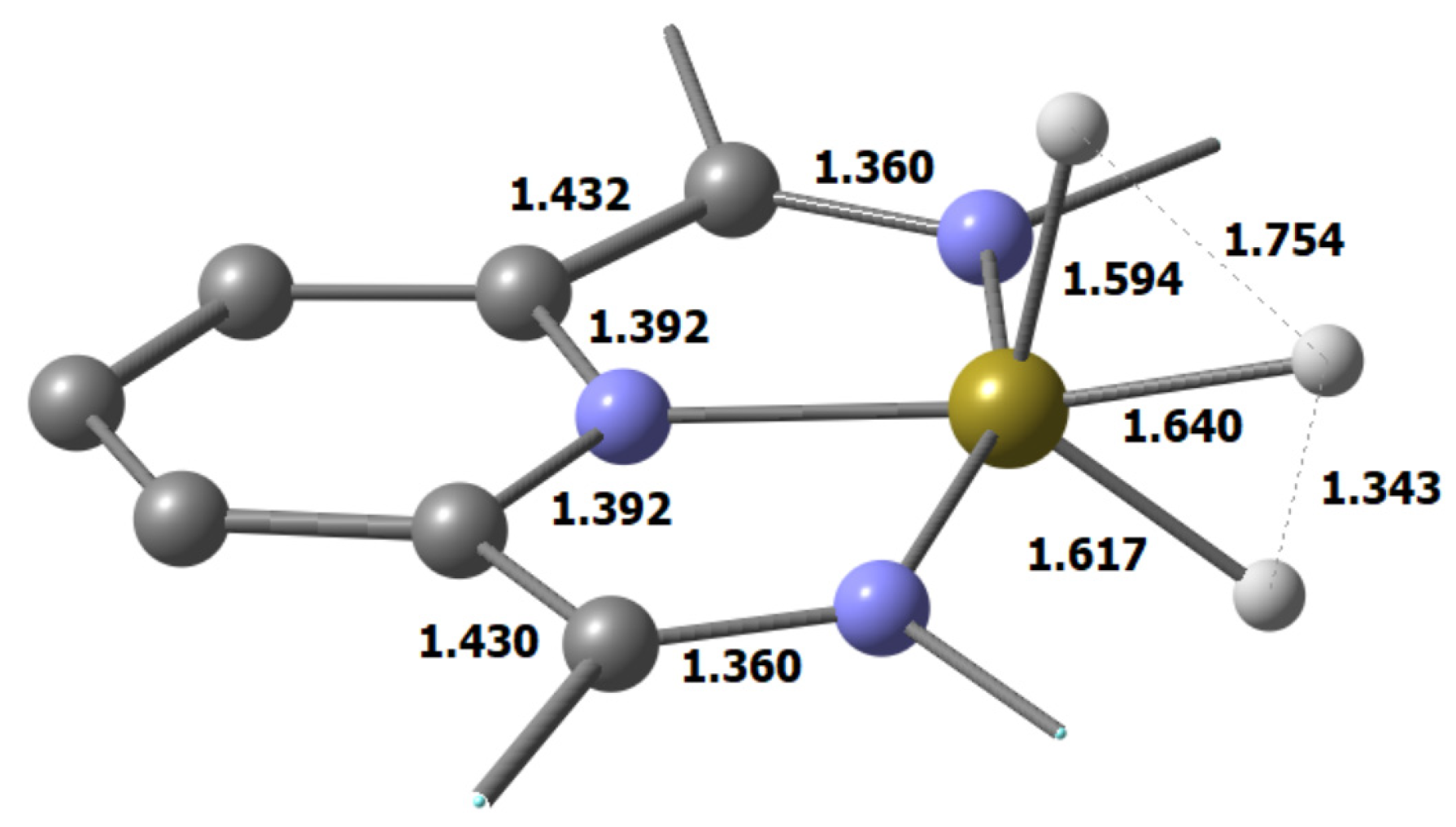
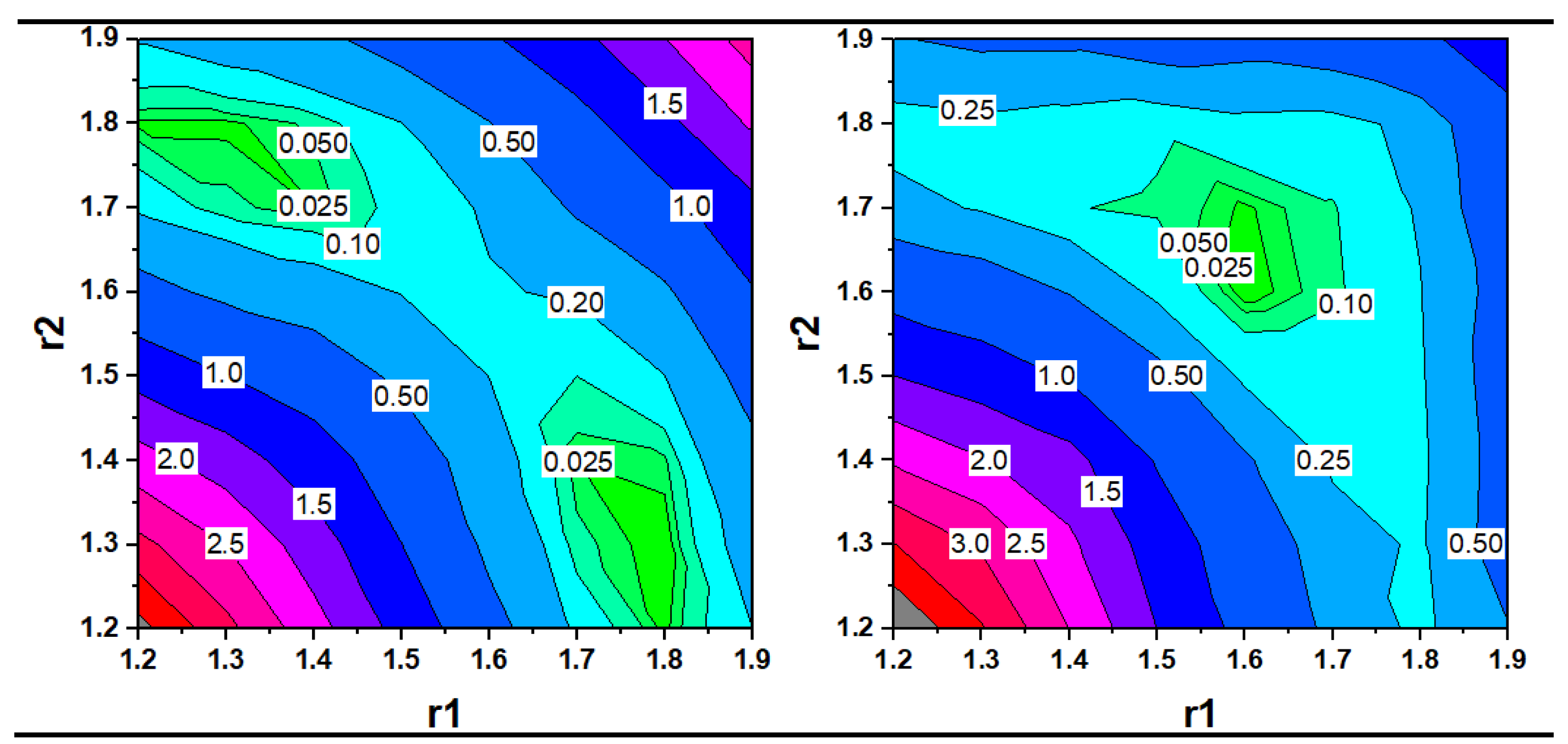
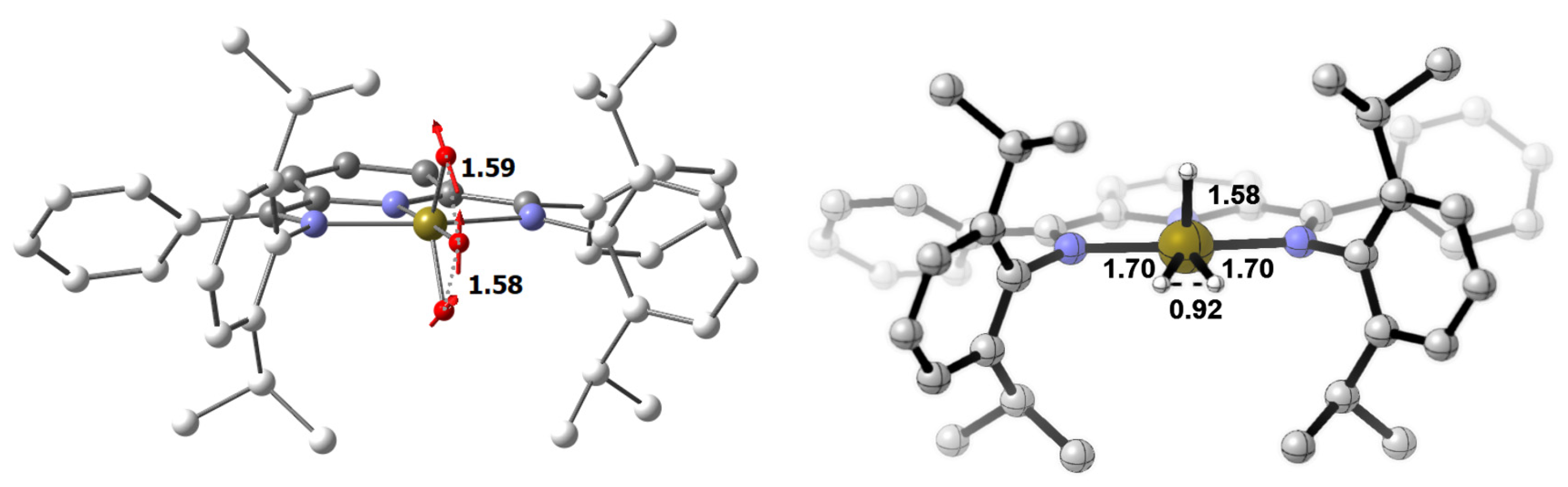
3.2. Characterization of the Trihydride Product
4. Thermodynamics and Mechanism of the Hydrogenation Reaction
4.1. Thermodynamics
Analysis of the Thermodynamic Data
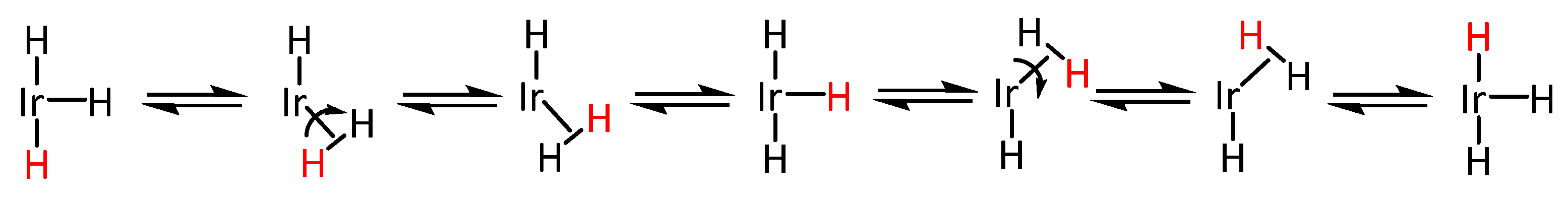
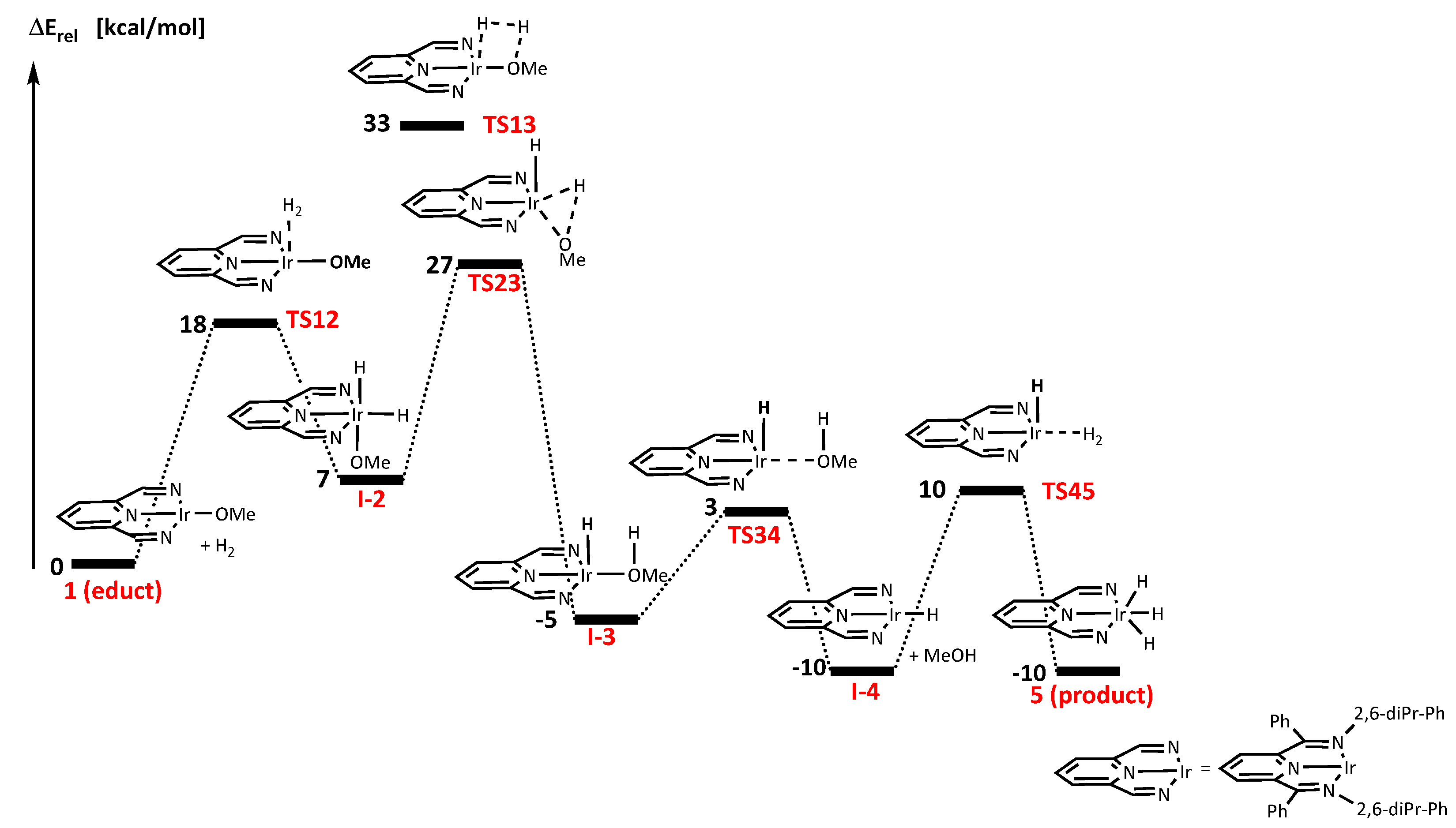
4.2. Hydrogenation Mechanism
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fulmer, G.R.; Herndon, A.N.; Kaminsky, W.; Kemp, R.A.; Goldberg, K.I. Hydrogenolysis of palladium(II) hydroxide, phenoxide, and alkoxide complexes. J. Am. Chem. Soc. 2011, 133, 17713–17726. [Google Scholar] [CrossRef] [PubMed]
- Waterman, R. σ-Bond Metathesis: A 30-Year Retrospective. Organometallics 2013, 32, 7249–7263. [Google Scholar] [CrossRef]
- Bailey, W.D.; Phearman, A.S.; Luconi, L.; Rossin, A.; Yakhvarov, D.G.; D’Accolti, L.; Flowers, S.E.; Kaminsky, W.; Kemp, R.A.; Giambastiani, G.; et al. Hydrogenolysis of Dinuclear PCNR Ligated PdII mu-Hydroxides and Their Mononuclear PdII Hydroxide Analogues. Chemistry 2019, 25, 9920–9929. [Google Scholar] [CrossRef] [PubMed]
- Parkes, M.V.; Bailey, W.D.; Goldberg, K.I.; Kemp, R.A. The Effect of the cis-donor in pincer ligands on hydrogenolysis of Pd-OH: A DFT study. J. Organomet. Chem. 2017, 845, 165–170. [Google Scholar] [CrossRef]
- Misumi, Y.; Seino, H.; Mizobe, Y. Heterolytic cleavage of hydrogen molecule by rhodium thiolate complexes that catalyze chemoselective hydrogenation of imines under ambient conditions. J. Am. Chem. Soc. 2009, 131, 14636–14637. [Google Scholar] [CrossRef]
- Seino, H.; Misumi, Y.; Hojo, Y.; Mizobe, Y. Heterolytic H2 activation by rhodium thiolato complexes bearing the hydrotris(pyrazolyl)borato ligand and application to catalytic hydrogenation under mild conditions. Dalton Trans. 2010, 39, 3072–3082. [Google Scholar] [CrossRef]
- Omann, L.; Konigs, C.D.F.; Klare, H.F.T.; Oestreich, M. Cooperative Catalysis at Metal-Sulfur Bonds. Acc. Chem. Res. 2017, 50, 1258–1269. [Google Scholar] [CrossRef]
- Zhu, D.; Thapa, I.; Korobkov, I.; Gambarotta, S.; Budzelaar, P.H. Redox-active ligands and organic radical chemistry. Inorg. Chem. 2011, 50, 9879–9887. [Google Scholar] [CrossRef]
- Zhu, D.; Budzelaar, P.H.M. A Measure for σ-Donor and π-Acceptor Properties of Diiminepyridine-Type Ligands. Organometallics 2008, 27, 2699–2705. [Google Scholar] [CrossRef]
- Stephan, M.; Völker, M.; Schreyer, M.; Burger, P. Syntheses, Crystal and Electronic Structures of Rhodium and Iridium Pyridine Di-Imine Complexes with O- and S-Donor Ligands: (Hydroxido, Methoxido and Thiolato). Chemistry 2023, 5, 1961–1989. [Google Scholar] [CrossRef]
- Sieh, D.; Schlimm, M.; Andernach, L.; Angersbach, F.; Nückel, S.; Schöffel, J.; Šušnjar, N.; Burger, P. Metal–Ligand Electron Transfer in 4d and 5d Group 9 Transition Metal Complexes with Pyridine, Diimine Ligands. Eur. J. Inorg. Chem. 2012, 2012, 444–462. [Google Scholar] [CrossRef]
- Römelt, C.; Weyhermüller, T.; Wieghardt, K. Structural characteristics of redox-active pyridine-1,6-diimine complexes: Electronic structures and ligand oxidation levels. Coord. Chem. Rev. 2019, 380, 287–317. [Google Scholar] [CrossRef]
- Knijnenburg, Q.; Gambarotta, S.; Budzelaar, P.H. Ligand-centred reactivity in diiminepyridine complexes. Dalton Trans. 2006, 46, 5442–5448. [Google Scholar] [CrossRef] [PubMed]
- Bart, S.C.; Chlopek, K.; Bill, E.; Bouwkamp, M.W.; Lobkovsky, E.; Neese, F.; Wieghardt, K.; Chirik, P.J. Electronic structure of bis(imino)pyridine iron dichloride, monochloride, and neutral ligand complexes: A combined structural, spectroscopic, and computational study. J. Am. Chem. Soc. 2006, 128, 13901–13912. [Google Scholar] [CrossRef] [PubMed]
- Angersbach-Bludau, F.; Schulz, C.; Schoffel, J.; Burger, P. Syntheses and electronic structures of mu-nitrido bridged pyridine, diimine iridium complexes. Chem. Commun. 2014, 50, 8735–8738. [Google Scholar] [CrossRef]
- Nückel, S.; Burger, P. Transition Metal Complexes with Sterically Demanding Ligands, 3.1 Synthetic Access to Square-Planar Terdentate Pyridine−Diimine Rhodium(I) and Iridium(I) Methyl Complexes: Successful Detour via Reactive Triflate and Methoxide Complexes. Organometallics 2001, 20, 4345–4359. [Google Scholar] [CrossRef]
- Schiller, C.; Sieh, D.; Lindenmaier, N.; Stephan, M.; Junker, N.; Reijerse, E.; Granovsky, A.A.; Burger, P. Cleavage of an Aromatic C–C Bond in Ferrocene by Insertion of an Iridium Nitrido Nitrogen Atom. J. Am. Chem. Soc. 2023, 145, 11392–11401. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2007, 64, 112. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456. [Google Scholar] [CrossRef]
- Franzke, Y.J.; Holzer, C.; Andersen, J.H.; Begusic, T.; Bruder, F.; Coriani, S.; Della Sala, F.; Fabiano, E.; Fedotov, D.A.; Furst, S.; et al. TURBOMOLE: Today and Tomorrow. J. Chem. Theory Comput. 2023, 19, 6859–6890. [Google Scholar] [CrossRef] [PubMed]
- ChemShell. A Computational Chemistry Shell. Available online: www.chemshell.org (accessed on 5 August 2024).
- Kallay, M.; Nagy, P.R.; Mester, D.; Rolik, Z.; Samu, G.; Csontos, J.; Csoka, J.; Szabo, P.B.; Gyevi-Nagy, L.; Hegely, B.; et al. The MRCC program system: Accurate quantum chemistry from water to proteins. J. Chem. Phys. 2020, 152, 074107. [Google Scholar] [CrossRef] [PubMed]
- Kanchanakungwankul, S.; JBao, J.L.; Zheng, J.; Alecu, I.M.; Lynch, B.J.; Zhao, Y.; Truhlar, D.G. Database of Frequency Scale Factors for Electronic Model Chemistries—Version 5. Available online: https://comp.chem.umn.edu/freqscale/ (accessed on 26 July 2024).
- Molpro 2022.3. 2022. Available online: www.molpro.net (accessed on 26 July 2024).
- Werner, H.J.; Knowles, P.J.; Manby, F.R.; Black, J.A.; Doll, K.; Hesselmann, A.; Kats, D.; Kohn, A.; Korona, T.; Kreplin, D.A.; et al. The Molpro quantum chemistry package. J. Chem. Phys. 2020, 152, 144107. [Google Scholar] [CrossRef] [PubMed]
- Nückel, S.; Burger, P. Transition-metal complexes with sterically demanding ligands: Facile thermal intermolecular C-H bond activation in a square-planar IrI complex. Angew. Chem. Int. Ed. Engl. 2003, 42, 1632–1636. [Google Scholar] [CrossRef] [PubMed]
- Albright, T.A.; Burdett, J.K.; Whangbo, M.-H. Orbital Interactions in Chemistry; Wiley: Hoboken, NJ, USA, 2013. [Google Scholar] [CrossRef]
- Kloek, S.M.; Heinekey, D.M.; Goldberg, K.I. C-H bond activation by rhodium(I) hydroxide and phenoxide complexes. Angew. Chem. Int. Ed. Engl. 2007, 46, 4736–4738. [Google Scholar] [CrossRef]
- Hanson, S.K.; Heinekey, D.M.; Goldberg, K.I. C−H Bond Activation by Rhodium(I) Phenoxide and Acetate Complexes: Mechanism of H−D Exchange between Arenes and Water. Organometallics 2008, 27, 1454–1463. [Google Scholar] [CrossRef]
- Tenn, W.J.; Young, K.J.; Bhalla, G.; Oxgaard, J.; Goddard, W.A., 3rd; Periana, R.A. CH activation with an O-donor iridium-methoxo complex. J. Am. Chem. Soc. 2005, 127, 14172–14173. [Google Scholar] [CrossRef]
- Tenn, W.J.; Young, K.J.H.; Oxgaard, J.; Nielsen, R.J.; Goddard, W.A.; Periana, R.A. Heterolytic CH activation and catalysis by an O-donor iridium-hydroxo complex. Organometallics 2006, 25, 5173–5175. [Google Scholar] [CrossRef]
- Schöffel, J.; Šušnjar, N.; Nückel, S.; Sieh, D.; Burger, P. 4d vs. 5d—Reactivity and Fate of Terminal Nitrido Complexes of Rhodium and Iridium. Eur. J. Inorg. Chem. 2010, 2010, 4911–4915. [Google Scholar] [CrossRef]
- Kubas, G.J. Metal Dihydrogen and σ-Bond Complexes; Springer Nature: Dordrecht, The Netherlands, 2001. [Google Scholar] [CrossRef]
- Crabtree, R.H. Dihydrogen Complexation. Chem. Rev. 2016, 116, 8750–8769. [Google Scholar] [CrossRef]
- Hebden, T.J.; Goldberg, K.I.; Heinekey, D.M.; Zhang, X.; Emge, T.J.; Goldman, A.S.; Krogh-Jespersen, K. Dihydrogen/dihydride or tetrahydride? An experimental and computational investigation of pincer iridium polyhydrides. Inorg. Chem. 2010, 49, 1733–1742. [Google Scholar] [CrossRef] [PubMed]
- Gelabert, R.; Moreno, M.; Lluch, J.M.; Lledos, A.; Heinekey, D.M. Determination of the temperature dependence of the H-D spin-spin coupling constant and the isotope effect on the proton chemical shift for the compressed dihydride complex [CpIr(P-P)H2]2+. J. Am. Chem. Soc. 2005, 127, 5632–5640. [Google Scholar] [CrossRef] [PubMed]
- Gusev, D.G. Effect of weak interactions on the H...H distance in stretched dihydrogen complexes. J. Am. Chem. Soc. 2004, 126, 14249–14257. [Google Scholar] [CrossRef] [PubMed]
- Polukeev, A.V.; Capelli, S.C.; Wendt, O.F. Unravelling strong temperature-dependence of J(HD) in transition metal hydrides: Solvation and non-covalent interactions versus temperature-elastic H-H bonds. Chem. Sci. 2023, 14, 12308–12320. [Google Scholar] [CrossRef] [PubMed]
- Taw, F.L.; Mellows, H.; White, P.S.; Hollander, F.J.; Bergman, R.G.; Brookhart, M.; Heinekey, D.M. Synthesis and investigation of [Cp(PMe3)Rh(H)(H2)]+ and its partially deuterated and tritiated isotopomers: Evidence for a hydride/dihydrogen structure. J. Am. Chem. Soc. 2002, 124, 5100–5108. [Google Scholar] [CrossRef] [PubMed]
- Heinekey, D.M.; Hinkle, A.S.; Close, J.D. Quantum Mechanical Exchange Coupling in Iridium Trihydride Complexes. J. Am. Chem. Soc. 1996, 118, 5353–5361. [Google Scholar] [CrossRef]
- Heinekey, D.M.; Millar, J.M.; Koetzle, T.F.; Payne, N.G.; Zilm, K.W. Structural and spectroscopic characterization of iridium trihydride complexes: Evidence for proton-proton exchange coupling. J. Am. Chem. Soc. 1990, 112, 909–919. [Google Scholar] [CrossRef]
- Gordon, B.M.; Lease, N.; Emge, T.J.; Hasanayn, F.; Goldman, A.S. Reactivity of Iridium Complexes of a Triphosphorus-Pincer Ligand Based on a Secondary Phosphine. Catalytic Alkane Dehydrogenation and the Origin of Extremely High Activity. J. Am. Chem. Soc. 2022, 144, 4133–4146. [Google Scholar] [CrossRef]
- Feller, M.; Gellrich, U.; Anaby, A.; Diskin-Posner, Y.; Milstein, D. Reductive Cleavage of CO2 by Metal-Ligand-Cooperation Mediated by an Iridium Pincer Complex. J. Am. Chem. Soc. 2016, 138, 6445–6454. [Google Scholar] [CrossRef]
- Millard, M.D.; Moore, C.E.; Rheingold, A.L.; Figueroa, J.S. Four-coordinate iridium(I) monohydrides: Reversible dinitrogen binding, bond activations, and deprotonations. J. Am. Chem. Soc. 2010, 132, 8921–8923. [Google Scholar] [CrossRef]
- Danopoulos, A.A.; Pugh, D.; Wright, J.A. “Pincer” pyridine-dicarbene-iridium complexes: Facile C-H activation and unexpected eta2-imidazol-2-ylidene coordination. Angew. Chem. Int. Ed. Engl. 2008, 47, 9765–9767. [Google Scholar] [CrossRef] [PubMed]
- Rocher-Casterline, B.E.; Ch’ng, L.C.; Mollner, A.K.; Reisler, H. Communication: Determination of the bond dissociation energy (D0) of the water dimer, (H2O)2, by velocity map imaging. J. Chem. Phys. 2011, 134, 211101. [Google Scholar] [CrossRef]
- Das, A.; Mandal, P.K.; Lovas, F.J.; Medcraft, C.; Walker, N.R.; Arunan, E. The H2S Dimer is Hydrogen-Bonded: Direct Confirmation from Microwave Spectroscopy. Angew. Chem. Int. Ed. Engl. 2018, 57, 15199–15203. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Werner, H.J. Scalable Electron Correlation Methods. 7. Local Open-Shell Coupled-Cluster Methods Using Pair Natural Orbitals: PNO-RCCSD and PNO-UCCSD. J. Chem. Theory Comput. 2020, 16, 3135–3151. [Google Scholar] [CrossRef] [PubMed]
- Neese, F.; Hansen, A.; Liakos, D.G. Efficient and accurate approximations to the local coupled cluster singles doubles method using a truncated pair natural orbital basis. J. Chem. Phys. 2009, 131, 064103. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, D.; Gunnoe, T.B.; Ess, D.H. Theory of late-transition-metal alkyl and heteroatom bonding: Analysis of Pt, Ru, Ir, and Rh complexes. Inorg. Chem. 2012, 51, 6710–6718. [Google Scholar] [CrossRef] [PubMed]
- Bryndza, H.E.; Fong, L.K.; Paciello, R.A.; Tam, W.; Bercaw, J.E. Relative metal-hydrogen, -oxygen, -nitrogen, and -carbon bond strengths for organoruthenium and organoplatinum compounds; equilibrium studies of Cp*(PMe3)2RuX and (DPPE)MePtX systems. J. Am. Chem. Soc. 2002, 109, 1444–1456. [Google Scholar] [CrossRef]
- Peebles, L.R.; Marshall, P. High-accuracy coupled-cluster computations of bond dissociation energies in SH, H2S, and H2O. J. Chem. Phys. 2002, 117, 3132–3138. [Google Scholar] [CrossRef]
- Cabral do Couto, P.; Costa Cabral, B.J.; Martinho Simões, J.A. S–H bond dissociation enthalpies: The importance of a complete basis set approach. Chem. Phys. Lett. 2006, 421, 504–507. [Google Scholar] [CrossRef]
- Ruscic, B. Active Thermochemical Tables: Sequential Bond Dissociation Enthalpies of Methane, Ethane, and Methanol and the Related Thermochemistry. J. Phys. Chem. A 2015, 119, 7810–7837. [Google Scholar] [CrossRef]
- Kesharwani, M.K.; Brauer, B.; Martin, J.M. Frequency and zero-point vibrational energy scale factors for double-hybrid density functionals (and other selected methods): Can anharmonic force fields be avoided? J. Phys. Chem. A 2015, 119, 1701–1714. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Angelici, R.J. Metal−Hydrogen Bond Dissociation Enthalpies in Series of Complexes of Eight Different Transition Metals. J. Am. Chem. Soc. 1996, 118, 935–942. [Google Scholar] [CrossRef]
- Stoutland, P.O.; Bergman, R.G.; Nolan, S.P.; Hoff, C.D. The thermodynamic driving force for C H activation at iridium. Polyhedron 1988, 7, 1429–1440. [Google Scholar] [CrossRef]
- Burgess, S.A.; Devarajan, D.; Bolano, T.; Ess, D.H.; Gunnoe, T.B.; Sabat, M.; Myers, W.H. 1,2-Addition of dihydrogen across rhodium(III)-OMe bonds. Inorg. Chem. 2014, 53, 5328–5340. [Google Scholar] [CrossRef]
- Hartwig, J.F. Organotransition Metal Chemistry: From Bonding to Catalysis; University Science Books: Herndon, VA, USA, 2010. [Google Scholar]
- Blum, O.; Milstein, D. Direkte Beobachtung der reduktiven O—H-Eliminierung aus IrIII-Komplexen. Angew. Chem. 1995, 107, 210–212. [Google Scholar] [CrossRef]
- Milstein, D. Carbon-hydrogen vs. oxygen-hydrogen reductive elimination of methanol from a metal complex. Which is a more likely process? J. Am. Chem. Soc. 2002, 108, 3525–3526. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method/ Basis Set | ΔΕrel = ΔE(Ir(H2)(H) − ΔE(Ir(H)3 [kcal/mol] | Ir(H2)(H) Distances in [Å] | Ir(H)3 Distances in [Å] | ||||
|---|---|---|---|---|---|---|---|
| Ir-H1 Ir-H2 Ir-H3 | H1-H2, r1 | H2-H3, r2 | Ir-H1 Ir-H2 Ir-H3 | H1-H2, r1 | H2-H3, r2 | ||
| PBE-D3BJ/ def2-TZVP | −1.0 | 1.594 1.640 1.617 | 1.754 | 1.343 | 1.598 1.638 1.602 | 1.579 | 1.584 |
| r2scan-3c/ def2-mTZVPP | −0.3 | 1.591 1.625 1.605 | 1.769 | 1.521 | 1.592 1.629 1.596 | 1.637 | 1.664 |
| BP86-D3BJ/ def2-TZVP | −0.2 | 1.596 1.636 1.614 | 1.751 | 1.411 | 1.600 1.637 1.603 | 1.597 | 1.619 |
| PBE0-D3BJ/ def2-TZVP | −1.3 | 1.574 1.669 1.653 | 1.822 | 1.009 | 1.584 1.625 1.586 | 1.539 | 1.573 |
| PBE0-D4/ def2-QZVPP | 1.573 1.667 1.651 | 1.799 | 1.005 | ||||
| B3LYP-D4/ def2-TZVP | -0.3 | 1.590 1.628 1.605 | 1.752 | 1.474 | 1.592 1.630 1.597 | 1.630 | 1.655 |
| WB97X-V/ def2-TZVP | n/a | 1.592 1.626 1.591 | 1.671 | 1.685 | |||
| WB97X-D4/ def2-TZVP | 1.591 1.626 1.590 | 1.663 | 1.596 | ||||
| PW6B95/ def2-TZVP | −1.8 | 1.575 1.688 1.677 | 1.861 | 0.952 | 1.585 1.625 1.590 | 1.609 | 1.587 |
| Method/Complex | (PDI)Ir-OH 3 | (PDI)Ir-OMe 4 | (PDI)Ir-SH 5 | (PDI)Ir-SMe 6 |
|---|---|---|---|---|
| ΔEhydrogenation DFT [kcal/mol] a | −10.48 | −10.33 | +8.97 | +8.19 |
| ΔEhydrogenation LNO-CCSD(T) b [kcal/mol] | −13.34 | −12.10 | +7.55 | +6.61 |
| ΔG298, hydrogenation LNO-CCSD(T) c [kcal/mol] | −0.78 | −2.07 | +16.08 | +14.14 |
| ΔG298 LNO-CCSD(T) COSMO d (ε = 7.6) [kcal/mol] | −4.19 | −5.81 | +15.22 | +13.27 |
| Bond/Method | PBED3BJ | PW6B95D3BJ | PNO-CCSD(T)//PBED3BJ/def2-TZVP | ||
|---|---|---|---|---|---|
| def2-TZVP/def2-QZVPP | def2-TZVPP | def2-QZVPP | CBS(3-4) | ||
| Ir: def2-ECP | Ir: def2-ECP | ||||
| (PDI)Ir-H | 58.58 | 63.29 | 63.20 | 63.33 | 63.44 |
| (PDI)Ir-Me | 53.07 | 51.24 | 55.64 | 55.42 | 55.36 |
| (PDI)Ir-OH | 90.09 | 84.80 | 87.91 | 87.91 | 87.99 |
| (PDI)Ir-SH | 81.41 | 78.22 | 79.08 | 80.21 | 81.20 |
| (PDI)Ir-OMe | 72.47 | 69.52 | 76.36 | 76.01 | 75.83 |
| (PDI)Ir-SMe | 75.06 | 72.34 | 74.36 | 75.10 | 75.82 |
| H-H | 99.35/99.44 | 102.71/103.01 | 103.01 | 103.74 | 104.20 (104.20) |
| H-CH3 | 102.62/102.65 | 104.76/104.87 | 103.54 | 104.34 | 104.91 (105.00) |
| H-OH | 115.98/117.62 | 114.85/116.51 | 116.07 | 117.96 | 119.12 (118.98) |
| H-SH | 89.77/90.07 | 90.60/90.99 | 89.55 | 90.85 | 91.72 (91.29) |
| H-OMe | 97.57/103.71 | 98.87/104.71 | 102.93 | 104.32 | 105.18 (105.32) |
| H-SMe | 83.76/85.05 | 85.05/86.81 | 85.18 | 86.31 | 87.08 (87.49) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Völker, M.; Schreyer, M.; Burger, P. Hydrogenation Studies of Iridium Pyridine Diimine Complexes with O- and S-Donor Ligands (Hydroxido, Methoxido and Thiolato). Chemistry 2024, 6, 1230-1245. https://doi.org/10.3390/chemistry6050071
Völker M, Schreyer M, Burger P. Hydrogenation Studies of Iridium Pyridine Diimine Complexes with O- and S-Donor Ligands (Hydroxido, Methoxido and Thiolato). Chemistry. 2024; 6(5):1230-1245. https://doi.org/10.3390/chemistry6050071
Chicago/Turabian StyleVölker, Max, Matthias Schreyer, and Peter Burger. 2024. "Hydrogenation Studies of Iridium Pyridine Diimine Complexes with O- and S-Donor Ligands (Hydroxido, Methoxido and Thiolato)" Chemistry 6, no. 5: 1230-1245. https://doi.org/10.3390/chemistry6050071
APA StyleVölker, M., Schreyer, M., & Burger, P. (2024). Hydrogenation Studies of Iridium Pyridine Diimine Complexes with O- and S-Donor Ligands (Hydroxido, Methoxido and Thiolato). Chemistry, 6(5), 1230-1245. https://doi.org/10.3390/chemistry6050071