Abstract
Glioblastoma (GBM) is a malignant cancer affecting the brain. As per the WHO classifications, it is a grade IV glioma and is characterized by heterogenous histopathology, high recurrence rates, and a high median age of diagnosis. Most individuals diagnosed with GBM are aged between 50 and 64 years, and the prognosis is often poor. Untreated GBM patients have a median survival of 3 months, while treatments with Temozolomide (TMZ) and radiotherapy can improve the survival to 10–14 months. Tumor recurrence is common, owing to the inefficiency of surgical resection in removing microscopic tumor formations in the brain. A crucial component of GBM-related research is understanding the tumor microenvironment (TME) and its characteristics. The various cellular interactions in the TME contribute to the higher occurrence of malignancy, resistance to treatments, and difficulty in tumor resection and preventative care. Incomplete pictures of the TME have been obtained in 2D cultures, which fail to incorporate the ECM and other crucial components. Identifying the hallmarks of the TME and developing ex vivo and in vitro models can help study patient-specific symptoms, assess challenges, and develop courses of treatment in a timely manner which is more efficient than the current methods. Microfluidic models, which incorporate 3D cultures and co-culture models with various channel patterns, are capable of stimulating tumor conditions accurately and provide better responses to therapeutics as would be seen in the patient. This facilitates a more refined understanding of the potential treatment delivery systems, resistance mechanisms, and metastatic pathways. This review collates information on the application of such microfluidics-based systems to analyze the GBM TME and highlights the use of such systems in improving patient care and treatment options.
1. Introduction
The manipulation of fluids at the submillimeter scales defines microfluidic technology. Microfluidics involves the analysis and application of extremely small liquid samples (~1 nL to 1 pL) on fabricated platforms [1]. At reduced volumes, fluid behaviors undergo several changes, as seen with capillary forces and surface tension, amongst others [2]. The altered properties of fluids at this scale include viscosity, surface tension, diffusion, wetting, capillary length, etc. [1,3]. The behavior of fluids on such platforms cannot be fully described by macroscale equations, such as the Navier–Stokes equation. Thus, the interactions between these fluid molecules can be divided into intermolecular interactions and interactions with the external surface. Effects pertaining to the external surface interactions, such as surface tension, are more important in microfluidics.
The selection of a material for such a platform is crucial. These platforms may be made of several materials, with the selection criteria including solvent reactivity, absorption or adsorption potential of the material, response to the chosen fabrication technique, cost, and, in the case of biological application, its ability to resist degradation due to heat and chemical sterilization [4,5]. Thus, the material choices include silicon, stainless steel, glass, polystyrene, elastomers such as PDMS, paper, epoxy resins, and polymers, amongst others [5]. The choice of material is specific to its application. Fluid behavior can thus be manipulated by altering the channels for flow, the flow rate, the platform material, and the nature of the fluid itself, i.e., viscosity, rendering an extremely useful advantage for various applications, as seen in Figure 1 [1].
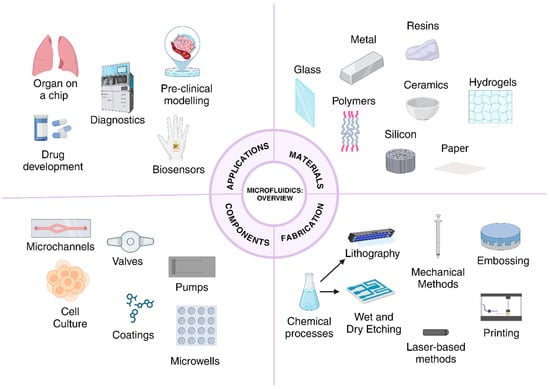
Figure 1.
Overview of the potential materials for chips, major fabrication techniques, components of microfluidic chips for disease-related applications, and applications in biology.
The WHO classification system defines glioblastoma (GBM) as a grade IV glioma. It affects the central nervous system (CNS). The median five-year survival rate is 7% for GBM as per the statistics provided by UT Southwestern. In the last decade, GBM has accounted for over 4.49 diagnosed cancer cases per 100,000, a significant increase from 0.73 per 100,000 in 2007 [6]. Patients are not expected to survive beyond 14–15 months, with the higher median age at the time of diagnosis [6] being an influencing factor. An increase in the age at the time of diagnosis is negatively correlated with the chances of survival. Conventional treatment regimens are surgical resection followed by chemotherapy [7]. The low survival rates even after treatment is due to the heterogeneity of GBM tumors, since no standard treatment will be effective for a large cohort. The tumor microenvironment (TME) is a complex structure defined by the presence of cancer stem cells, epithelial cells, fibroblasts, immune cells, cancer cells, amongst other components present in the extracellular matrix (ECM) and is heavily involved in maintaining the tumor as well as aiding in metastasis and growth [8]. Ineffective resection of the original tumor, accompanied by chemoresistance conferred by the TME contribute to the high rates of recurrence [6]. This heterogeneity can be attributed to the diversity of the tumor microenvironment, which itself is highly immunosuppressive [9].
Since GBM is a CNS-associated cancer, a model that studies the behavior of its TME must be able to accurately stimulate the blood–brain barrier (BBB), the stromal tissue, and cancerous tissue, along with the ECM, for the purpose of accurately depicting patterns of maintenance, metastasis, migration, and drug resistance [10]. The aggressive nature of GBM, coupled with insufficient technologies used to draw conclusions, give rise to a diagnosis and treatment landscape that is largely incapable of producing results that are accurate to the in vivo observations.
Microfluidics-based technology has revolutionized the field of oncology, playing a significant role in the development of products and understanding the underlying mechanisms of cancer [11]. Studies of the TME are largely improved and more precise at the microfluidics scale. The proliferation, maintenance, and metastasis of cancerous cells in the body are characteristics attributed to processes associated with the TME. Organ-on-a-chip models are important technologies that enable the study of these complex biological environments and have emerged as a potential breakthrough in the past decade [12]. They integrate the fundamental principles that govern microfluidics with novel technologies, such as 3D culturing and co-culturing methods to build systems that are able to accurately mimic the natural conditions in the human body [13,14]. They eliminate the need for using ethically dubious and inaccurate means, such as mouse models, or inconsistent models, such as xenograft models. These platforms possess several advantages, including potentially larger accessibility, lower time constraints, lower costs, multiplexing, integration with advanced analysis technologies for swifter diagnoses, and the production of patient-specific results (Figure 2 and Table 1) [15]. In this review, we begin by outlining the nature of GBM, its physical characteristics and presentation, as well as the associated biomarkers used to quantify observations in a laboratory setting. The limitations of current pre-clinical models, treatment regimens and diagnosis are discussed, followed by a look at the several microfluidic platforms that have emerged in the last decade to combat the issues highlighted previously.
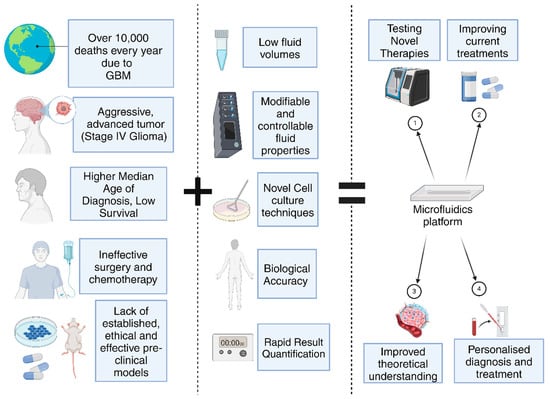
Figure 2.
Integration of microfluidics in GBM research has several important applications (Created with BioRender.com).

Table 1.
Overview of the aim of the device, biomarkers used, target drug and the advantages and disadvantages of several microfluidics-based devices.
2. Understanding GBM
2.1. Physical Presentation of GBM
GBM is a highly heterogenous tumor. It tends to present at advanced stages. Physical symptoms that define GBM may not be exclusive to the cancer itself. They appear at different stages of tumor progression, with specific symptoms appearing towards terminal stages, such as cognitive deterioration. Non-specific symptoms appear earlier on, like headaches, eye pain, nausea, insomnia, stress, and tiredness [37]. These symptoms are often unreported to medical professionals and thus lead to more severe symptoms. Specific systems may include epilepsy, strokes, motor impairment, cognitive deterioration, visual impairment (e.g., double vision), and processing issues [9,37]. The irregular progress of symptoms and the lack of early specific symptoms may be the reason for the higher median age of diagnosis in GBM patients. A correlation between symptoms and prognosis has been hypothesized, for example, epilepsy in GBM indicated a more favorable prognosis owing to the possible location of the tumor. GBM lesions can be identified in the cerebellum, spinal cord, supra-tentorial space, frontal lobe, brainstem, and spinal cord [9]. Lesions may be symmetric or asymmetric, depending on the primary location of tumor initiation and whether the tumor spreads across the corpus callosum, which is common due to the aggressive nature of tumor infiltration [38]. GBM tumors are diagnosed by mapping the frequency of external symptoms and biopsies (refer Figure 3).
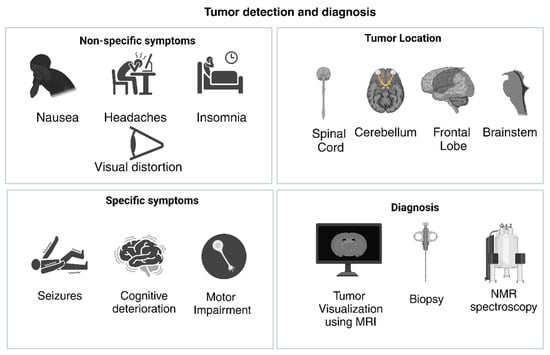
Figure 3.
Symptoms associated with GBM, with corresponding tumor locations and diagnosis methods. (Created with BioRender.com).
2.2. Tumor Structure in Glioblastoma
GBM is composed of a solid origin with differentiated regions which contain glioblastoma stem cells (GSCs). They are also highly heterogenous with highly variable phenotypes, indicated by the large differences in cellular developmental stages and types found in different tumors [38]. The presence of tumor niches is a hallmark of GBM, and at least three separate niches exist, i.e., the perivascular, hypoxic and invasive niche, each serving a different purpose for tumor maintenance, growth, and migration [39]. These niches are discussed with respect to the microenvironment in subsequent sections. More specifically, the tumor is differentiated into a central necrotic region, surrounded by dense regions of proliferating cells and surrounding vasculature, and a dominance of one particular cell type is common to GBM, although the dominant type differs in each tumor [38].
2.3. Tumor Microenvironment in Glioblastoma
The TME is highly immunosuppressive and thus accounts for the increased resistance of GBM to chemotherapy and radiotherapy, leading to the high recurrence rate and the low survival rate post-resection. The components of the GBM TME are the parenchyma, which contains the astrocytes, oligodendrocytes, and neurons, the primary tumor foci, which contains the heterogenous tumor population, containing GSCs and multiple types of poorly differentiated cells, the vasculature, which consists of the blood vessels, extracellular matrix (ECM), and the blood–brain barrier (BBB), the local immune cell population, consisting of T-cells, NK cells, dendritic cells, microglial cells, glioma-associated macrophages (GAMs), monocytes, neutrophils, tumor-infiltrating lymphocytes (TILs), and myeloid-derived suppressor cells (MDSCs), the cellular communication components and the chemical components, such as pH and oxygen concentration [40,41]. The defining features of GBM, such as immunosuppression, therapeutic resistance, metastasis, tumor maintenance, etc., are all attributed to the components discussed above, as seen in Figure 4. For example, the increased uptake of glucose helps induce cellular proliferation in some tumor cell subtypes [41], while favorable interactions between the tumor vasculature and immune cell populations can help develop a perivascular tumor niche, resulting in BBB disruption and tumor maintenance, and the development of hypoxic regions induces and helps the development of invasive tumor subtypes [39]. This highly complex nature poses challenges in both the treatment and detection processes, and its features are summarized in Figure 4. These challenges are discussed in subsequent sections.
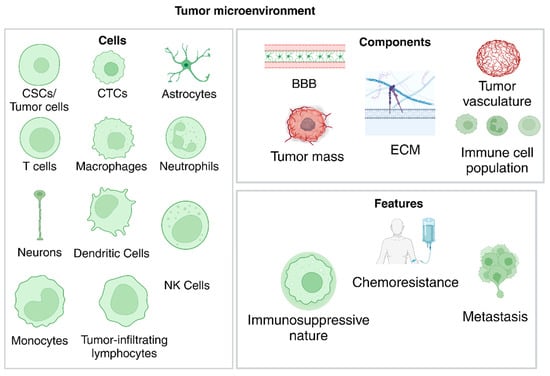
Figure 4.
Cells found in the TME, along with the main components needed for niche formation and the resulting features that define GBM. (Created with BioRender.com).
2.4. Biomarkers in Glioblastoma
The highly heterogenous nature of GBM contributes significantly to the large diversity in the expressed genes and subsequent biomarkers that can be identified for diagnosis and treatment. These biomarkers are of importance in tumor identification and classification and in developing therapeutic targets for specific niches. In addition to the implications in treatment, glioblastoma subtypes are identified based on the subset of genetic signatures that are chosen, for example, the Veerhak classification divides GBM into proneural, neural, classical, and mesenchymal subtypes on the basis of expressions and mutations identified in the EGFR, NF1, and PDGFRA/IDH1 genes [42]. Other systems include the Philips classification and the Jiao classification [43]. For the purposes of this review, only certain biomarkers will be discussed with reference to their purpose in this paper (Figure 5A,B).
2.4.1. TME and Tumor-Specific Biomarkers
Major histocompatibility complex-1 (MHC-1) and CD45 are cell surface biomarkers associated with the immunologically cold nature of GBM and are often highly downregulated in the tumor and its TME [16]. It enables the tumor to evade attacks by immune cells, as MHC downregulation causes the ineffective binding between cytotoxic cells that may be able to eradicate the tumor mass. Programmed death ligand-1 (PD-L1) is another upregulated biomarker that helps tumors evade immune detection by inactivating cytotoxic T-cells [16]. The upregulation of blood vessel formation in tumors is promoted by an increase in the levels of reactive oxygen species (ROS). Mechanisms of apoptosis are altered in cancer cells, with natural cell death due to injury or lack of nutrition being downregulated. Associated biomarkers include glutathione (GSH).
Isocitrate dehydrogenase (IDH) genes are being investigated for their applications in determining the prognosis of patients and are found in various regions of the cell, including the cytoplasm, peroxisomes, and mitochondria. The nature of IDH isoforms and their mutations with respect to the stage of tumor progression is an important prognostic factor for gliomas. The glial fibrillary acidic protein (GFAP) can be used to differentiate CTCs from other tumor cells.
Circulating immune cells are also significant in determining immune infiltration rates and developing therapeutics. Markers associated with these cells include CD86, CD163, CD154, CD8, IL-1β, IL-6, and TNF-α.
2.4.2. Biomarkers Associated with Therapeutics
CD64 is a biomarker that, when coupled with other entities, is capable of enhancing the docking potential for other antibodies or targeted molecules on sEVs [16]. Vimentin is a protein that is associated with maintaining cell stability and integrity and is often used as a biomarker for testing the effect of chemotherapeutic treatment regimens [18,20,27]. Chemoresistance is a mechanism developed by tumors to resist the cytotoxic activity of certain drugs. The main cellular drivers of chemoresistance are cancer stem cells (CSCs) and the associated markers include 6-O-methylguanine (6-O-MeG), 7-methylguanine (7-MeG), APNG, GSTπ1, ERCC1, ERCC2, MVP, ABCC3, CASP8, and IGFBP2 [36], while markers such as Bmi-1, CD133, Nestin, SOX2, CD44, and MGMT can be used to tract GSC pluripotency and proliferation [21]. Immunoreactivity can be assessed through identifying the levels of GFAP and VEGFR2. CD71 is a biomarker used to detect tumors by means of antigen–antibody interactions. Annexin V (or Annexin A5) is a protein that is usually detected on cell surfaces and binds to surface polysaccharides, inhibiting apoptosis in several tumors. However, this protein may be upregulated or downregulated in the TME depending on the cancer and is an identifiable biomarker for motility, invasiveness, and proliferative rate of tumor cells [44,45,46]. Caspases are a family of proteins responsible for initiating apoptosis pathways and are employed as biomarkers for testing the efficacy of cytotoxic drugs and drug delivery systems. Dysregulated isoforms include Caspase 3, 7, and 8 [47], with Caspase 8 being upregulated and contributing to chemoresistance in GBM [48].
2.4.3. Biomarkers Associated with Tumorigenic Processes
Cell adhesion is a process by which the neighboring cells are able to adhere to each other and carry out effective intercellular communication. These processes help regulate the rate of proliferation and growth in normal cells but are disrupted in tumors, leading to cellular aggregation. Biomarkers associated with these processes include PECAM-1, CDH5, and TJP1, whose levels are highly elevated in cell junctions [19]. EGFR is a biomarker that has been implicated in tumor proliferation, invasiveness, and resistance to therapeutics. A similar biomarker used is PDPN [36]. Tumor migration is also a crucial process for tumor survival and can be simulated or tracked by means of mapping glucose gradients. Actin filaments are also stained using phalloidin, as they are involved in several cellular processes that may or may not be carcinogenic, including cell death.
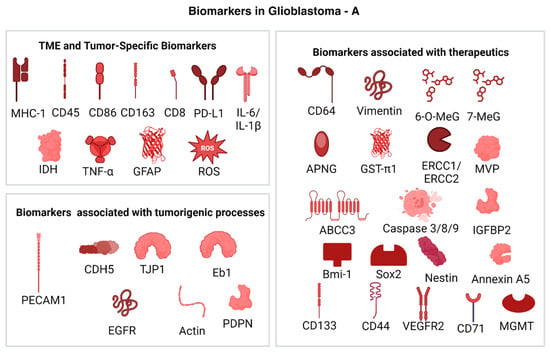
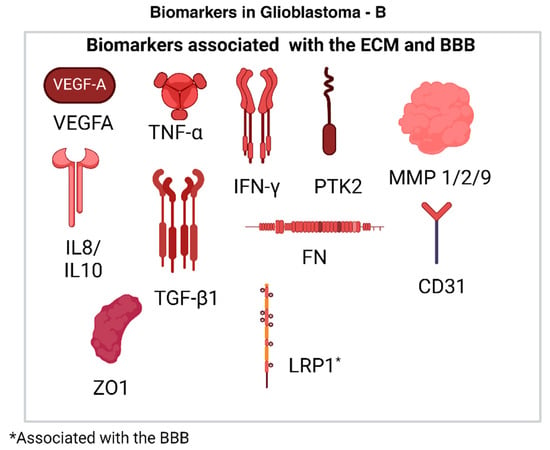
Figure 5.
(A): Biomarkers in GBM that are associated with different processes related to tumor maintenance and proliferation. They are used to quantify effects such as metastasis, proliferation and tumor recurrence [49]. (Created with BioRender.com). (B): Biomarkers in GBM that are associated with different processes related to tumor maintenance and proliferation. They are used to quantify effects such as metastasis, proliferation, chemoresistance, and tumor recurrence [49]. (Created with BioRender.com).
2.4.4. Biomarkers Associated with the ECM
The ECM plays an important role in tumor proliferation and maintenance. In laboratory studies, the brain-derived decellularized extracellular matrix (bdECM) is used to evaluate the therapeutic effects of drugs. The ECM is altered in a cancerous environment, as excessive production and cross-linking of collagen promotes immune cell evasion, angiogenesis, and tumor survival, amongst other functions. Biomarkers associated with the bdECM include VEGFA and IL-8. An increase in the expression of VEGFA is associated with increased blood vessel formation and tumor proliferation. IL-8, TNF-α, and IFN-γ are pro-inflammatory cytokines, and elevated levels of IL-8 are indicative of increased angiogenesis in tumor cells [19]. Anti-inflammatory cytokines used as biomarkers include TGF-β1 and IL-10. The formation of a tumor niche is essential to tumor survival and is often preceded and supported by a process called ECM remodeling, wherein the base structure of the ECM is altered through enzyme-mediated activity and promotes the progression and metastasis of the cancer as discussed earlier. Biomarkers used to map ECM remodeling include focal adhesion kinase (PTK2), human fibronectin (FN), and matrix metalloproteinases (MMP1, MMP2, and MMP9) [19]. Tubule formation in cancerous ECM regions is also affected by tubule formations, which is directly related to the nature of the experimental ECM. CD31 is a marker for endothelial cells and is used to map tubule formation [19]. Cellular proliferation markers include ZO-1.
2.4.5. Biomarkers Associated with the BBB
The BBB is a common roadblock encountered when attempting to develop effective therapies for the BBB. Proteins such as LRP1 have been found to be dysregulated in GBM and may play an important role in developing carrier systems for drugs [50].
2.5. Challenges Associated with Detection and Treatment
Conventional methods for detection include radio imaging (MRIs, MRS, etc.), biopsies, and other laboratory methods [9,51]. These methods have an important drawback, i.e., they are not able to diagnose lower-grade gliomas using risk factors and clinical presentation before the tumor progresses to become a high-grade glioma. This is largely due to the lack of established correlations between physiological changes and the onset or establishment of GBM, for example, increased cerebral blood flow [9], frequent headaches, strokes, or epileptic shocks [37] are identifiable as symptoms that arise due to GBM only after the tumor has progressed considerably. Thus, pre-clinical models of GBM are extremely important in modeling effective therapies and correlating cellular behavior with specific diagnoses. 2D cell cultures are commonly used across diseases to establish pre-clinical models for drug testing in cancer. Cell lines used for these cultures are often immortal or can be patient-derived but have several shortcomings, such as the acquiring of genetic variation through generations, the change in morphology when cultured in Petri dishes and a loss of several crucial interactions, such as those between the cells in the tumor niche and the external matrix [52]. These types of cell lines are difficult to maintain and standardize, ultimately reducing their real-time applicability in diagnosis and treatment. The standard of care (SoC) protocol for GBM is surgical resection followed by radiotherapy or Temozolomide (TMZ) therapy or both [9]. The presence of residual circulating tumor cells (CTCs) as microscopic tumors often renders these approaches ineffective in the long term, as survival rates rarely exceed 15 months post diagnosis. Recurrence is an irreversible event and is inevitable in GBM. Diagnostic methods have evolved considerably in the last decade, with biomarker-based detection coming to the fore to establish personalized treatment modules and improve the specificity of both treatment and diagnosis. Emerging diagnostic methods improve upon existing methods, such as MRIs, for example, diffusion-weighted imaging (DWI), and MR spectroscopy may be used, or they establish novel protocols such as biomarker-based diagnostics or proteomic analysis. Common biomarkers that can be detected include RNA sequences, CTCs, cell surface markers, DAMPs, etc. [51]. CTCs are of particular importance in identifying both the primary tumor and recurrence events. It has been hypothesized that CTCs are more invasive and possess a higher potential to initiate tumor formation as compared to the resected regions of the GBM tumor, and they even possess differential responses to the SoC treatment using TMZ [53]. The biomarkers evaluated with respect to CTCs include CD133, Nestin, Sox2, and Masashi 1 [53]. These results all highlight the need for early detection methods with distinct and reliable cellular targets in pre-clinical or clinical models that are capable of stimulating biologically accurate models in GBM.
2.6. Challenges with Drug Development for GBM
2.6.1. Disadvantages of Current Pre-Clinical Models for Drug Development
As discussed in previous sections, pre-clinical models remain underdeveloped in GBM, particularly when being used for drug development and testing. The various types of pre-clinical models currently in use include xenograft models, genetically engineered mouse models, and chemically induced models [54]. These models bear a significant advantage over traditional 2D cultures as the required vasculature and endothelial interactions can be stimulated in vivo. The development of novel therapeutics and the proving of their clinical applications are challenging. Take the instance of using genetically engineered mouse models (GEM), where the tumor cell line is usually grown and then transplanted to a suitable area of the mouse model, which may not be the CNS [54]. These are challenges that directly impact drug testing as essential interactions between the BBB, ECM, and the other cell populations are not observed. Other disadvantages of GEM models are differences in homologous genes, variations in inheritance patterns between the mice and humans, insufficient tumorigenesis due to weak oncogene transfer (RCAS-tVA model), expensive and time-consuming methods (CRe-LoxP system), latency of tumor formation, and small tumor volume (Sleeping beauty transposon method) and ethical concerns [55]. Xenograft models are significantly cheaper and are also highly disadvantageous. Their main drawback is their inability to completely replicate the desired tumor volume and microenvironment in the host [55,56]. In addition to this, using immunosuppressed mice can alter the underlying cellular process to stimulate a biologically accurate picture of GBM [56]. Thus, these 3D models are unable to fully encapsulate the required conditions to effectively understand GBM and in drug development, as recapitulated in Figure 6.
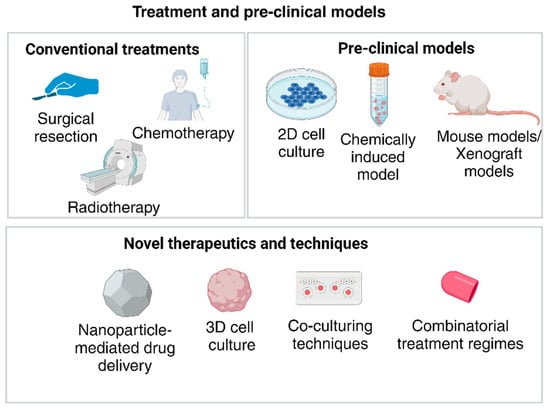
Figure 6.
Current treatment and pre-clinical models being used contrasted to the emerging techniques for GBM. (Created with BioRender.com).
2.6.2. Emerging Role of Organoids in GBM
Organoids or spheroids are 3D cell cultures which are emerging as one of the new-age pre-clinical models for GBM research. They are highly customizable, as the required cells can be derived directly from the patient and cultured to accurately mimic the patient’s TME. Their applications include TME studies, GSC and biomarker research, along with drug development and screening [57]. These models are often implemented ex vivo. They may be generated by several methods which differ in the cells used to culture the tumor/TME and their modes of implementation. The types of organoids thus generated are patient-derived organoids (PDO), iPSC (Induced pluripotent stem cell)-derived organoids, fusion organoids, and bioprinted organoids [58]. Bioprinted GBM models may make use of patient-derived vascular and tumor components thus enhancing the similarity between drug effects in the patient and the lab [58]. PDOs are capable of maintaining the original tumor characteristics and may be preferred in the drug development process. iPSC models are more commonly used to study GSCs [57]. While such organoid models may be limited by their lack of standardization and varying quality, they are exceptional in their ability to mimic tumor heterogeneity, making them extremely important pre-clinical models in GBM [59]. The TME can be replicated by establishing co-cultures with the GBM spheroids, as seen in bioprinting. However, it has been reported that patient-derived cell lines can be cultured in an appropriate matrix to generate a TME, and the cells may be harvested post-resection. While certain cell subpopulations, e.g., lymphocytes, do not survive for longer periods of time, the genotypic and phenotypic makeup of the PDO accurately mimics the behaviors seen in vivo [57,59]. These organoids may be cultured in several models or platforms, including mouse models, which is flawed. This flaw is the effect that the genetic and phenotypic makeup of the mouse, the location of tumor infiltration and immune response will have on the tumor [55]. Thus, microfluidic platforms are emerging as a cheap, controllable, customizable, and effective option to be used for GBM research (Refer Table 1 for different applications).
3. Microfluidics-Based Systems for the Study of Tumors/Microfluidics in Cancer
With regards to cancer specifically, studies of the tumor microenvironment are of significant importance to emulate the accurate TME characteristics for establishing safe and effective standard of care (SOC) protocols. The process of designing a microfluidic chip/system for TME studies of solid tumors include culturing spheroids of a viable size, design of microfluidic channels, incorporation of an ECM/culture medium, establishing a regular source of energy/feed, detection/visualization protocols, etc. [60]. The applications of such models include the estimation of nanoparticle penetration, efficacy of drug delivery, real-time response to chemotherapeutic drugs, visualization of circulating cell populations, etc. [60]. The choice of culture is also an important factor. While 2D cells were used in earlier protocols, the adherence of 2D cells to the culture medium, the lack of cell diversity, and the absence of cell–cell interactions and cell–extracellular matrix (ECM) interactions have made the implementation of 3D cultures far more suited to cancer studies [13]. However, 3D cultures are expensive and difficult to both maintain and cultivate. Despite these drawbacks, they are able to stimulate the TME more accurately than 2D cultures. An example is the increased cell tolerance to cisplatin when administered in a 2D culture when compared to a 3D culture, of which the latter is more accurate to the response elicited in the human body [61]. As stated previously, spheroids are masses of cancer cells cultured from pre-existing or pre-harvested aggregates. They consist of diverse cell populations, with varying levels of receptor expression, e.g., ALDH+/CD44++ expression [13]
Spheroids can be synthesized with or without microfluidic systems. When they are synthesized without microfluidic systems, scaffolds may or may not be used; in the latter case, the processes are referred to as scaffold-free syntheses. Supporting scaffolds can induce binding of the cells to the surface, thereby preventing spheroid formation and removal, but they are highly successful in forming large cellular aggregates [60]. Scaffold-free synthesis methods include hanging drops, magnetic levitation, liquid overlay method and force-driven method [14,60]. Methods requiring a scaffold include rotary systems, nanofiber-containing cell suspensions, matrix-embedded spheroid formation, etc. [14,60] Microfluidics-based methods include droplet formation, electrowetting, dielectrophoresis, the formation of microstructures and microwells, acoustic methods, and the formation of hanging drops [62].
While microfluidic platforms have vast applicability and can be used for several studies related to cancer, for the purposes of this review, TME and tumor modeling as well as drug development and testing are the two major categories of devices being discussed. Tumor and TME modeling platforms focus on providing biologically accurate models of the tumor mass and surrounding cells, such as immune cells, ECM cells, and CTCs. They also attempt to establish the role of these cells in various tumorigenic processes [11,63]. Platforms used for drug delivery focus on improving the accuracy of ex vivo/in vitro simulations for drug testing by implementing more accurate features, such as using spheroid cultures, etc. They also may be differentiated into those that propose novel treatment protocols, e.g., using nanoparticle-based delivery and activation systems, or propose improvements to existing chemotherapeutic treatment regimens, for example, improving the efficacy of TMZ.
3.1. General Features of Microfluidic Devices Used in Cancer Studies
The nature and arrangement of the microfluidic channels along with the associated components also plays an important role in the fabrication of such systems. Since the tumor niche is composed of both cancerous and non-cancerous cell populations, in addition to matrix proteins and circulating cells, the channel layout is important to ensure accuracy. Pumps are used to introduce the desired flow patterns, which may be recirculating or perfusion, with the common choices being syringe pumps, microvalve-driven actuator pumps, peristaltic pumps, and hydrostatic pressure-driven pumps. In the absence of pumps, a gravity-mediated flow can be introduced [61]. Common materials used are PDMS and PMMA for the fabrication of the chip itself, and the latter is easier to work with despite the former being used more widely [64]. This can be attributed to the inability of PMMA to withstand continued heat sterilization, which is necessary for biological systems [65]. However, PDMS cannot be considered as the best possible alternative, owing to multiple issues with its reactivity to organic solvents, difficulties in sealing and large-scale production, and detrimental surface properties, such as selective reactivity. An additional challenge that is important when considering lab-on-a-chip models is the highly complex nature of sample preparation. Samples for these applications may be derived from patients, necessitating the introduction of steps to purify, dilute, or concentrate the samples as required, thereby driving up costs and the time required to carry out any studies. Fabrication methods include lithography, wet and dry etching, and possibly even embossing and imprinting. Soft lithography remains the most common mode of fabrication for PDMS-based devices. ECM materials include collagen and Matrigel, depending on the nature of the cancer and the required interactions [61].
The choice of fabrication method relies largely on the application for which a chip is designed. For chips that need to be compatible with biological materials, it is imperative that the cost to output ratio is optimized. Thus, the techniques of interest include soft lithography, bioprinting, hot embossing, injection molding, laser cutting, and etching [65,66]. Soft lithography remains the preferred technique for the manufacture of GBM models. It is less expensive and has more controllable parameters for fabrication, for example, the thickness of a chip, which must be optimized to accurately mimic cellular interactions [67]. However, as with most other fabrication methods, it requires a clean room and high-precision equipment, which may render the chip inaccessible on a larger scale. Newer techniques, such as 3D bioprinting, are emerging to the fore for chip fabrication. Bioprinting bears several advantages, such as the minimization of human interference, thereby reducing the risk of cross-contamination [68]. Other advantages include the precision of implementing the desired design features, decrease in the time from conception to fabrication and reproducibility [68,69]. Bioprinting can also be integrated with lithography approaches to improve biocompatibility, as seen in the case of stereolithography, which makes use of optical-based 3D bioprinting techniques [68].
3.2. Progression of Microfluidics in Disease Modeling, Diagnosis, and Treatment
The first organ-on-a-chip model was constructed in 2010 and stimulated the functions of human lungs on a microfluidic chip [12]. Since then, several advances have been made in the construction of organ-like models on microscales for various purposes, including toxicology reports, drug delivery and design, tumor-related studies, etc. [70]. The main aim of rapid development in developing organ-on-a-chip models lies in their ability to replace animal testing, which is currently the most reliable method to model in vivo disease characteristics. These microfluidic models have undergone multiple advancements. For example, the initial models were incapable of maintaining organoids or 3D cultures for extended periods of time, in addition to stimulating the ECM and vascular interactions, and the dynamic cellular conditions were also difficult to maintain and design [70]. However, microfluidic systems have evolved to stimulate hydrostatic and hydrodynamic conditions, and multiple organoid interactions can be stimulated as well. The inclusion of various modifications, including multiple wells, sampling ports, diversity of cellular samples, and even microporous channels can help in the development of more biologically accurate models that may eventually replace animal models [71]. The highly controllable nature of these models also offers them a distinct advantage [70].
3.3. Microfluidic Platforms for the Study of GBM TME and Tumor Modeling
In previous sections, the role of establishing reliable pre-clinical models for GBM was discussed, and the disadvantages of various models were illustrated. In addition to the mentioned technical difficulties, a large portion of these methods tend to be ethically dubious and expensive. Thus, microfluidic platforms have emerged as an inexpensive alternative with highly controllable characteristics. CTCs play an important role in the recurrence of GBM (Figure 7). While their existence has not been a matter of debate, their detection is not advanced and not much is understood with regards to their exact role in GBM. A chip using spiral microfluidic technology was able to establish a correlation between progression-free survival (PFS) with the estimated CTC count in PDOs [27]. In another study conducted in 2018, the CTCs in patient blood samples were counted using the Parsortix® PC1 system (Manufactured by ANGLE pLc, United Kingdom) and it was established that CTCs might be the primary reason for metastasis in GBM, as they are capable of moving across the BBB; thus, a correlation between the tumor volume assessed from MRIs and the CTC counts was proposed, although the latter was not proven [32]. Both these studies, however, suffered from a significant disadvantage of a relatively small patient cohort.
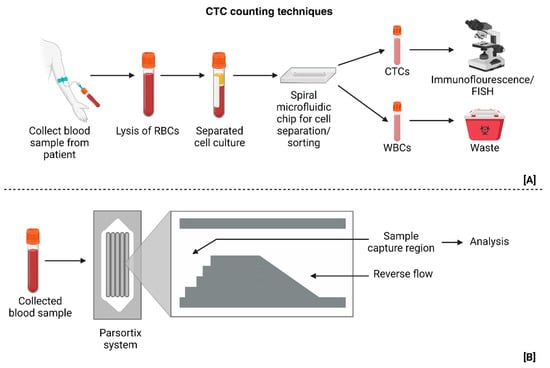
Figure 7.
(A) Sorting technique that utilizes forces generated in spiral microfluidic technology to isolate CTCS from blood samples [27]. (B) Parsortix system used to separate and collect chosen samples, which are CTCs in this case, for analysis and testing [32]. (Created with BioRender.com).
GSCs are another important cell subpopulation in GBM, having been implicated in the tumor maintenance, progression, recurrence, and, more recently, in the development of chemoresistance. They are also capable of influencing the effectivity of immune therapy in GBM, as the different subtypes of GSCs (mesenchymal and proneural) influence immune infiltration across the BBB [72]. This makes them an important biomarker for immunotherapy. In 2018, Lin et al. constructed an artificial perivascular niche on a PDMS chip [21]. The perivascular niche (PVN) plays a significant role in generating GSCs [73]. The chip established a link between the presence of GSCs and the development of chemoresistance to TMZ and was able to successfully mimic the in vivo biological conditions due to the presence of a co-culture model. Monitoring the real-time activity in GBM is important, and a concentric co-culture chip fabricated to host a triculture model mimicked the PVN, with three distinct tumor, stroma, and vascular regions [30].
As discussed earlier, the GBM TME is highly complex and often has hypoxic regions which are believed to promote tumor metastasis. Pseudopalisade formation is also a prominent characteristic of GBM. In a 2017 study conducted by Ayuso et al., restricted and unrestricted conditions were implemented on the channels of the chip to mimic cellular migration patterns with pseudopalisade formation and overall aggressiveness, and these correlated with the simulations to validate their findings, which suggested that GBM tends to proliferate more aggressively in unfavorable conditions [28]. Migration velocities of individual cells is an emerging parameter, as GBM is highly metastatic. An analysis of single-cell migration velocities using an aggressive cell line (U87) was carried out in 2021 by Sengul et al. [35]. They were able to establish that glioma cells remain unaffected by the nature of obstacles encountered during migration, and that the nature of the culture medium affects the velocity measurements as well. These conclusions suggest that GBM is heterogenous and its behavior is heavily influenced by the surrounding cells and structures.
3.4. Microfluidic Platforms for Drug Development
A significant challenge with drug development or treatment protocol design in GBM is the lack of personalization. Personalization is an important aspect of GBM treatment owing to the highly heterogenous nature of the cancer. Microfluidic platforms designed for this purpose may be used to test drug efficacy, develop patient-specific treatment protocols, or in the production of drug delivery and manufacturing systems. In 2023, Dong et al. constructed a novel microfluidic model, wherein electroporation was used to produce large amounts of extracellular vesicles [16]. They used electroporation techniques on a microfluidic platform to manufacture small endothelial vesicles (sEVs) loaded with mRNA from modified mouse fibroblasts and human kidney cells. The use of electroporation enhanced the cells’ ability to produce sEVs and was implemented for the immunotherapeutic treatment of GBM. This method is also customizable to target specific cells and barriers. While SoC protocols for GBM use TMZ, its efficiency as a standalone chemotherapeutic drug is quite low. The disadvantages of using only TMZ for treatment include the development of chemoresistance before or after the initial dose, the development of other diseases, such as thrombocytopenia, lymphopenia, etc., non-specific cell cytotoxicity, poor delivery efficiency, and issues with solubility [74,75]. Thus, emerging treatment protocols seek to use other chemotherapy and radiotherapy drugs to improve the efficiency of such treatments. Drug combinations tested on microfluidic platforms have tested various such combinations, in which tumor regression was almost certain. These combinations include TMZ, along with Bevacizumab (BEV), Resveratrol, CCRT, CPT-11, and CP [19,20,22,24,30]. The microfluidic platforms used here possessed several advantages and employed unique techniques, such as the one seen in Figure 8, especially when PDOs were used. These advantages are the use of multiple chambers to culture spheroids, use of micropore channels and structures to stimulate vascular interactions, and the use of co-culture methods to mimic the TME and rapid rate of testing.
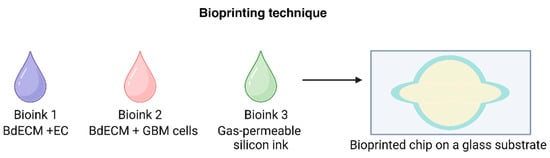
Figure 8.
Bioprinting techniques are an emerging method of creating microfluidic chips with co-culture systems for drug testing and development [19]. (Created with BioRender.com).
Other drugs used in such studies depart from the standard protocols, with substances such as BAL101553, a microtubule inhibitor, α-lipoic acid, ascorbic acid, catechins, AP2, Colchicine, and CDDP being tested on microfluidic platforms [26,29,32,34]. These platforms use dynamic culture methods, are able to stimulate the BBB, are capable of mimicking endothelial interactions and detecting CTCs, and have a prolonged survival time in vitro, all of which enhance the results of drug screening. Co-cultures are used commonly throughout these systems and are thus more biologically accurate than other pre-clinical models. Underlying biological characteristics that influence drug uptake by cells, such as cellular adhesion, are suitably analyzed when microfluidic platforms are used. A single-cell study employed a microfluidic probe that covered a cell in trypsin solution and tested the effect of TMZ, 5-FU, Act D, and Allicin on cellular adhesion [17]. A multichannel microfluidic device attempted to investigate the effects of TMZ and Simva on cell apoptosis and autophagy [18]. These studies have made startling observations, highlighting the advantage of introducing hyaluronic acid into the chip to simulate the acidic TME. The latter study established that 3D models showed less sensitivity to these drugs, thereby validating previous conclusions on the defects of TMZ treatment. Apart from the conventional chemotherapy treatments, alternatives such as antioxidants, for example, α-lipoic acid, ascorbic acid, and catechins have been proposed to be capable of reducing cell invasion and slowing down accelerated growth, and they were validated by Liu et al. in 2017, when a PDMS chip capable of mimicking GBM vasculature showed lower levels of ROS and GSH on treatment with antioxidants [23].
Microfluidic platforms have also been employed to study drug delivery mechanisms, such as using nanoparticles (NPs) to effectively deliver chemotherapeutic medication, e.g., paclitaxel (Figure 9) [33]. This study looked at macrophages loaded with NP-bound Paclitaxel on a microfluidic platform using a co-culture model containing endothelial and vascular cells, with a conclusive result—an increase in targeted cell death. Magnetic nanoparticles have also been shown to be capable of reducing cell proliferation by inducing hyperthermia. This was tested on a simple microfluidic model containing a central chamber with multiple connecting channels and validated the aforementioned hypothesis. However, the model failed to account for the tumor vasculature or the BBB, rendering the results clinically inapplicable [31].
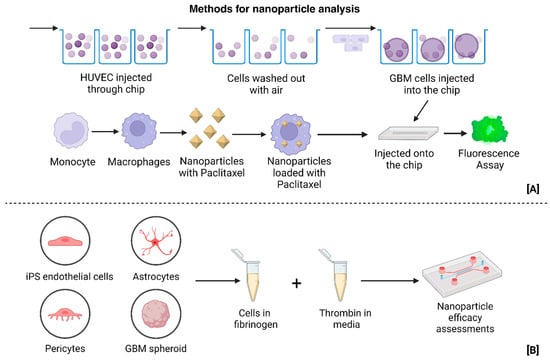
Figure 9.
(A) Construction of microfluidic platform for nanoparticle-loaded macrophage delivery to tumors, serving the dual function of tumor clearance/killing, as well as personalized and targeted treatment delivery [33]. (B) Construction of TME and Tumor model for the study of GBM using ECM components [26]. (Created with BioRender.com).
A concentric-channel model also attempted to test the efficacy of an immune-therapeutic treatment, BLZ945 (CSF-1R inhibitor) and Nivolumab, on PDOs [34]. While this study elucidated the applications of microfluidic chips to test different protocols ex vivo, it also suffered a significant setback in its lack of ECM and BBB, both of which are crucial to stimulate accurate immune responses to treatment. As mentioned earlier, the quantification and analysis of results must be rapid in order to solidify the position of microfluidic devices in these applications. A microfluidic device constructed by Shao et al. in 2015 was able to identify various levels of 13 GBM-related biomarkers in over 10 patient-derived cell lines, in addition to establishing their sensitivity to TMZ treatment when the genes were modified to render the cell lines more responsive to treatment. A unique feature of this device is the rapid nature of results, owing to qPCR, which was performed on the chip itself [36].
Other applications of microfluidic chips include diagnostics, where established GBM markers such as MGMT, Ki-67, and IDH1 are used to predict patient survival and provide a prognosis. This study mapped the expression patterns of RNAs associated with the biomarkers mentioned above and were able to predict tumor recurrence time as well, with patient-specific factors not being a disadvantage to diagnosis [25]. However, the choice of biomarkers must be improved to be associated purely with GBM. These devices thus prove that the intersection of GBM-related studies and microfluidics is an important area of development with wide-ranging applications, as detailed in Table 1 and Figure 10.
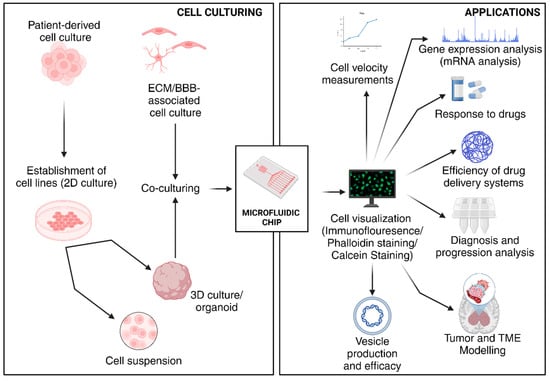
Figure 10.
Applications of various microfluidic devices used in GBM-related studies. (Created with BioRender.com).
4. Conclusions and Future Perspectives
Microfluidic platforms provide a promising alternative to GEMs and xenograft models as pre-clinical models for GBM. The complexity of the GBM TME, lack of specific external symptoms, and its intra-tumoral and inter-tumoral heterogeneity make it a difficult cancer to diagnose and treat. Personalized medicine is also extremely important in this case, as no two tumors are the same. Microfluidic platforms, when integrated with novel cell culture methods, for example, dynamic cultures, co-cultures, tri-cultures, 3D models, etc., provide an excellent method to mimic the biological conditions and stimulate the appropriate drug response. In addition to drug development, microfluidic platforms can be used to study the pathogenesis and progression of GBM. Important advantages in this aspect include the ability to control the nature and composition of the cellular components and accurately simulate the in vivo tumor model.
These platforms possess several advantages, including their relative inexpensiveness, portability, reusability, and lack of ethical roadblocks in implementation, unlike the current standards, which primarily focus on GEMs and Xenograft models. The integration of 3D cell cultures also improves upon the existing 2D culture method, which fails to mimic the required TME conditions to accurately predict results. However, several limitations must be overcome before these models are accepted. One of these is the standardization of patient-derived cell lines. A lack of standardization may cause cell lines to develop mutations during the culturing process which eventually impact the results of any studies or screening tests. Other challenges include the lack of a biologically accurate BBB and the lack of large patient cohorts to validate drug screening studies and subsequently obtain approval (as detailed in Table 1). A major concern in this area is the prevalent lack of standardization for chip measurements. These measurements include the permeability of the stimulated BBB, stiffness of the ECM, choice of biomarkers, nature and quality of the cell cultures used, and even the effect of several forces acting on the microchannels. Currently, there are no uniform measurement standards established that enable the comparison of these models accurately. The criterion for standardization may include minimally invasive or destructive techniques, accessibility, standard margins of error, and, as a consequence, homogenous modes of chip design, manufacture, and testing. These are mammoth tasks and thus constitute significant drawbacks in the implementation of such microfluidic devices for glioma detection and treatment.
Despite these disadvantages, microfluidic platforms will play a significant role in the development of accurate pre-clinical models in GBM for the applications of drug delivery, design, simulations, tumor modeling, TME studies, and potentially even diagnostics, making them one of the frontrunners in the race to establish a standard GBM model and diagnostic and treatment protocol, which may serve to improve patient survival. Advancements that support this include the acceptance of known parameters, such as transepithelial resistance (TEER), permeability coefficients, chip specifications, such as channel size and overall thickness, and established cell cultures derived from patients [76,77]. Another improvement that will be required to facilitate the integration of these technologies is the choice of biomarkers. As evident from the previous sections, the choice of biomarkers used to study any single effect in GBM, such as angiogenesis or drug resistance is extremely diverse, and thus the impact of any single proposal or device cannot be fairly compared to others with similar applications. Two aspects must be considered when referring to biomarkers in GBM studies. One of these is choosing a standard set of biomarkers, and the other is the establishment of novel biomarkers with greater efficacy. One such novel biomarker would be ion channels, particularly Sodium–Calcium exchangers (NCX), which have been proven to have an effect on accelerated wound healing and proliferation in cancerous tissues, especially in highly malignant cancers, such as GBM [78,79,80]. The implications of this discovery are particularly important in the context of developing microfluidic chips for GBM analysis. This being the proven presence of alternate, dynamic biomarkers which are often overlooked and the potential for an improved SoC protocol for patients affected by GBM. This opens up avenues for improved drug targeting and testing, in addition to providing deeper insights into the functionality of GBM cells and the processes that govern them. While the role of such biomarkers may be disputed, their emergence presents a larger scope of clinical applications for GBM treatment, as they are capable of inhibiting the core processes that drive malignancy and perhaps even recurrence, pointing towards a potentially revolutionary technology that can permanently alter the field of cancer diagnosis and treatment.
Author Contributions
Conceptualization, P.K. and N.K.M.; methodology, V.P., P.K. and N.K.M.; software R.R.; validation, V.P., P.K. and N.K.M.; formal analysis, R.R.; investigation, R.R., V.P., P.K. and N.K.M.; resources, P.K. and N.K.M.; data curation, R.R., V.P., P.K. and N.K.M.; writing—original draft preparation, R.R., P.K.; writing—review and editing, V.P., P.K. and N.K.M.; visualization, P.K. and N.K.M.; supervision, P.K. and N.K.M.; project administration, V.P., P.K. and N.K.M.; funding acquisition, P.K. and N.K.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Malloggi, F. Microfluidics: From Basic Principles to Applications. In Soft Matter at Aqueous Interfaces; Lang, P., Liu, Y., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 515–546. [Google Scholar]
- Sackmann, E.K.; Fulton, A.L.; Beebe, D.J. The present and future role of microfluidics in biomedical research. Nature 2014, 507, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Hajam, M.I.; Khan, M.M. Microfluidics: A concise review of the history, principles, design, applications, and future outlook. Biomater. Sci. 2023, 12, 218–251. [Google Scholar] [CrossRef] [PubMed]
- Yeo, L.Y.; Chang, H.C.; Chan, P.P.Y.; Friend, J.R. Microfluidic devices for bioapplications. Small 2011, 7, 12–48. [Google Scholar] [CrossRef] [PubMed]
- Niculescu, A.G.; Chircov, C.; Bîrcă, A.C.; Grumezescu, A.M. Fabrication and applications of microfluidic devices: A review. Int. J. Mol. Sci. 2021, 22, 2011. [Google Scholar] [CrossRef] [PubMed]
- Grech, N.; Dalli, T.; Mizzi, S.; Meilak, L.; Calleja, N.; Zrinzo, A. Rising Incidence of Glioblastoma Multiforme in a Well-Defined Population. Cureus 2020, 12, e8195. [Google Scholar] [CrossRef]
- Davis, M.E. Glioblastoma: Overview of Disease and Treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef]
- Weber, C.E.; Kuo, P.C. The tumor microenvironment. Surg. Oncol. 2012, 21, 172–177. [Google Scholar] [CrossRef]
- Gilard, V.; Tebani, A.; Dabaj, I.; Laquerrière, A.; Fontanilles, M.; Derrey, S.; Marret, S.; Bekri, S. Diagnosis and management of glioblastoma: A comprehensive perspective. J. Pers. Med. 2021, 11, 258. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Z.-X.; Huang, G.H.; Xiang, Y.; Yang, L.; Pei, Y.C.; Yang, W.; Lv, S.-Q. A systematic review of multifocal and multicentric glioblastoma. J. Clin. Neurosci. 2021, 83, 71–76. [Google Scholar] [CrossRef]
- Regmi, S.; Poudel, C.; Adhikari, R.; Luo, K.Q. Applications of Microfluidics and Organ-on-a-Chip in Cancer Research. Biosensors 2022, 12, 459. [Google Scholar] [CrossRef]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting Organ-Level Lung Functions on a Chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [PubMed]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, L.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef]
- Białkowska, K.; Komorowski, P.; Bryszewska, M.; Miłowska, K. Spheroids as a type of three-dimensional cell cultures—Examples of methods of preparation and the most important application. Int. J. Mol. Sci. 2020, 21, 6225. [Google Scholar] [CrossRef] [PubMed]
- Ravi, M.; Paramesh, V.; Kaviya, S.R.; Anuradha, E.; Paul Solomon, F.D. 3D Cell Culture Systems: Advantages and Applications. J. Cell. Physiol. 2015, 230, 16–26. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Liu, X.; Bi, Y.; Wang, Y.; Antony, A.; Lee, D.; Huntoon, K.; Jeong, S.; Ma, Y.; Li, X.; et al. Adaptive design of mRNA-loaded extracellular vesicles for targeted immunotherapy of cancer. Nat. Commun. 2023, 14, 6610. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Mao, S.; Li, W.; Huang, Q.; Feng, S.; Hong, Z.; Lin, J. Microfluidic adhesion analysis of single glioma cells for evaluating the effect of drugs. Sci. China Chem. 2020, 63, 865–870. [Google Scholar] [CrossRef]
- Samiei, E.; Seyfoori, A.; Toyota, B.; Ghavami, S.; Akbari, M. Investigating programmed cell death and tumor invasion in a three-dimensional (3d) microfluidic model of glioblastoma. Int. J. Mol. Sci. 2020, 21, 3162. [Google Scholar] [CrossRef]
- Yi, H.-G.; Jeong, Y.H.; Kim, Y.; Choi, Y.; Moon, H.; Park, S.; Kang, K.; Bae, M.; Jang, J.; Youn, H. A bioprinted human-glioblastoma-on-a-chip for the identification of patient-specific responses to chemoradiotherapy. Nat. Biomed. Eng. 2019, 3, 509–519. [Google Scholar] [CrossRef]
- Ma, J.; Li, N.; Wang, Y.; Wang, L.; Wei, W.; Shen, L.; Sun, Y.; Jiao, Y.; Chen, W.; Liu, J. Engineered 3D tumour model for study of glioblastoma aggressiveness and drug evaluation on a detachably assembled microfluidic device. Biomed. Microdevices 2018, 20, 80. [Google Scholar] [CrossRef]
- Lin, C.; Lin, L.; Mao, S.; Yang, L.; Yi, L.; Lin, X.; Wang, J.; Lin, Z.; Lin, J. Reconstituting Glioma Perivascular Niches on a Chip for Insights into Chemoresistance of Glioma. Anal. Chem. 2018, 90, 10326–10333. [Google Scholar] [CrossRef]
- Akay, M.; Hite, J.; Avci, N.G.; Fan, Y.; Akay, Y.; Lu, G.; Zhu, J. Drug Screening of Human GBM Spheroids in Brain Cancer Chip. Sci. Rep. 2018, 8, 15423. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Jie, M.; He, Z.; Li, H.; Lin, J. Study of antioxidant effects on malignant glioma cells by constructing a tumor-microvascular structure on microchip. Anal. Chim. Acta 2017, 978, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Jie, M.; Mao, S.; Liu, H.; He, Z.; Li, H.; Lin, J. Evaluation of drug combination for glioblastoma based on an intestine–liver metabolic model on microchip. Analyst 2017, 142, 3629–3638. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.S.; Shah, S.R.; Yankaskas, C.L.; Bajpai, V.; Wu, P.; Chin, D.; Ifemembi, B.; ReFaey, K.; Schiapparelli, P.; Zheng, X.; et al. A microfluidic cell-migration assay for the prediction of progression-free survival and recurrence time of patients with glioblastoma. Nat. Biomed. Eng. 2021, 5, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Straehla, J.P.; Hajal, C.; Safford, H.C.; Offeddu, G.; Boehnke, N.; Dacoba, T.; Wyckoff, J.; Kamm, R.; Hammond, P. A predictive microfluidic model of human glioblastoma to assess trafficking of blood–brain barrier-penetrant nanoparticles. Proc. Natl. Acad. Sci. 2022, 119, e2118697119. [Google Scholar] [CrossRef]
- Müller Bark, J.; Kulasinghe, A.; Hartel, G.; Leo, P.; Warkiani, M.; Jeffree, R.; Chua, B.; Day, B.; Punyadeera, C. Isolation of Circulating Tumour Cells in Patients with Glioblastoma Using Spiral Microfluidic Technology—A Pilot Study. Front. Oncol. 2021, 11, 681130. [Google Scholar] [CrossRef]
- Ayuso, J.M.; Monge, R.; Martínez-González, A.; Virumbrales-Muñoz, M.; Llamazares, G.; Berganzo, J.; Hernández-Laín, A.; Santolaria, J.; Doblaré, M.; Hubert, C.; et al. Glioblastoma on a microfluidic chip: Generating pseudopalisades and enhancing aggressiveness through blood vessel obstruction events. Neuro Oncol. 2017, 19, 503–513. [Google Scholar] [CrossRef]
- Liu, W.; Sun, P.; Yang, L.; Wang, J.; Li, L.; Wang, J. Assay of glioma cell responses to an anticancer drug in a cell-based microfluidic device. Microfluid. Nanofluidics 2010, 9, 717–725. [Google Scholar] [CrossRef]
- Adjei-Sowah, E.A.; O’Connor, S.A.; Veldhuizen, J.; Lo Cascio, C.; Plaisier, C.; Mehta, S.; Nikkhah, M. Investigating the Interactions of Glioma Stem Cells in the Perivascular Niche at Single-Cell Resolution using a Microfluidic Tumor Microenvironment Model. Adv. Sci. 2022, 9, 2201436. [Google Scholar] [CrossRef]
- Mamani, J.B.; Marinho, B.S.; Rego, G.N.; Nucci, M.P.; Alvieri, F.; Santos, R.S.; Ferreira, J.V.; Oliveira, F.A.; Gamarra, L.F. Magnetic hyperthermia therapy in glioblastoma tumor on-a-Chip model. Einstein 2020, 18, eAO4954. [Google Scholar] [CrossRef]
- Krol, I.; Castro-Giner, F.; Maurer, M.; Gkountela, S.; Szczerba, B.; Scherrer, R.; Coleman, N.; Carreira, S.; Bachmann, F.; Anderson, S.; et al. Detection of circulating tumour cell clusters in human glioblastoma. Br. J. Cancer 2018, 119, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Ye, J.; Gao, X.; Chen, H.; Chen, M.; Lian, J.; Ma, J.; Wang, H. Evaluation of nanoparticle albumin-bound paclitaxel loaded macrophages for glioblastoma treatment based on a microfluidic chip. Front. Bioeng. Biotechnol. 2024, 12, 1361682. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Ma, C.; Vasudevaraja, V.; Serrano, J.; Tong, J.; Peng, Y.; Delorenzo, M.; Shen, G.; Frenster, J.; Morales, R.; et al. Dissecting the immunosuppressive tumor microenvironments in Glioblastoma-on-a-Chip for optimized PD-1 immunotherapy. Elife 2020, 9, e52253. [Google Scholar] [CrossRef] [PubMed]
- Sengul, E.; Elitas, M. Long-term migratory velocity measurements of single glioma cells using microfluidics. Analyst 2021, 146, 5143–5149. [Google Scholar] [CrossRef] [PubMed]
- Shao, H.; Chung, J.; Lee, K.; Balaj, L.; Min, C.; Carter, B.; Hochberg, F.; Breakerfield, X.; Lee, H.; Weissleder, R. Chip-based analysis of exosomal mRNA mediating drug resistance in glioblastoma. Nat. Commun. 2015, 6, 6999. [Google Scholar] [CrossRef]
- Peeters, M.C.M.; Dirven, L.; Koekkoek, J.A.F.; Gortmaker, E.G.; Fritz, L.; Vos, M.J.; Taphoorn, M.J.B. Prediagnostic symptoms and signs of adult glioma: The patients’ view. J. Neurooncol. 2020, 146, 293–301. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestier, O.D.; Cavenee, W.; Ellison, D.; Figarella-Branger, D.; Perry, A.; Reifenberger, G.; Von Deimling, A. WHO Classification of Tumours of the Central Nervous System. Neurol. Med. Chir. 2016, 57, 301–311. [Google Scholar]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef]
- Himes, B.T.; Geiger, P.A.; Ayasoufi, K.; Bhargav, S.; Brown, D.; Parney, I. Immunosuppression in Glioblastoma: Current Understanding and Therapeutic Implications. Front. Oncol. 2021, 11, 770561. [Google Scholar] [CrossRef]
- Sharma, P.; Aaroe, A.; Liang, J.; Puduvalli, V.K. Tumor microenvironment in glioblastoma: Current and emerging concepts. Neurooncol. Adv. 2023, 5, vdad009. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.; Miller, C.; Ding, L.; Golub, T.; Mesirov, J.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- McNamara, M.G.; Sahebjam, S.; Mason, W.P. Emerging biomarkers in glioblastoma. Cancers 2013, 5, 1103–1119. [Google Scholar] [CrossRef] [PubMed]
- Hein, T.; Krammer, P.H.; Weyd, H. Molecular analysis of Annexin expression in cancer. BMC Cancer 2022, 22, 994. [Google Scholar] [CrossRef] [PubMed]
- Gerke, V.; Moss, S.E. Annexins: From Structure to Function. Physiol. Rev. 2002, 82, 331–371. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huo, L.; Jin, H.; Han, Y.; Wang, J.; Zhang, Y.; Lai, X.; Le, Z.; Zhang, J.; Hua, Z. Anti-cancer activity of Annexin V in murine melanoma model by suppressing tumor angiogenesis. Oncotarget 2017, 8, 42602–42612. [Google Scholar] [CrossRef]
- Valdés-Rives, S.A.; Casique-Aguirre, D.; Germán-Castelán, L.; Velasco-Velázquez, M.A.; González-Arenas, A. Apoptotic Signaling Pathways in Glioblastoma and Therapeutic Implications. Biomed. Res. Int. 2017, 2017, 7403747. [Google Scholar] [CrossRef]
- Fianco, G.; Mongiardi, M.P.; Levi, A.; De Luca, T.; Desideri, M.; Trisciuoglio, D.; Bufalo, D.; Cinà, I.; Benedetto, A.; Mottolese, M. Caspase-8 contributes to angiogenesis and chemotherapy resistance in glioblastoma. Elife 2017, 6, e22593. [Google Scholar] [CrossRef]
- Szopa, W.; Burley, T.A.; Kramer-Marek, G.; Kaspera, W. Diagnostic and therapeutic biomarkers in glioblastoma: Current status and future perspectives. Biomed. Res. Int. 2017, 2017, 8013575. [Google Scholar] [CrossRef]
- Boyé, K.; Pujol, N.; DAlves, I.; Chen, Y.; Daubon, T.; Lee, Y.; Dedieu, S.; Constantin, M.; Bello, L.; Rossi, M.; et al. The role of CXCR3/LRP1 cross-talk in the invasion of primary brain tumors. Nat. Commun. 2017, 8, 1571. [Google Scholar] [CrossRef]
- Hosseini, A.; Ashraf, H.; Rahimi, F.; Alipourfard, I.; Alivirdiloo, V.; Hashemi, B.; Yazdani, Y.; Ghazi, F.; Eslami, M.; Ameri Shah Reza, M.; et al. Recent advances in the detection of glioblastoma, from imaging-based methods to proteomics and biosensors: A narrative review. Cancer Cell Int. 2023, 23, 98. [Google Scholar] [CrossRef]
- Flores Ledur, P.; Onzi, G.R.; Zong, H.; Lenz, G. Oncotarget 69185. Culture Conditions Defining Glioblastoma Cells Behavior: What Is the Impact for Novel Discoveries? Available online: www.impactjournals.com/oncotarget (accessed on 1 July 2024).
- Glas, M.; Rath, B.H.; Simon, M.; Reinartz, R.; Schramme, A.; Trageser, D.; Eisenreich, R.; Leinhaas, A.; Keller, M.; Schildhaus, H.; et al. Residual tumor cells are unique cellular targets in glioblastoma. Ann. Neurol. 2010, 68, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Ganipineni, L.P.; Danhier, F.; Préat, V. Drug delivery challenges and future of chemotherapeutic nanomedicine for glioblastoma treatment. J. Control. Release 2018, 281, 42–57. [Google Scholar] [CrossRef] [PubMed]
- Akter, F.; Simon, B.; de Boer, N.L.; Redjal, N.; Wakimoto, H.; Shah, K. Pre-clinical tumor models of primary brain tumors: Challenges and opportunities. Biochim. Et Biophys. Acta (BBA)—Rev. Cancer 2021, 1875, 188458. [Google Scholar] [CrossRef]
- Varna, M.; Bertheau, P.; Legres, L. Tumor Microenvironment in Human Tumor Xenografted Mouse Models. J. Anal. Oncol. 2014, 3, 159–166. [Google Scholar] [CrossRef]
- Xu, C.; Yuan, X.; Hou, P.; Li, Z.; Wang, C.; Fang, C.; Tan, Y. Development of glioblastoma organoids and their applications in personalized therapy. Cancer Biol. Med. 2023, 20, 353–368. [Google Scholar] [CrossRef]
- Wang, X.; Sun, Y.; Zhang, D.Y.; Ming, G.; Song, H. Glioblastoma modeling with 3D organoids: Progress and challenges. Oxf. Open Neurosci. 2023, 2, kvad008. [Google Scholar] [CrossRef]
- Rybin, M.J.; Ivan, M.E.; Ayad, N.G.; Zeier, Z. Organoid Models of Glioblastoma and Their Role in Drug Discovery. Front. Cell. Neurosci. 2021, 15, 605255. [Google Scholar] [CrossRef]
- Trujillo-de Santiago, G.; Flores-Garza, B.G.; Tavares-Negrete, J.A.; Lara-Mayorga, I.; González-Gamboa, I.; Zhang, Y.; Rojas-Martínez, A.; Ortiz-López, R.; Álvarez, M.M. The tumor-on-chip: Recent advances in the development of microfluidic systems to recapitulate the physiology of solid tumors. Materials 2019, 12, 2945. [Google Scholar] [CrossRef]
- Leung, C.M.; de Haan, P.; Ronaldson-Bouchard, K.; Kim, G.; Ko, J.; Rho, H.; Chen, Z.; Habibovic, P.; Jeon, N.; Takayama, S.; et al. A guide to the organ-on-a-chip. Nat. Rev. Methods Primers 2022, 2, 33. [Google Scholar] [CrossRef]
- Tevlek, A.; Kecili, S.; Ozcelik, O.S.; Kulah, H.; Tekinm, H. Spheroid Engineering in Microfluidic Devices. ACS Omega 2022, 8, 3630–3649. [Google Scholar] [CrossRef]
- Preetam, S.; Nahak, B.K.; Patra, S.; Toncu, D.; Park, S.; Syväjärvi, M.; Orive, G.; Tiwari, A. Emergence of microfluidics for next generation biomedical devices. Biosens. Bioelectron. X 2022, 10, 100106. [Google Scholar] [CrossRef]
- Prakash, S.; Kumar, S. Fabrication of microchannels: A review. Proc. Inst. Mech. Eng. B J. Eng. Manuf. 2014, 229, 1273–1288. [Google Scholar] [CrossRef]
- Cao, U.M.; Zhang, Y.; Chen, J.; Sayson, D.; Pillai, S.; Tran, S.D. Microfluidic Organ-on-A-chip: A Guide to Biomaterial Choice and Fabrication. Int. J. Mol. Sci. 2023, 24, 3232. [Google Scholar] [CrossRef] [PubMed]
- Tajeddin, A.; Mustafaoglu, N. Design and fabrication of organ-on-chips: Promises and challenges. Micromachines 2021, 12, 1443. [Google Scholar] [CrossRef] [PubMed]
- Rasponi, M. Organ-on-a-Chip Methods and Protocols; Spinger: Berlin/Heidelberg, Germany, 2022. [Google Scholar]
- Miri, A.K.; Mostafavi, E.; Khorsandi, D.; Hu, S.K.; Malpica, M.; Khademhosseini, A. Bioprinters for organs-on-chips. Biofabrication 2019, 11, 042002. [Google Scholar] [CrossRef] [PubMed]
- Chliara, M.A.; Elezoglou, S.; Zergioti, I. Bioprinting on Organ-on-Chip: Development and Applications. Biosensors 2022, 12, 1135. [Google Scholar] [CrossRef]
- Bhatia, S.N.; Ingber, D.E. Microfluidic organs-on-chips. Nat. Biotechnol. 2014, 32, 760–772. [Google Scholar] [CrossRef]
- Ingber, D.E. Human organs-on-chips for disease modelling, drug development and personalized medicine. Nat. Rev. Genet. 2022, 23, 467–491. [Google Scholar] [CrossRef]
- Beier, C.P.; Kumar, P.; Meyer, K.; Leukel, P.; Bruttel, V.; Aschenbrenner, I.; Riemenschneider, M.; Fragoulis, A.; Rümmele, P.; Lamszus, K.; et al. The cancer stem cell subtype determines immune infiltration of Glioblastoma. Stem Cells Dev. 2012, 21, 2753–2761. [Google Scholar] [CrossRef]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.; Fuller, C.; Hamner, B.; Oh, E.; Gaber, M.; Finkelstein, D.; Allen, M.; et al. A Perivascular Niche for Brain Tumor Stem Cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef]
- Liu, J.; Yang, F.; Hu, J.; Zhang, X. Nanoparticles for efficient drug delivery and drug resistance in glioma: New perspectives. CNS Neurosci. Ther. 2024, 30, e14715. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, M.C. Temozolomide: Therapeutic limitations in the treatment of adult high-grade gliomas. Expert. Rev. Neurother. 2010, 10, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Manini, I.; Caponnetto, F.; Bartolini, A.; Ius, T.; Mariuzzi, L.; Loreto, C.; Beltrami, A.; Cesselli, D. Role of microenvironment in glioma invasion: What we learned from in vitro models. Int. J. Mol. Sci. 2018, 19, 147. [Google Scholar] [CrossRef] [PubMed]
- van der Helm, M.W.; van der Meer, A.D.; Eijkel, J.C.T.; van den Berg, A.; Segerink, L. Microfluidic organ-on-chip technology for blood-brain barrier research. Tissue Barriers 2016, 4, e1142493. [Google Scholar] [CrossRef] [PubMed]
- Brandalise, F.; Ramieri, M.; Pastorelli, E.; Priori, E.; Ratto, D.; Venuti, M.; Roda, E.; Talpo, F.; Rossi, P. Role of Na+/Ca2+ Exchanger (NCX) in Glioblastoma Cell Migration (In Vitro). Int. J. Mol. Sci. 2023, 24, 12673. [Google Scholar] [CrossRef]
- Zhang, Y. Ion Channels and Transporters in the Regulation of Cancer Cell Migration and Metastasis. Doctoral Dissertation, Johns Hopkins University, Baltimore, MD, USA, 2022. [Google Scholar]
- Hu, H.-J.; Wang, S.-S.; Wang, Y.-X.; Liu, Y.; Feng, X.; Shen, Y.; Zhu, L.; Chen, H.; Song, M. Blockade of the forward Na+/Ca2+ exchanger suppresses the growth of glioblastoma cells through Ca2+-mediated cell death. Br. J. Pharmacol. 2019, 176, 2691–2707. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).