Theoretical Evaluation of the Properties of Nitrogen-Doped C24 Fullerenes and Their Interactions with Two Adamantane-Derived Antivirals


Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussions
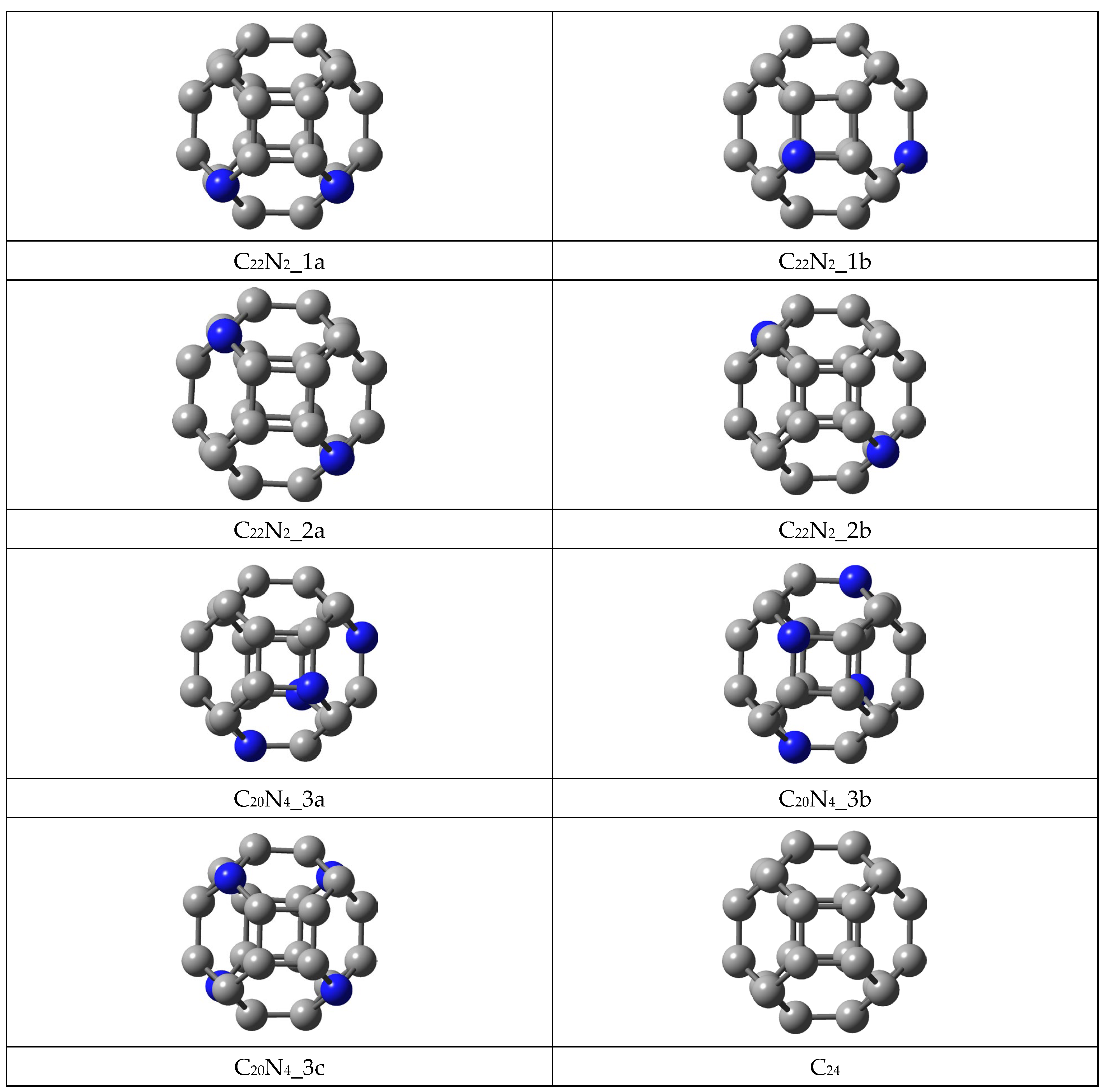
3.1. Frontier Molecular Orbital (FMO) Analysis, Energetic Characterization and Local Aromaticity of 24-Membered Fullerenes
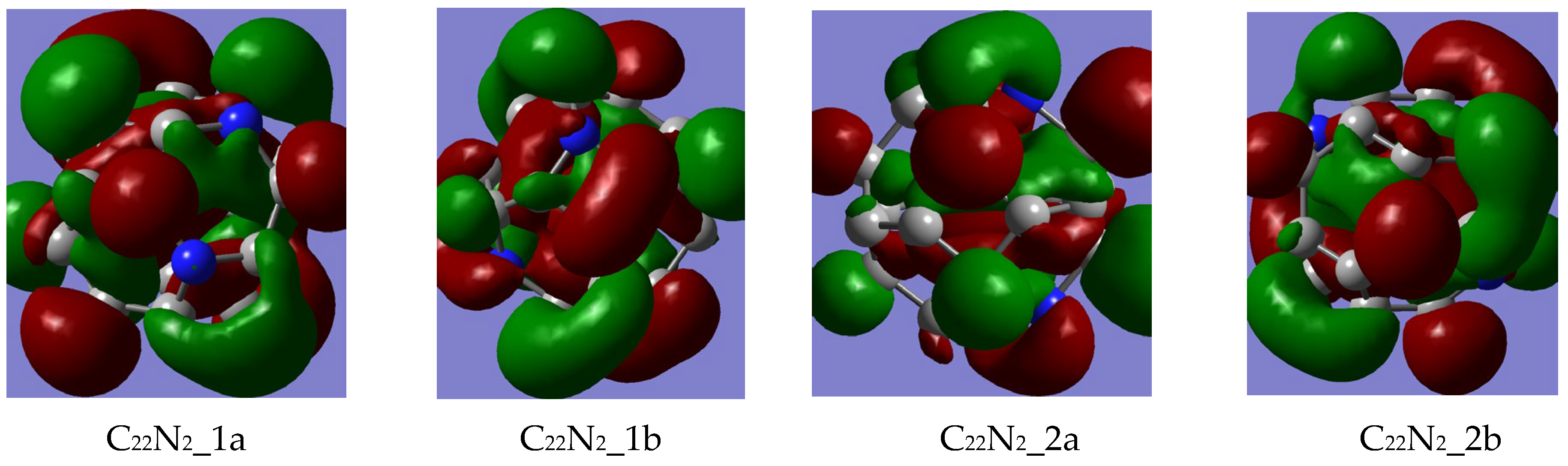
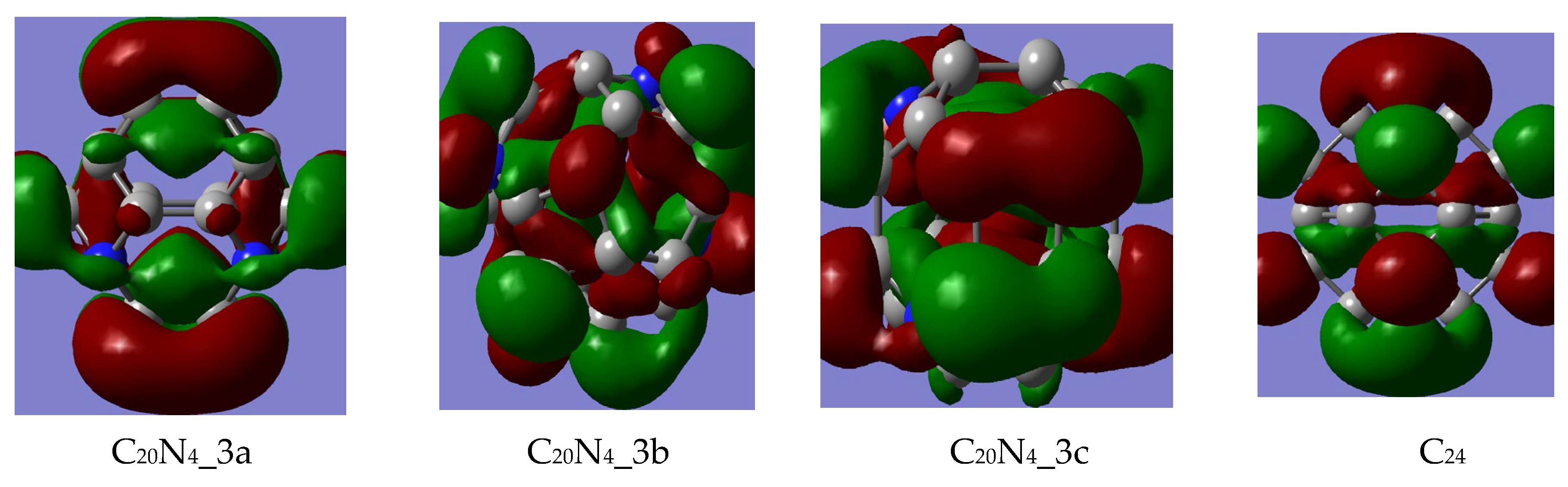
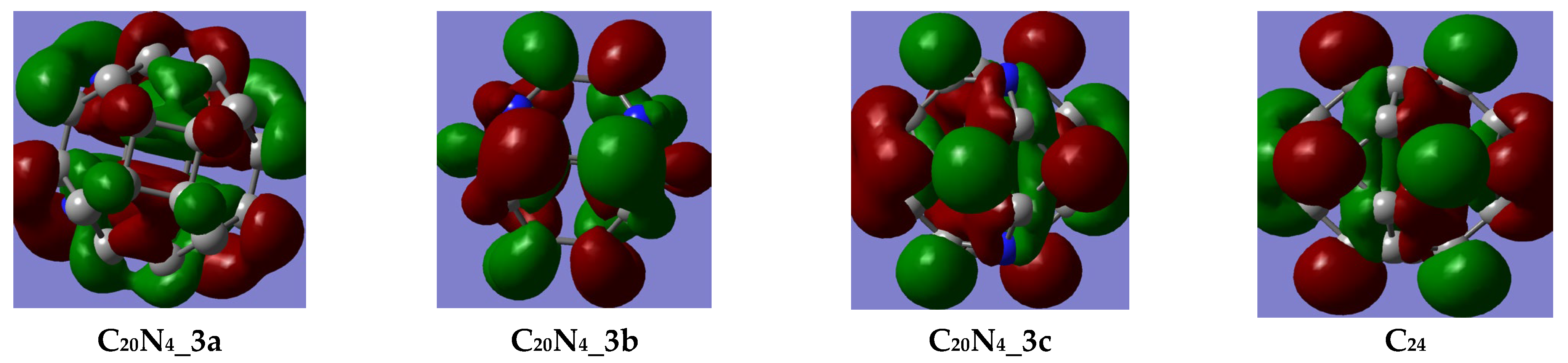
3.1.1. FMO Analysis
3.1.2. Energetic Characterization: Total Binding Energy and Singlet–Triplet Gap
3.1.3. Local Aromaticity Evaluation of Nitrogen-Doped Fullerenes
3.2. Evaluation of the Interactions between Fullerenes and Adamantane-Derived Structures (Amantadine and Rimantadine)
4. Discussion
5. Conclusions
- (i)
- The stability of the newly designed aza-fullerenes is influenced both by the number and the position of the heteroatoms, given that better values of the HOMO–LUMO gap, chemical potential and hardness were obtained for two of the C22N2 fullerenes (1a and 1b) and one of the C20N4 fullerene (3a);
- (ii)
- The aromaticity evaluation suggests that the six-membered and four-membered rings of the fullerenes present antiaromatic character;
- (iii)
- The steric parameters of the fullerenes, calculated prior to the molecular docking study, show little differences among them; the most significant influence of the number and position of the nitrogen atoms is reflected in the logP values;
- (iv)
- All of the fullerenes presented better binding affinities towards rimantadine than amantadine; among them, the most hydrophobic aza-fullerenes C22N2 1a and 1b gave better or equal results compared to pristine C24.
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Aschberger, K.; Johnston, H.J.; Stone, V.; Aitken, R.J.; Tran, C.L.; Hankin, S.M.; Peters, S.A.; Christensen, F.M. Review of fullerene toxicity and exposure—Appraisal of a human health risk assessment, based on open literature. Regul. Toxicol. Pharmacol. 2010, 58, 455–473. [Google Scholar] [CrossRef] [PubMed]
- Bakry, R.; Vallant, R.M.; Najam-Ul-Haq, M.; Rainer, M.; Szabo, Z.; Huck, C.W.; Bonn, G.K. Medicinal applications of fullerenes. Int. J. Nanomed. 2007, 2, 639–649. [Google Scholar]
- Kazemzadeh, H.; Mozafari, M. Fullerene-based delivery systems. Drug Discov. Today 2019, 24, 898–905. [Google Scholar] [CrossRef] [PubMed]
- László, F.; László, M. Electronic properties of doped fullerenes. Rep. Prog. Phys. 2001, 64, 649–699. [Google Scholar]
- Antonietti, M.; Lopez Salas, N.; Primo, A. Adjusting the Structure and Electronic Properties of Carbons for Metal-Free Carbocatalysis of Organic Transformations. Adv. Mater. 2019, 31, 1805719. [Google Scholar] [CrossRef]
- Chattopadhyay, J.; Pathak, T.S.; Pak, D. Heteroatom-Doped Metal-Free Carbon Nanomaterials as Potential Electrocatalysts. Molecules 2022, 27, 670. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wang, Q.; Ji, G.; Li, A.; Niu, J. Doping strategy, properties and application of heteroatom-doped ordered mesoporous carbon. RSC Adv. 2021, 11, 5361–5383. [Google Scholar] [CrossRef] [PubMed]
- Zoua, V.d.P.; Fouegue, A.D.T.; Bouba, M.O.; Ntieche, R.A.; Abdoul, W. Adsorption of juglone on pure and boron-doped C24 fullerene-like nano-cage: A density functional theory investigation. Comput. Theor. Chem. 2023, 1222, 114077. [Google Scholar] [CrossRef]
- Çatal, E.; Bağlayan, Ö.; Köroğlu, A.; Parlak, C.; Alver, Ö. Assessing a double silicon decorated fullerene for the delivery of interacting flurbiprofen and salicylic acid drugs: A DFT approach. J. Ind. Chem. Soc. 2023, 100, 101046. [Google Scholar] [CrossRef]
- Vatanparast, M.; Shariatinia, Z. AlN and AlP doped graphene quantum dots as novel drug delivery systems for 5-fluorouracil drug: Theoretical studies. J. Fluor. Chem. 2018, 211, 81–93. [Google Scholar] [CrossRef]
- Karimzadeh, S.; Safaei, B.; Jen, T.C. Theorical investigation of adsorption mechanism of doxorubicin anticancer drug on the pristine and functionalized single-walled carbon nanotube surface as a drug delivery vehicle: A DFT study. J. Mol. Liq. 2021, 322, 114890. [Google Scholar] [CrossRef]
- Celaya, C.A.; Hernández-Ayala, L.F.; Zamudio, F.B.; Vargas, J.A.; Reina, M. Adsorption of melphalan anticancer drug on C24, B12N12, B12C6N6, B6C12N12 and B6C6N12 nanocages: A comparative DFT study. J. Mol. Liq. 2021, 329, 15528. [Google Scholar] [CrossRef]
- Perveen, M.; Nazir, S.; Arshad, A.W.; Shamim, M.; Ayub, K.; Khan, M.A.; Iqbal, J. Therapeutic potential of graphitic carbon nitride as a drug delivery system for cisplatin (anticancer drug): A DFT approach. Biophys. Chem. 2020, 267, 106461. [Google Scholar] [CrossRef]
- Jana, S.K.; Som, N.N.; Jha, P.K. Theoretical appraisements on the interaction behaviour of amphetamine, ketamine and mercaptopurine drug molecules over C24 fullerene: A combined dispersion corrected DFT and MD simulation study. J. Mol. Liq. 2023, 383, 122084. [Google Scholar] [CrossRef]
- Jana, S.K.; Chodvadiya, D.; Som, N.N.; Jha, P.K. A quantum mechanical prediction of C24 fullerene as a DNA nucleobase biosensor. Diam. Rel. Mater. 2022, 129, 109305. [Google Scholar] [CrossRef]
- Soliman, K.A.; Abdel Aal, S. Theoretical investigation of favipiravir antiviral drug based on fullerene and boron nitride nanocages. Diam. Rel. Mater. 2021, 117, 108458. [Google Scholar] [CrossRef]
- Tukadiya, N.A.; Jana, S.K.; Chakraborty, B.; Jha, P.K. C24 Fullerene and its derivatives as a viable glucose sensor: DFT and TD-DFT studies. Surf. Interfaces 2023, 41, 103220. [Google Scholar] [CrossRef]
- Da Ros, T. Twenty Years of Promises: Fullerene in Medicinal Chemistry. In Medicinal Chemistry and Pharmacological Potential of Fullerenes and Carbon Nanotubes. Carbon Materials: Chemistry and Physics; Cataldo, F., Da Ros, T., Eds.; Springer: Berlin/Heidelberg, Germany, 2008; Volume 1, pp. 1–21. [Google Scholar]
- Grebowski, J.; Kazmierska, P.; Krokosz, A. Fullerenols as a new therapeutic approach in nanomedicine. Biomed. Res. Int. 2013, 2013, 751913. [Google Scholar] [CrossRef]
- Majumder, J.; Taratula, O.; Minko, T. Nanocarrier-based systems for targeted and site specific therapeutic delivery. Adv. Drug Deliv. Rev. 2019, 144, 57–77. [Google Scholar] [CrossRef] [PubMed]
- Madannejad, R.; Shoaie, N.; Jahanpeyma, F.; Darvishi, M.H.; Azimzadeh, M.; Javadi, H. Toxicity of carbon-based nanomaterials: Reviewing recent reports in medical and biological systems. Chem. Biol. Interact. 2019, 307, 206–222. [Google Scholar] [CrossRef]
- Li, Z.; Wang, L.; Li, Y.; Feng, Y.; Feng, W. Carbon-based functional nanomaterials: Preparation, properties and applications. Compos. Sci. Tech. 2019, 179, 10–40. [Google Scholar] [CrossRef]
- Pop, R.; Andoni, M.; Van Staden, J.; Păuşescu, I.; Medeleanu, M. Theoretical considerations regarding the aromaticity of λ3-heterobenzenes containing 15-group elements. Dig. J. Nanomater. Bios. 2013, 8, 1739–1750. [Google Scholar]
- Păuşescu, I.; Medeleanu, M.; Ştefănescu, M.; Peter, F.; Pop, R. A DFT Study on The Stability and Aromaticity of Heterobenzenes Containing 15-Group Elements. Heteroatom. Chem. 2015, 26, 206–214. [Google Scholar] [CrossRef]
- Pop, R.; van Staden, J.; Diudea, M. Nitrogen-Containing Coronenes: Theoretical Evaluation of the Influence of Aza-substitution on their Aromaticity. Z. Für Naturforschung A 2015, 70, 171–175. [Google Scholar] [CrossRef]
- Pop, R.; van Staden, J. Theoretical evaluation of the interactions between met-al-phthalocyanins and various fullerenes as delivery systems. Chemistry 2022, 4, 1016–1027. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision a01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
- Wolinski, K.; Hilton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Pearson, R.J. Chemical hardness and the electronic chemical potential. Inorg. Chim. Acta 1992, 198–200, 781–786. [Google Scholar] [CrossRef]
- Poater, J.; Fradera, X.; Duran, M.; Solà, M. The Delocalization Index as an Electronic Aromaticity Criterion: Application to a Series of Planar Polycyclic Aromatic Hydrocarbons. Chem. Eur. J. 2003, 9, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Matito, E.; Duran, M.; Solà, M. The aromatic fluctuation index (FLU): A new aromaticity index based on electron delocalization. J. Chem. Phys. 2005, 122, 014109. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.J. Multiwfn: A multifunctional wavefunction analyzer. Comp. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Zyrianov, Y. Distribution-Based Descriptors of the Molecular Shape. J. Chem. Inf. Model. 2005, 45, 657–672. [Google Scholar] [CrossRef] [PubMed]
- Richmond, T.J. Solvent accessible surface area and excluded volume in proteins: Analytical equations for overlapping spheres and implications for the hydrophobic effect. J. Mol. Biol. 1984, 178, 63–89. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Zevalos, J.; Toro-Labbes, A. A theoretical analysis of the Kohn-Sham and Hartree-Fock orbitals and their use in the determination of electronic properties. J. Chil. Chem. Soc. 2003, 48, 39–47. [Google Scholar] [CrossRef]
- Rostami, Z.; Hosseinian, A.; Monfared, A. DFT results against experimental data for electronic properties of C60 and C70 fullerene derivatives. J. Mol. Graph. Model. 2018, 81, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, Z.; Liu, H.; Gong, H.; Zhang, J.; Yao, Y.; Guo, Q. Organic molecules with inverted singlet-triplet gaps. Front. Chem. 2022, 10, 999856. [Google Scholar] [CrossRef]
- Pedersen, J.; Mikkelsen, K.V. A benchmark study of aromaticity indexes for benzene, pyridine and the diazines—I. Ground state aromaticity. RSC Adv. 2022, 12, 2830–2842. [Google Scholar] [CrossRef] [PubMed]
- Feixas, F.; Matito, E.; Poater, J.; Solà, M. Quantifying aromaticity with electron delocalisation measures. Chem. Soc. Rev. 2015, 44, 6434–6451. [Google Scholar]
- Schleyer, P.v.R.; Maerker, C.; Dransfeld, A.; Jiao, H.; Hommes, N.J.R.v.E. Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef]
- Krygowski, T.M.; Szatylowicz, H. Aromaticity: What does it mean? ChemTexts 2015, 1, 12. [Google Scholar] [CrossRef] [PubMed]


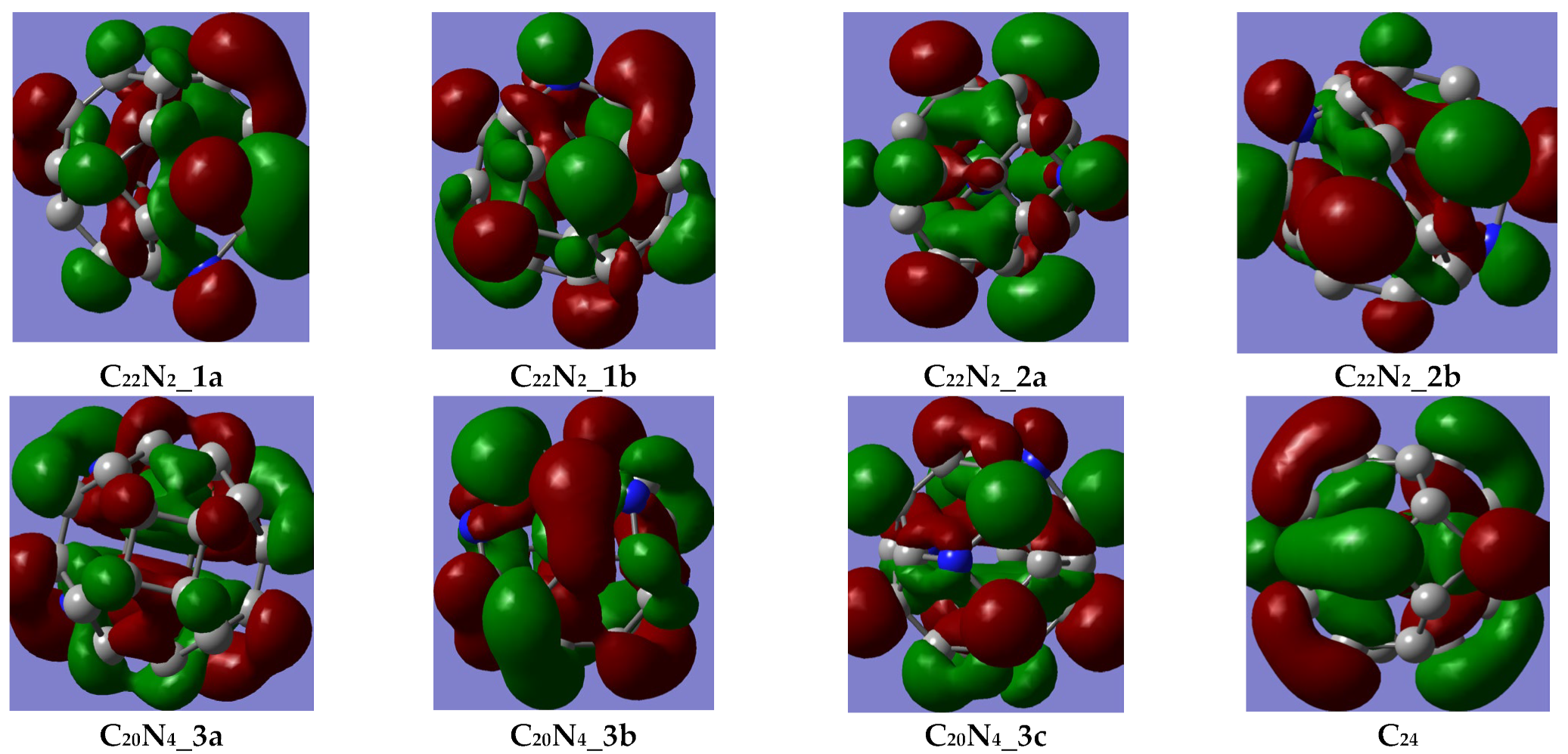



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
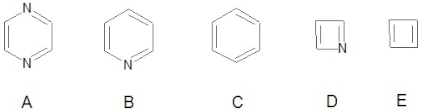 | |||||
|---|---|---|---|---|---|
| A (Cycles) | B (Cycles) | C (Cycles) | D (Cycles) | E (Cycles) | |
| C22N2_1a | 1 | 2 | 5 | 2 | 4 |
| C22N2_1b | - | 4 | 4 | 2 | 4 |
| C22N2_2a | - | 4 | 4 | 2 | 4 |
| C22N2_2b | - | 4 | 4 | 2 | 4 |
| C20N4_3a | 4 | - | 4 | 4 | 2 |
| C20N4_3b | 2 | 4 | 2 | 4 | 2 |
| C20N4_3c | - | 8 | - | 4 | 2 |
| C24 | - | - | 8 | - | 6 |
| Compound | HOMO (Eh) | LUMO (Eh) |
|---|---|---|
| C22N2_1a | −0.261/−0.231 | −0.039/−0.148 |
| C22N2_1b | −0.243/−0.219 | −0.043/−0.150 |
| C22N2_2a | −0.232/−0.213 | −0.047/−0.153 |
| C22N2_2b | −0.231/−0.207 | −0.042/−0.148 |
| C20N4_3a | −0.247/−0.217 | −0.044/−0.145 |
| C20N4_3b | −0.217/−0.199 | −0.049/−0.152 |
| C20N4_3c | −0.222/−0.204 | −0.052/−0.151 |
| C24 | −0.310/−0.243 | −0.017/−0.142 |
| Compound | HL Gap (eV) | Hardness η (eV) | Chemical Potential μ (eV) | Electrophilicity ω (eV) |
|---|---|---|---|---|
| C22N2_1a | 6.04/2.26 | 3.02/1.13 | −4.08/−5.15 | 2.76/11.73 |
| C22N2_1b | 5.44/1.88 | 2.72/0.94 | −3.89/−5.02 | 2.78/13.40 |
| C22N2_2a | 5.03/1.63 | 2.51/0.82 | −3.79/−4.98 | 2.86/15.12 |
| C22N2_2b | 5.14/1.60 | 2.57/0.80 | −3.71/−4.83 | 2.68/14.58 |
| C20N4_3a | 5.52/1.96 | 2.76/0.98 | −3.96/−4.92 | 2.84/12.35 |
| C20N4_3b | 4.57/1.28 | 2.28/0.64 | −3.62/−4.77 | 2.87/17.78 |
| C20N4_3c | 4.62/1.44 | 2.31/0.72 | −3.73/−4.83 | 3.01/16.20 |
| C24 | 7.97/2.75 | 3.98/1.37 | −4.45/−5.24 | 2.49/10.02 |
| Compound | E Rel (eV) | TBE/Atom (Eh) |
|---|---|---|
| C22N2_1a | 37.482/33.265 | −39.196/−39.458 |
| C22N2_1b | 37.509/33.275 | −39.195/−39.458 |
| C22N2_2a | 37.509/33.281 | −39.195/−39.457 |
| C22N2_2b | 37.509/33.283 | −39.195/−39.457 |
| C20N4_3a | 0/0 | −40.574/−40.844 |
| C20N4_3b | 0.054/0.036 | −40.572/−40.842 |
| C20N4_3c | 0.027/0.019 | −40.573/−40.843 |
| C24 | 74.800/66.541 | −37.824/−38.071 |
| Compound | Singlet (Eh) | Triplet (Eh) | ΔEST (eV) |
|---|---|---|---|
| C22N2_1a | −940.700/−946.990 | −940.685/−946.779 | −0.408/−5.739 |
| C22N2_1b | −940.681/−946.980 | −940.673/−946.782 | −0.218/−5.385 |
| C22N2_2a | −940.672/−946.974 | −940.764/−946.791 | 2.502/−4.977 |
| C22N2_2b | −940.680/−946.972 | −940.709/−946.768 | 0.788/−5.549 |
| C20N4_3a | −973.780/−980.255 | −973.823/−980.053 | 1.170/−5.494 |
| C20N4_3b | −973.738/−980.219 | −973.773/−980.047 | 0.952/−4.678 |
| C20N4_3c | −973.751/−980.236 | −973.888/−980.127 | 3.726/−2.965 |
| C24 | −907.769/−913.714 | −907.602/−913.438 | −4.542/−7.507 |
| Rings | |||
|---|---|---|---|
| Compound | A | B | C |
| C22N2_1a | 0.025/0.031 | 0.025/0.027 | 0.047/0.051 |
| C22N2_1b | - | 0.036/0.038 | 0.035/0.042 |
| C22N2_2a | - | 0.030/0.032 | 0.043/0.050 |
| C22N2_2b | - | 0.027/0.033 | 0.043/0.045 |
| C20N4_3a | 0.017/0.030 | - | 0.039/0.052 |
| C20N4_3b | 0.024/0.028 | 0.019/0.025 | 0.041/0.033 |
| C20N4_3c | - | 0.028/0.029 | - |
| C24 | - | - | 0.048/0.051 |
| Rings | |||||
|---|---|---|---|---|---|
| Compound | A | B | C | D | E |
| C22N2_1a | 0.063/0.079 | 0.054/0.072 | 0.028/0.035 | 0.087/0.111 | 0.050/0.080 |
| C22N2_1b | - | 0.044/0.056 | 0.032/0.039 | 0.079/0.110 | 0.057/0.078 |
| C22N2_2a | - | 0.046/0.058 | 0.033/0.035 | 0.088/0.115 | 0.049/0.084 |
| C22N2_2b | - | 0.050/0.054 | 0.029/0.037 | 0.077/0.099 | 0.053/0.082 |
| C20N4_3a | 0.071/0.074 | - | 0.031/0.031 | 0.074/0.104 | 0.049/0.070 |
| C20N4_3b | 0.056/0.051 | 0.054/0.052 | 0.027/0.037 | 0.070/0.093 | 0.047/0.062 |
| C20N4_3c | - | 0.041/0.052 | - | 0.046/0.090 | 0.072/0.063 |
| C24 | - | - | 0.041/0.039 | - | 0.079/0.078 |
| Rings | |||
|---|---|---|---|
| Compound | A | B | C |
| C22N2_1a | 22.246/41.508 | 18.548/36.527 | 7.972/24.891 |
| C22N2_1b | - | 20.504/45.047 | 18.933/42.597 |
| C22N2_2a | - | 75.947/11.023 | 43.153/4.211 |
| C22N2_2b | - | 58.520/9.985 | 44.415/7.521 |
| C20N4_3a | −15.060/−16.067 | - | −9.998/−9.355 |
| C20N4_3b | −12.877/−2.600 | −32.293/−20.088 | −17.452/−7.142 |
| C20N4_3c | - | −18.981/−4.754 | - |
| C24 | - | - | 26.763/33.053 |
| Compound | logP | Ovality | CAA (Å2) | CSEV (Å3) |
|---|---|---|---|---|
| C22N2_1a | 1.166 | 1.221 | 228 | 414 |
| C22N2_1b | 0.715 | 1.221 | 228 | 414 |
| C22N2_2a | 0.054 | 1.221 | 228 | 414 |
| C22N2_2b | −1.296 | 1.221 | 228 | 414 |
| C20N4_3a | −0.672 | 1.213 | 220 | 408 |
| C20N4_3b | −2.614 | 1.213 | 220 | 408 |
| C20N4_3c | −2.377 | 1.219 | 226 | 414 |
| C24 | 8.448 | - | - | - |
| Compound | logP | Ovality | CAA (Å2) | CSEV (Å3) |
|---|---|---|---|---|
| Amantadine | 2.236 | 1.132 | 336.179 | 159.145 |
| Rimantadine | 3.513 | 1.162 | 376.252 | 192.175 |
| Compound | 1a | 1b | 2a | 2b | 3a | 3b | 3c | 24 |
|---|---|---|---|---|---|---|---|---|
| Amantadine | −2.23 | −2.22 | −2.1 | −2.1 | −2.0 | −1.9 | −1.7 | −2.2 |
| Rimantadine | −2.65 | −2.6 | −2.5 | −2.44 | −2.35 | −2.2 | −1.91 | −2.6 |
| Compound | C22N2_1a | C22N2_1b | C22N2_2a | C22N2_2b | C20N4_3a | C20N4_3b | C20N4_3c | C24 |
|---|---|---|---|---|---|---|---|---|
| Amantadine | 15.705 | 15.550 | 13.791 | 13.791 | 12.479 | 11.291 | 9.245 | 15.241 |
| Rimantadine | 23.904 | 22.738 | 20.574 | 19.376 | 17.708 | 15.241 | 11.405 | 22.738 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pop, O.-R.; Căta, A.; Ienașcu, I.M.C. Theoretical Evaluation of the Properties of Nitrogen-Doped C24 Fullerenes and Their Interactions with Two Adamantane-Derived Antivirals. Chemistry 2023, 5, 2376-2391. https://doi.org/10.3390/chemistry5040157
Pop O-R, Căta A, Ienașcu IMC. Theoretical Evaluation of the Properties of Nitrogen-Doped C24 Fullerenes and Their Interactions with Two Adamantane-Derived Antivirals. Chemistry. 2023; 5(4):2376-2391. https://doi.org/10.3390/chemistry5040157
Chicago/Turabian StylePop, Oana-Raluca, Adina Căta, and Ioana Maria Carmen Ienașcu. 2023. "Theoretical Evaluation of the Properties of Nitrogen-Doped C24 Fullerenes and Their Interactions with Two Adamantane-Derived Antivirals" Chemistry 5, no. 4: 2376-2391. https://doi.org/10.3390/chemistry5040157
APA StylePop, O.-R., Căta, A., & Ienașcu, I. M. C. (2023). Theoretical Evaluation of the Properties of Nitrogen-Doped C24 Fullerenes and Their Interactions with Two Adamantane-Derived Antivirals. Chemistry, 5(4), 2376-2391. https://doi.org/10.3390/chemistry5040157