
Analysis of Local and Global Aromaticity in Si3C5 and Si4C8 Clusters. Aromatic Species Containing Planar Tetracoordinate Carbon


 and
and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
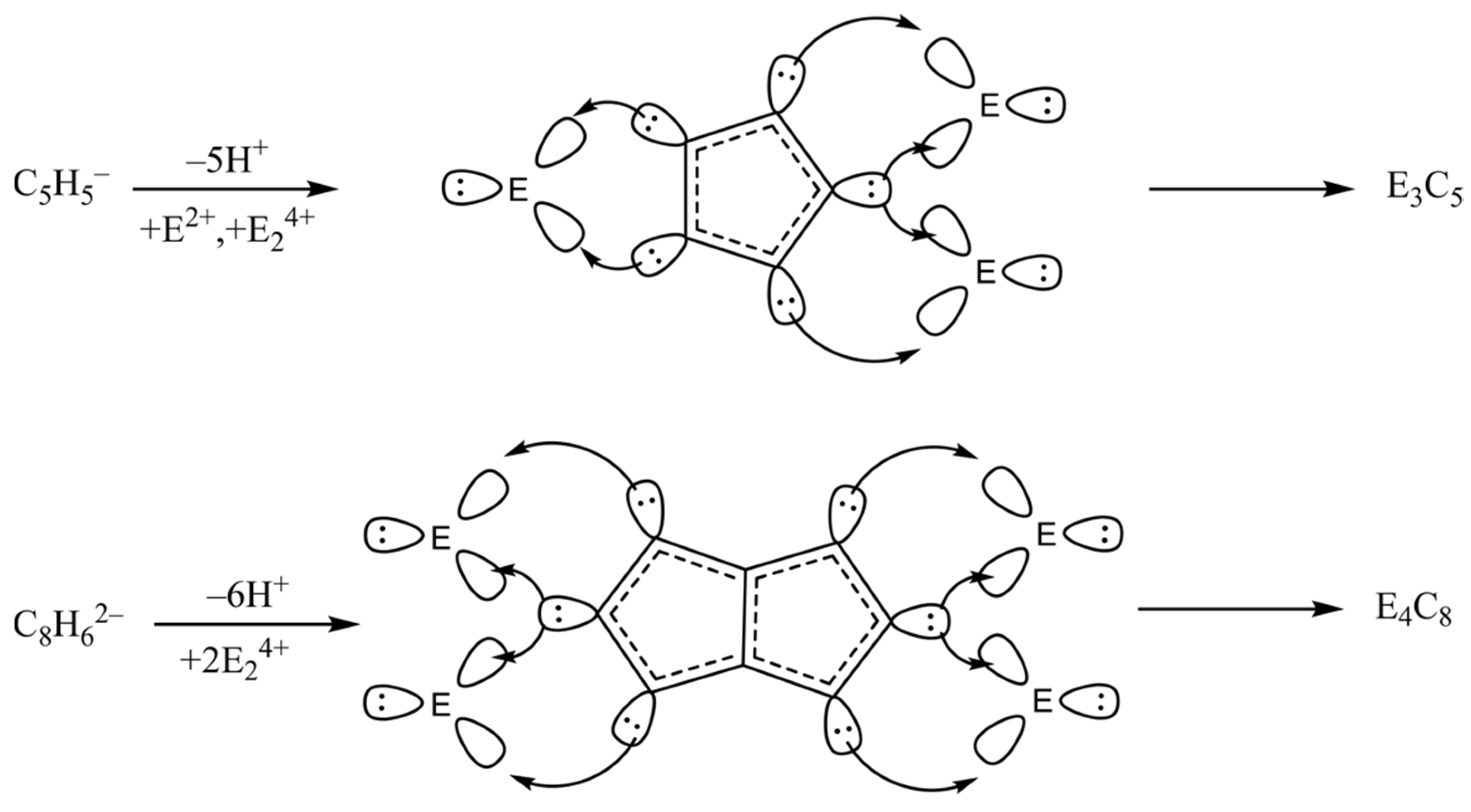
1. Introduction
2. Computational Methods
3. Results
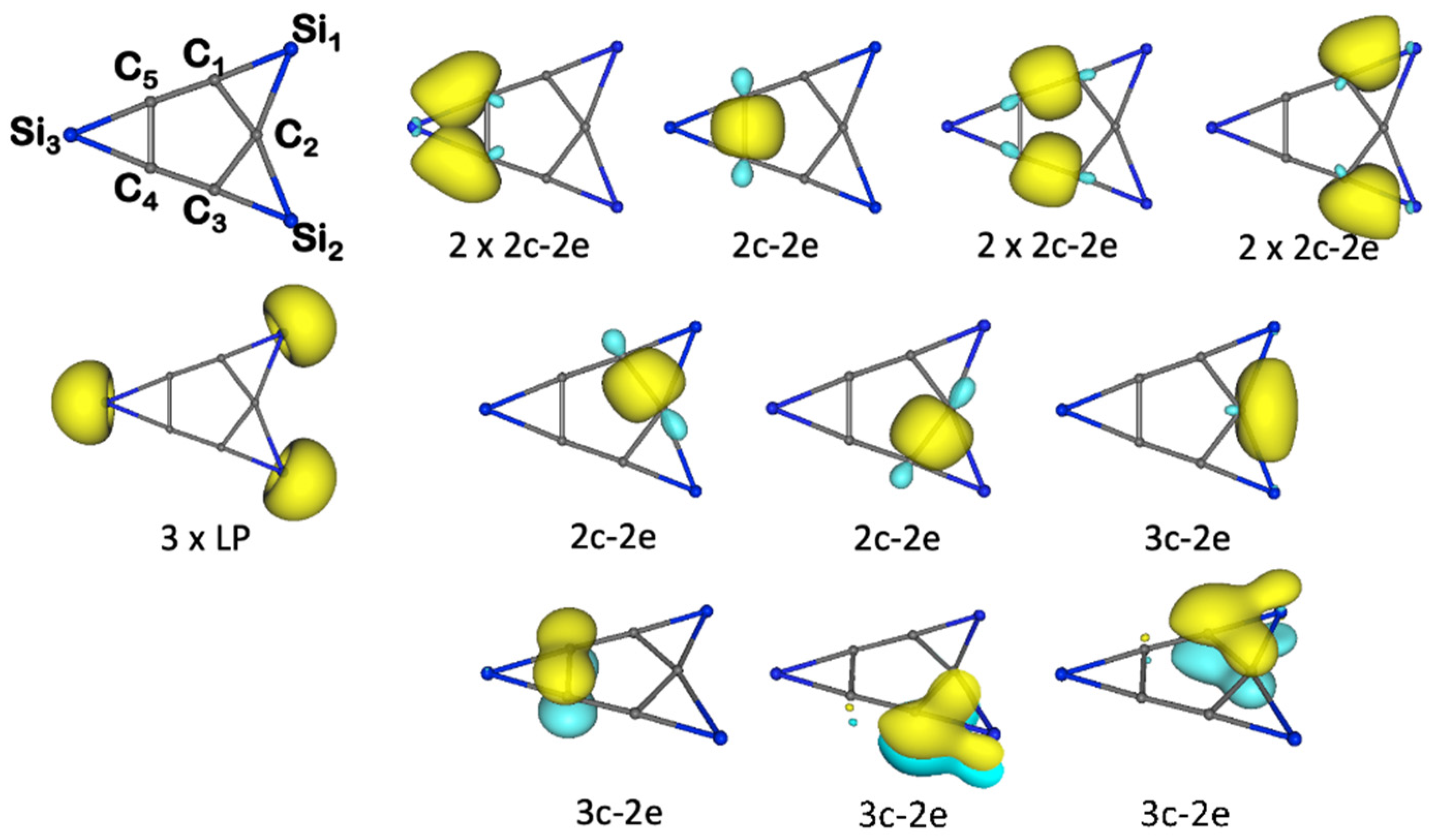
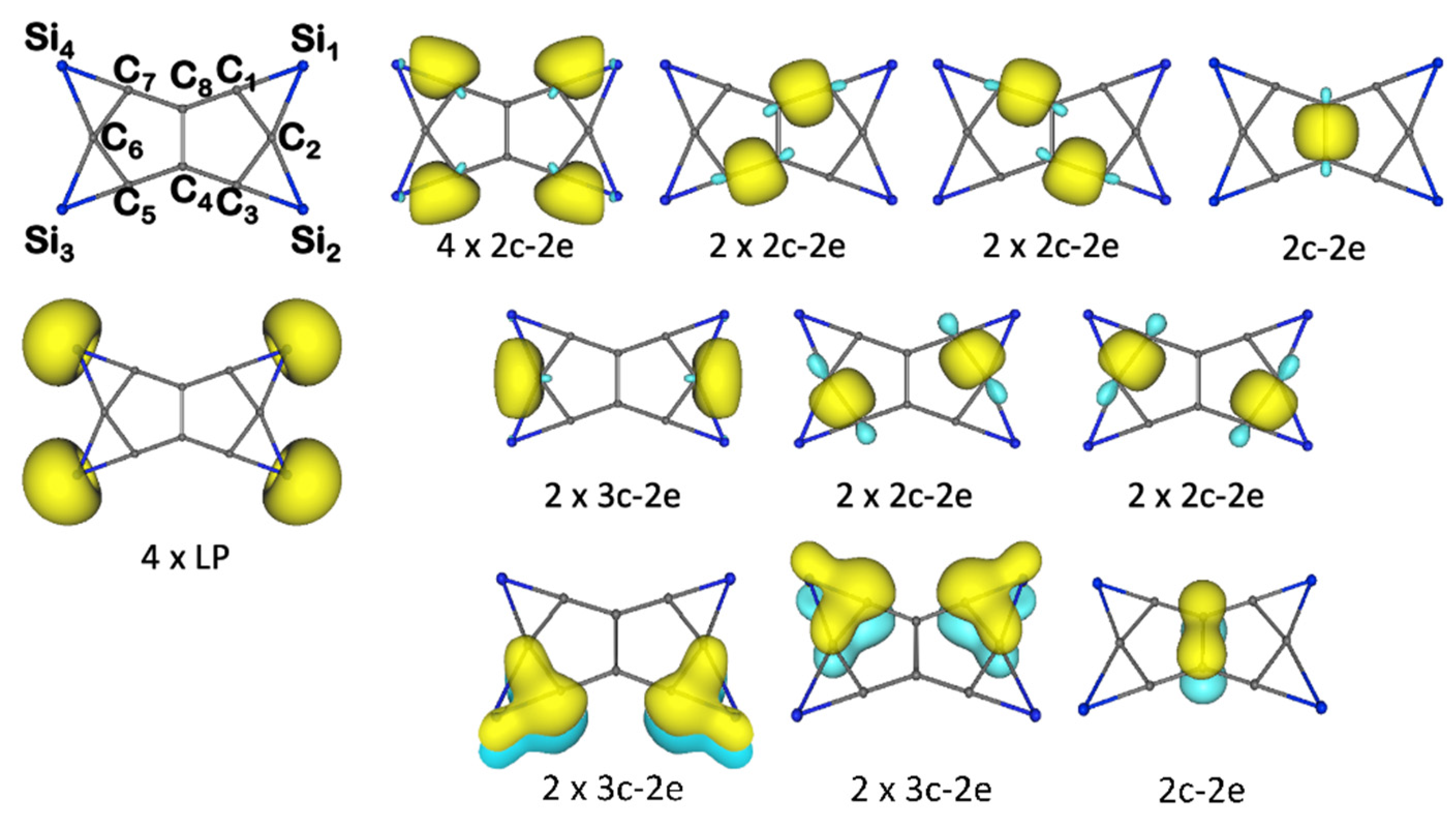
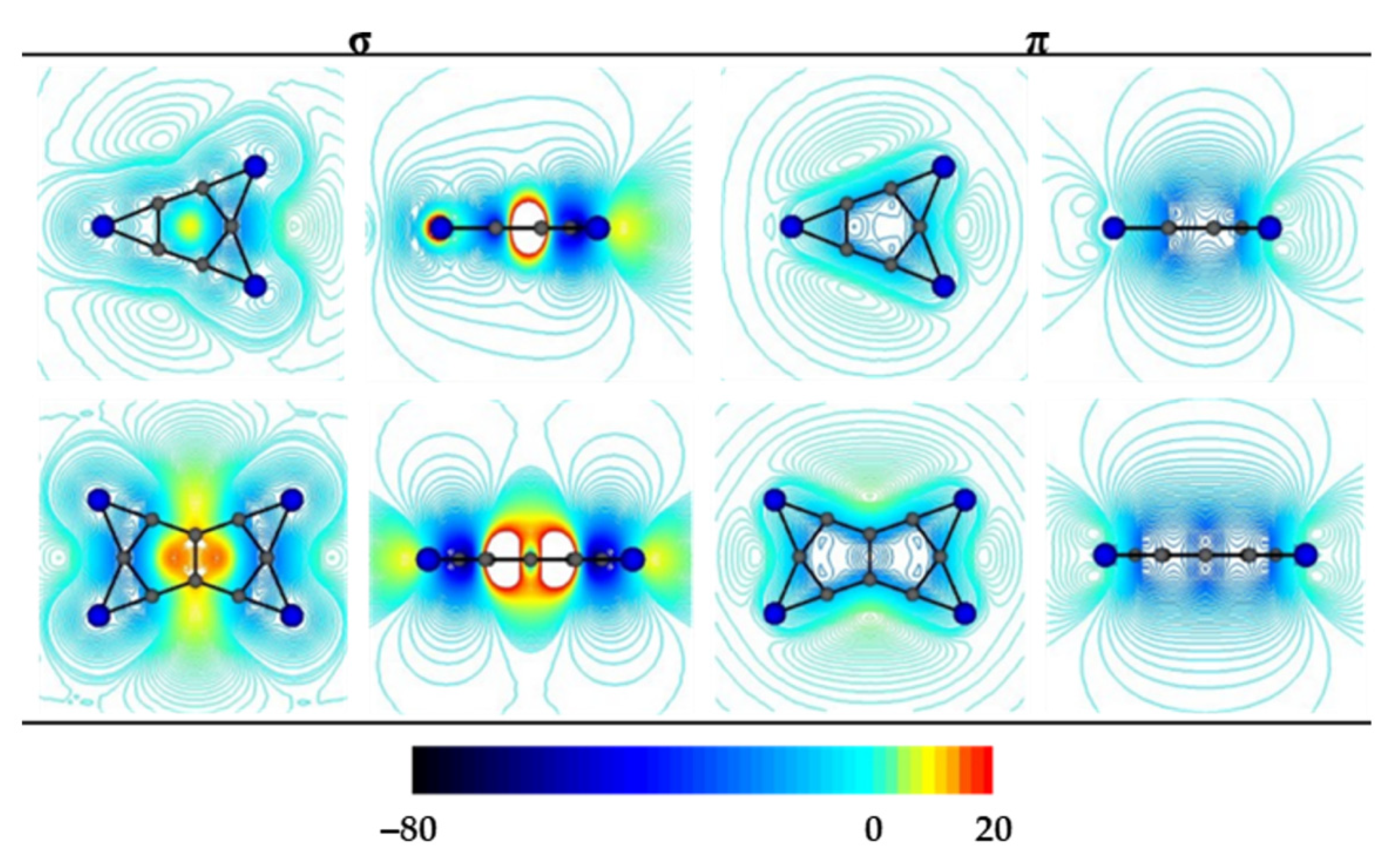
3.1. Chemical Bonding Analysis According to the ELF-LOC Method
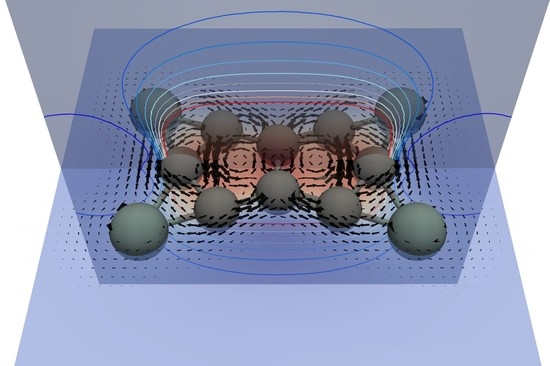
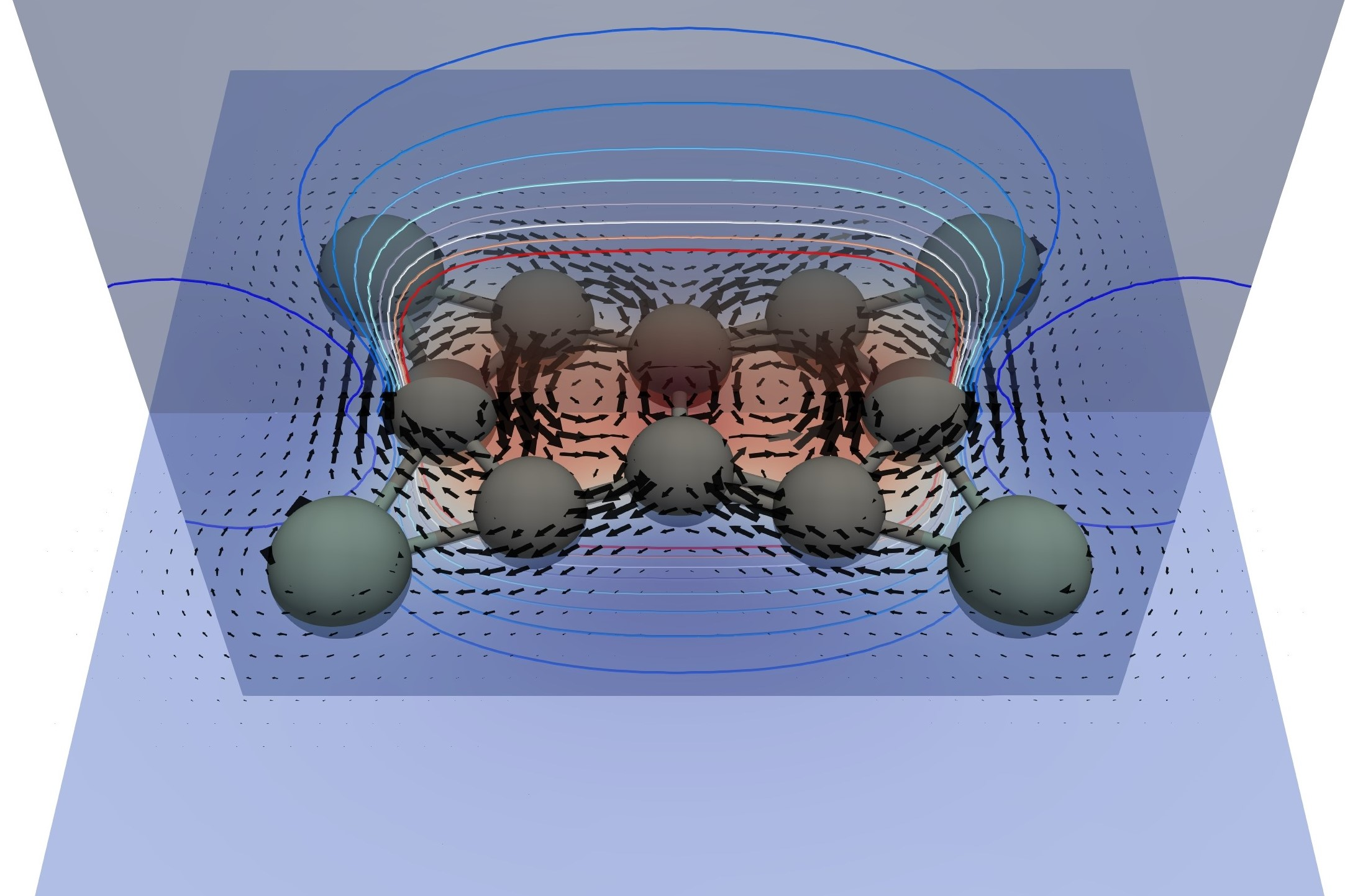
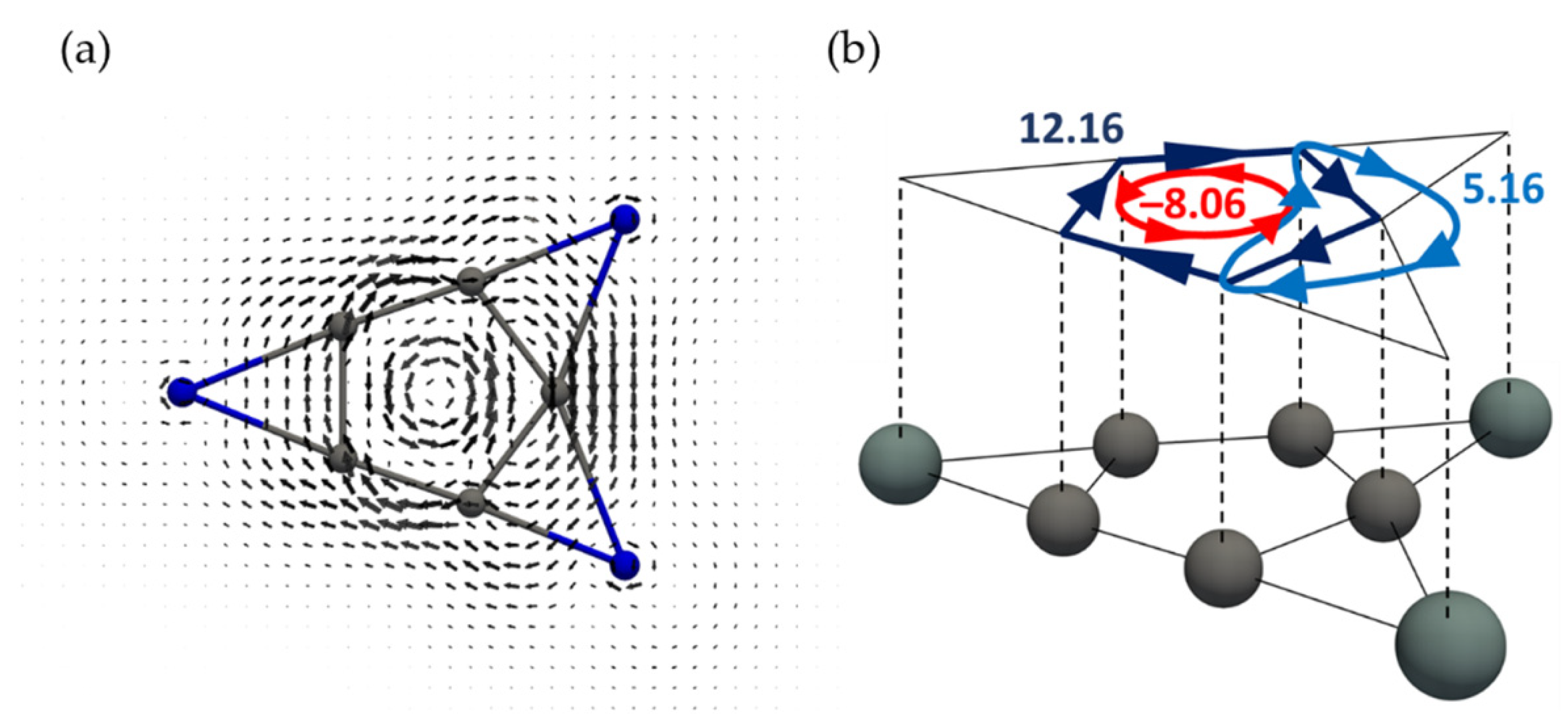
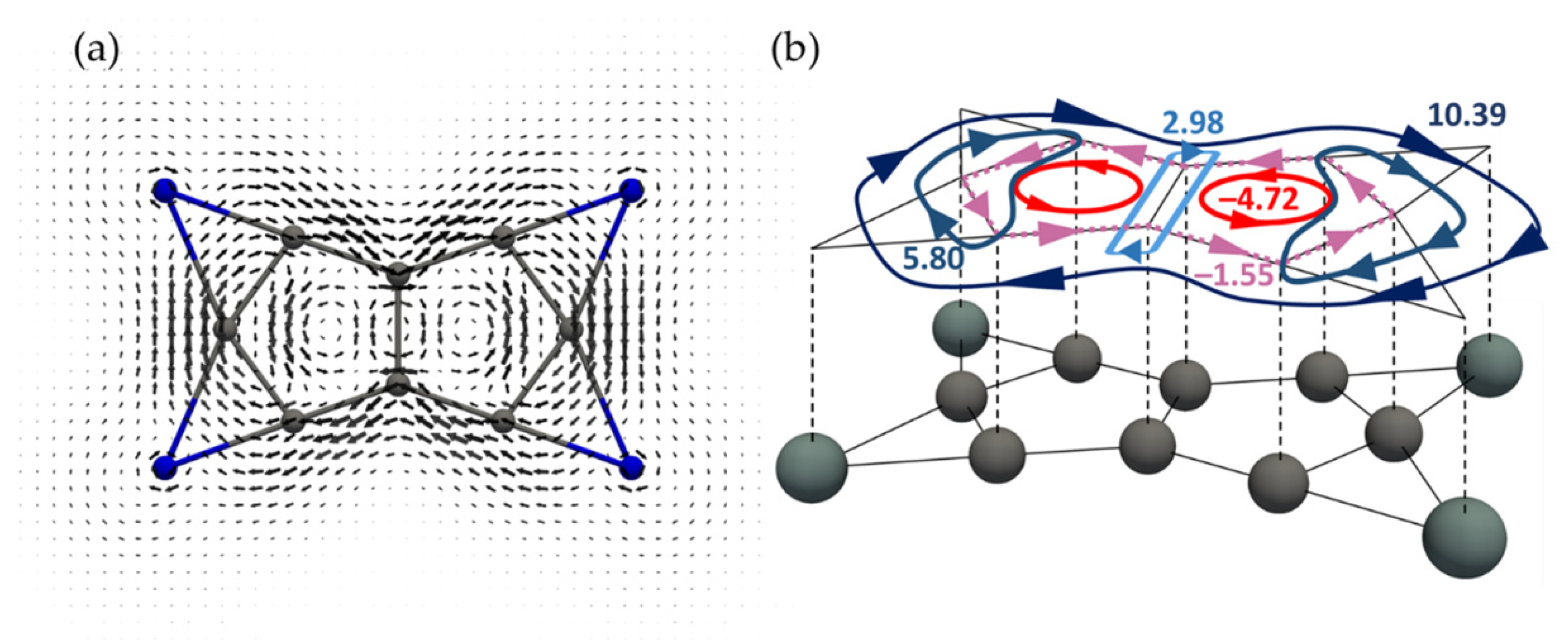
3.2. Current Density Analysis
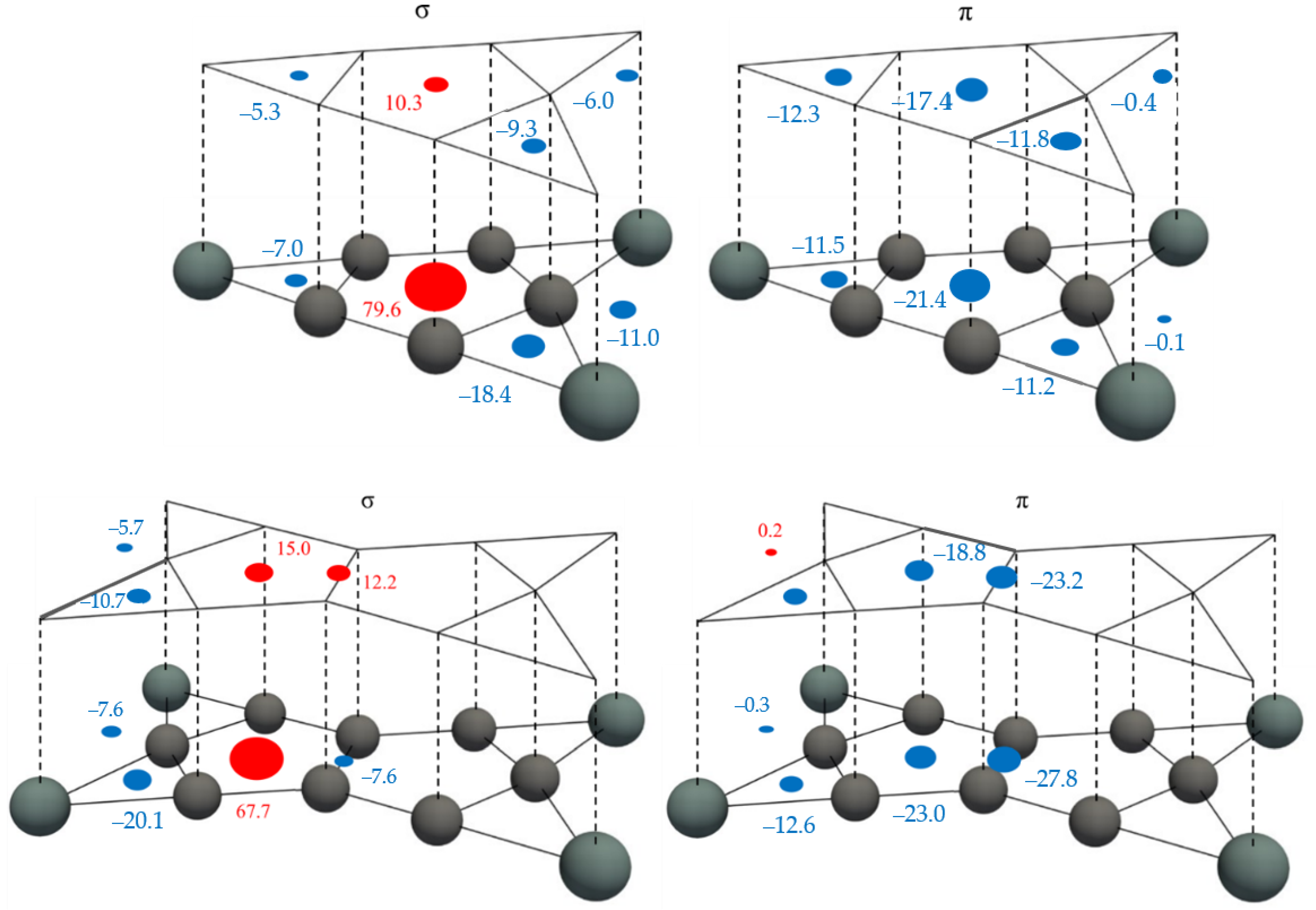
3.3. NICS Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- von Schleyer, P.R.; Jiao, H. What is aromaticity? Pure Appl. Chem. 1996, 68, 209–218. [Google Scholar] [CrossRef]
- Minkin, V.I.; Glukhovtsev, M.N.; Simkin, B.Y. Aromaticity and Antiaromaticity; John Wiley & Sons, Incorporated: Hoboken, NJ, USA, 1994; ISSN 0471593826. [Google Scholar]
- Krygowski, T.M.; Cyrański, M.K. Structural Aspects of Aromaticity. Chem. Rev. 2001, 101, 1385–1420. [Google Scholar] [CrossRef]
- Schleyer, P.V.R. Introduction: Aromaticity. Chem. Rev. 2001, 101, 1115–1118. [Google Scholar] [CrossRef]
- Boldyrev, A.I.; Wang, L.-S. All-Metal Aromaticity and Antiaromaticity. Chem. Rev. 2005, 105, 3716–3757. [Google Scholar] [CrossRef]
- Liu, C.; Popov, I.A.; Chen, Z.; Boldyrev, A.I.; Sun, Z. Aromaticity and antiaromaticity in Zintl clusters. Chem. Eur. J. 2018, 24, 14583–14597. [Google Scholar] [CrossRef]
- Boldyrev, A.I.; Wang, L.-S.S. Beyond organic chemistry: Aromaticity in atomic clusters. Phys. Chem. Chem. Phys. 2016, 18, 11589–11605. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, A.N.; Boldyrev, A.I.; Zhai, H.-J.J.; Wang, L.-S.S. All-boron aromatic clusters as potential new inorganic ligands and building blocks in chemistry. Coord. Chem. Rev. 2006, 250, 2811–2866. [Google Scholar] [CrossRef]
- Sergeeva, A.P.; Popov, I.A.; Piazza, Z.A.; Li, W.-L.; Romanescu, C.; Wang, L.-S.; Boldyrev, A.I. Understanding boron through size-selected clusters: Structure, chemical bonding, and fluxionality. Acc. Chem. Res. 2014, 47, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Mercero, J.M.; Boldyrev, A.I.; Merino, G.; Ugalde, J.M. Recent developments and future prospects of all-metal aromatic compounds. Chem. Soc. Rev. 2015, 44, 6519–6534. [Google Scholar] [CrossRef]
- Huang, X.; Zhai, H.; Kiran, B.; Wang, L. Observation of d-orbital aromaticity. Angew. Chemie 2005, 117, 7417–7420. [Google Scholar] [CrossRef]
- Tsipis, C.A. DFT study of “all-metal” aromatic compounds. Coord. Chem. Rev. 2005, 249, 2740–2762. [Google Scholar] [CrossRef]
- Rabanal-León, W.; Vásquez-Espinal, A.; Yañez, O.; Pino-Rios, R.; Arratia-Pérez, R.; Alvarez-Thon, L.; Torres-Vega, J.; Tiznado, W. Aromaticity of [M3(µ-X)3X6] 0/2-(M= Re and Tc, X= Cl, Br, I) Clusters Confirmed by Ring Current Analysis and Induced Magnetic Field. Eur. J. Inorg. Chem. 2018, 2018, 3312–3319. [Google Scholar] [CrossRef]
- Vásquez-Espinal, A.; Pino-Rios, R.; Alvarez-Thon, L.; Rabanal-León, W.A.; Torres-Vega, J.J.; Arratia-Perez, R.; Tiznado, W. New Insights into Re3(μ-Cl)3Cl6 Aromaticity. Evidence of σ-and π-Diatropicity. J. Phys. Chem. Lett. 2015, 6, 4326–4330. [Google Scholar] [CrossRef]
- Bleeke, J.R.; Behm, R.; Xie, Y.-F.; Chiang, M.Y.; Robinson, K.D.; Beatty, A.M. Synthesis, Structure, Spectroscopy, and Reactivity of a Metallabenzene1. Organometallics 1997, 16, 606–623. [Google Scholar] [CrossRef]
- Bleeke, J.R.; Xie, Y.F.; Peng, W.J.; Chiang, M. Metallabenzene: Synthesis, structure, and spectroscopy of a 1-irida-3, 5-dimethylbenzene complex. J. Am. Chem. Soc. 1989, 111, 4118–4120. [Google Scholar] [CrossRef]
- Bleeke, J.R. Metallabenzenes. Chem. Rev. 2001, 101, 1205–1228. [Google Scholar] [CrossRef] [PubMed]
- Fernández, I.; Frenking, G.; Merino, G. Aromaticity of metallabenzenes and related compounds. Chem. Soc. Rev. 2015, 44, 6452–6463. [Google Scholar] [CrossRef] [PubMed]
- Vásquez-Espinal, A.; Poater, J.; Solà, M.; Tiznado, W.; Islas, R. Testing the effectiveness of the isoelectronic substitution principle through the transformation of aromatic osmathiophene derivatives into their inorganic analogues. New J. Chem. 2017, 41, 1168–1178. [Google Scholar] [CrossRef]
- Popov, I.A.; Bozhenko, K.V.; Boldyrev, A.I. Is graphene aromatic? Nano Res. 2012, 5, 117–123. [Google Scholar] [CrossRef]
- Zubarev, D.Y.; Boldyrev, A.I. Revealing intuitively assessable chemical bonding patterns in organic aromatic molecules via adaptive natural density partitioning. J. Org. Chem. 2008, 73, 9251–9258. [Google Scholar] [CrossRef]
- Tkachenko, N.V.; Boldyrev, A.I. Multiple local σ-aromaticity of nonagermanide clusters. Chem. Sci. 2019, 10, 5761–5765. [Google Scholar] [CrossRef]
- Hoffmann, R.; Alder, R.W.; Wilcox, C.F. Planar tetracoordinate carbon. J. Am. Chem. Soc. 1970, 92, 4992–4993. [Google Scholar] [CrossRef]
- Erker, G. Planar-Tetracoordinate Carbon: Making Stable Anti-van′ t Hoff/LeBel Compounds. Comments Inorg. Chem. 1992, 13, 111–131. [Google Scholar] [CrossRef]
- Röttger, D.; Erker, G. Compounds Containing Planar-Tetracoordinate Carbon. Angew. Chemie Int. Ed. English 1997, 36, 812–827. [Google Scholar] [CrossRef]
- Siebert, W.; Gunale, A. Compounds containing a planar-tetracoordinate carbon atom as analogues of planar methane. Chem. Soc. Rev. 1999, 28, 367–371. [Google Scholar] [CrossRef]
- Keese, R. Carbon flatland: Planar tetracoordinate carbon and fenestranes. Chem. Rev. 2006, 106, 4787–4808. [Google Scholar] [CrossRef] [PubMed]
- Merino, G.; Méndez-Rojas, M.A.; Vela, A.; Heine, T. Recent advances in planar tetracoordinate carbon chemistry. J. Comput. Chem. 2007, 28, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ding, Y. Computational prediction of a global planar penta-coordinate carbon structure CAl4Ga+. Comput. Theor. Chem. 2014, 1048, 18–24. [Google Scholar] [CrossRef]
- Li, X.; Zhang, H.; Wang, L.; Geske, G.D.; Boldyrev, A.I. Pentaatomic tetracoordinate planar carbon,[CAl4] 2−: A new structural unit and its salt complexes. Angew. Chemie Int. Ed. 2000, 39, 3630–3632. [Google Scholar] [CrossRef]
- Wang, Z.-X.; von Ragué Schleyer, P. Construction principles of ”hyparenes”: Families of molecules with planar pentacoordinate carbons. Science 2001, 292, 2465–2469. [Google Scholar] [CrossRef]
- Wang, Y.; Li, F.; Li, Y.; Chen, Z. Semi-metallic Be5C2 monolayer global minimum with quasi-planar pentacoordinate carbons and negative Poisson’s ratio. Nat. Commun. 2016, 7, 11488. [Google Scholar] [CrossRef]
- Pan, S.; Cabellos, J.L.; Orozco-Ic, M.; Chattaraj, P.K.; Zhao, L.; Merino, G. Planar pentacoordinate carbon in CGa5+ derivatives. Phys. Chem. Chem. Phys. 2018, 20, 12350–12355. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; An, W.; Ito, K.; Schleyer, P.V.R.; Zeng, X.C. Planar pentacoordinate carbon in CAl5+: A global minimum. J. Am. Chem. Soc. 2008, 130, 10394–10400. [Google Scholar] [CrossRef] [PubMed]
- Vassilev-Galindo, V.; Pan, S.; Donald, K.J.; Merino, G. Planar pentacoordinate carbons. Nat. Rev. Chem. 2018, 2, 114. [Google Scholar] [CrossRef]
- Grande-Aztatzi, R.; Cabellos, J.L.; Islas, R.; Infante, I.; Mercero, J.M.; Restrepo, A.; Merino, G. Planar pentacoordinate carbons in CBe54− derivatives. Phys. Chem. Chem. Phys. 2015, 17, 4620–4624. [Google Scholar] [CrossRef]
- Zhao, X.-F.; Bian, J.-H.; Huang, F.; Yuan, C.; Wang, Q.; Liu, P.; Li, D.; Wang, X.; Wu, Y.-B. Stabilization of beryllium-containing planar pentacoordinate carbon species through attaching hydrogen atoms. RSC Adv. 2018, 8, 36521–36526. [Google Scholar] [CrossRef]
- Cui, Z.; Vassilev-Galindo, V.; Cabellos, J.L.; Osorio, E.; Orozco, M.; Pan, S.; Ding, Y.; Merino, G. Planar pentacoordinate carbon atoms embedded in a metallocene framework. Chem. Commun. 2017, 53, 138–141. [Google Scholar] [CrossRef]
- Wu, Y.-B.; Duan, Y.; Lu, G.; Lu, H.-G.; Yang, P.; von Rague Schleyer, P.; Merino, G.; Islas, R.; Wang, Z.-X. D3h CN3Be3+ and CO3Li3+: Viable planar hexacoordinate carbon prototypes. Phys. Chem. Chem. Phys. 2012, 14, 14760–14763. [Google Scholar] [CrossRef]
- Exner, K.; von Ragué Schleyer, P. Planar hexacoordinate carbon: A viable possibility. Science. 2000, 290, 1937–1940. [Google Scholar] [CrossRef]
- Parra, L.L.; Diego, L.; Yañez, O.; Inostroza, D.; Barroso, J.; Espinal, A.V.; Merino, G.; Tiznado, W. Planar Hexacoordinate Carbons: Half Covalent, Half Ionic. Angew. Chem. Int. Ed. Engl. 2021, 60, 8700–8704. [Google Scholar] [CrossRef]
- Li, Y.; Liao, Y.; Chen, Z. Be2C monolayer with quasi-planar hexacoordinate carbons: A global minimum structure. Angew. Chemie 2014, 126, 7376–7380. [Google Scholar] [CrossRef]
- Feixas, F.; Matito, E.; Poater, J.; Sola, M. Quantifying aromaticity with electron delocalisation measures. Chem. Soc. Rev. 2015, 44, 6434–6451. [Google Scholar] [CrossRef] [PubMed]
- Gomes, J.; Mallion, R.B. Aromaticity and ring currents. Chem. Rev. 2001, 101, 1349–1384. [Google Scholar] [CrossRef]
- Mitchell, R.H. Measuring aromaticity by NMR. Chem. Rev. 2001, 101, 1301–1316. [Google Scholar] [CrossRef]
- Cyranski, M.K.; Krygowski, T.M.; Katritzky, A.R.; Schleyer, P.V.R. To what extent can aromaticity be defined uniquely? J. Org. Chem. 2002, 67, 1333–1338. [Google Scholar] [CrossRef] [PubMed]
- Randic, M. Aromaticity of polycyclic conjugated hydrocarbons. Chem. Rev. 2003, 103, 3449–3606. [Google Scholar] [CrossRef]
- Balaban, A.T.; Oniciu, D.C.; Katritzky, A.R. Aromaticity as a cornerstone of heterocyclic chemistry. Chem. Rev. 2004, 104, 2777–2812. [Google Scholar] [CrossRef]
- McWeeny, R. Ring currents and proton magnetic resonance in aromatic molecules. Mol. Phys. 1958, 1, 311–321. [Google Scholar] [CrossRef]
- Pople, J.A. Molecular orbital theory of aromatic ring currents. Mol. Phys. 1958, 1, 175–180. [Google Scholar] [CrossRef]
- Pauling, L. The diamagnetic anisotropy of aromatic molecules. J. Chem. Phys. 1936, 4, 673–677. [Google Scholar] [CrossRef]
- Kumar, C.; Fliegl, H.; Sundholm, D. Relation Between Ring Currents and Hydrogenation Enthalpies for Assessing the Degree of Aromaticity. J. Phys. Chem. A 2017, 121, 7282–7289. [Google Scholar] [CrossRef]
- Ligabue, A.; Pincelli, U.; Lazzeretti, P.; Zanasi, R. Current density maps, magnetizability, and nuclear magnetic shielding tensors for anthracene, phenanthrene, and triphenylene. J. Am. Chem. Soc. 1999, 121, 5513–5518. [Google Scholar] [CrossRef]
- Monaco, G.; Fowler, P.W.; Lillington, M.; Zanasi, R. Designing paramagnetic circulenes. Angew. Chemie 2007, 119, 1921–1924. [Google Scholar] [CrossRef]
- Lazzeretti, P. Assessment of aromaticity via molecular response properties. Phys. Chem. Chem. Phys. 2004, 6, 217–223. [Google Scholar] [CrossRef]
- Schleyer, P.V.R.; Maerker, C.; Dransfeld, A.; Jiao, H.; van Eikema Hommes, N.J.R. Nucleus-independent chemical shifts: A simple and efficient aromaticity probe. J. Am. Chem. Soc. 1996, 118, 6317–6318. [Google Scholar] [CrossRef]
- Haddon, R.C. The application of the Biot-Savart law to the ring current analysis of proton chemical shifts—II: An approach to aromatic character in the annulenes. Tetrahedron 1972, 28, 3635–3655. [Google Scholar] [CrossRef]
- Báez-Grez, R.; Rabanal-León, W.A.; Alvarez-Thon, L.; Ruiz, L.; Tiznado, W.; Pino-Rios, R. Aromaticity in heterocyclic analogues of benzene: Dissected NICS and current density analysis. J. Phys. Org. Chem. 2019, 32, e3823. [Google Scholar] [CrossRef]
- Báez-Grez, R.; Ruiz, L.; Pino-Rios, R.; Tiznado, W. Which NICS method is most consistent with ring current analysis? Assessment in simple monocycles. RSC Adv. 2018, 8, 13446–13453. [Google Scholar] [CrossRef]
- Poater, J.; Solà, M.; Viglione, R.G.; Zanasi, R. Local aromaticity of the six-membered rings in pyracylene. A difficult case for the NICS indicator of aromaticity. J. Org. Chem. 2004, 69, 7537–7542. [Google Scholar] [CrossRef] [PubMed]
- Sundholm, D.; Berger, R.J.F.; Fliegl, H. Analysis of the magnetically induced current density of molecules consisting of annelated aromatic and antiaromatic hydrocarbon rings. Phys. Chem. Chem. Phys. 2016, 18, 15934–15942. [Google Scholar] [CrossRef][Green Version]
- Yañez, O.; Vásquez-Espinal, A.; Báez-Grez, R.; Rabanal-León, W.A.; Osorio, E.; Ruiz, L.; Tiznado, W. Carbon rings decorated with group 14 elements: New aromatic clusters containing planar tetracoordinate carbon. New J. Chem. 2019, 43, 6781–6785. [Google Scholar] [CrossRef]
- Yañez, O.; Báez-Grez, R.; Garza, J.; Pan, S.; Barroso, J.; Vásquez-Espinal, A.; Merino, G.; Tiznado, W. Embedding a Planar Hypercoordinate Carbon Atom into a [4n+ 2] π-System. ChemPhysChem 2020, 21, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Yañez, O.; Vásquez-Espinal, A.; Pino-Rios, R.; Ferraro, F.; Pan, S.; Osorio, E.; Merino, G.; Tiznado, W. Exploiting electronic strategies to stabilize a planar tetracoordinate carbon in cyclic aromatic hydrocarbons. Chem. Commun. 2017, 53, 12112–12115. [Google Scholar] [CrossRef]
- Becke, A.D. Becke’s three parameter hybrid method using the LYP correlation functional. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent molecular-orbital methods. IX. An extended Gaussian-type basis for molecular-orbital studies of organic molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self—consistent molecular orbital methods. XII. Further extensions of Gaussian—type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revis. C.01; Gaussian, Inc.: Wallingford, CT, USA.
- Oña, O.B.; Alcoba, D.R.; Tiznado, W.; Torre, A.; Lain, L. An orbital localization criterion based on the topological analysis of the electron localization function. Int. J. Quantum Chem. 2013, 113, 1401–1408. [Google Scholar] [CrossRef]
- Oña, O.B.; Alcoba, D.R.; Torre, A.; Lain, L.; Torres-Vega, J.J.; Tiznado, W. Orbital Localization Criterion as a Complementary Tool in the Bonding Analysis by Means of Electron Localization Function: Study of the Sin(BH)5-n2- (n = 0–5) Clusters. J. Phys. Chem. A 2013, 117, 12953–12958. [Google Scholar] [CrossRef]
- Alcoba, D.R.; Oña, O.B.; Torre, A.; Lain, L.; Tiznado, W. An orbital localization criterion based on the topological analysis of the electron localization function at correlated level. Int. J. Quantum Chem. 2018, 118, e25588. [Google Scholar] [CrossRef]
- Oña, O.B.; Torres-Vega, J.J.; Torre, A.; Lain, L.; Alcoba, D.R.; Vásquez-Espinal, A.; Tiznado, W. Chemical bonding analysis in boron clusters by means of localized orbitals according to the electron localization function topology. Theor. Chem. Acc. 2015, 134, 1–9. [Google Scholar] [CrossRef]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Noury, S.; Krokidis, X.; Fuster, F.; Silvi, B. Computational tools for the electron localization function topological analysis. Comput. Chem. 1999, 23, 597–604. [Google Scholar] [CrossRef]
- Fliegl, H.; Taubert, S.; Lehtonen, O.; Sundholm, D. The gauge including magnetically induced current method. Phys. Chem. Chem. Phys. 2011, 13, 20500–20518. [Google Scholar] [CrossRef] [PubMed]
- Jusélius, J.; Sundholm, D.; Gauss, J. Calculation of current densities using gauge-including atomic orbitals. J. Chem. Phys. 2004, 121, 3952–3963. [Google Scholar] [CrossRef]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Ahrens, J.; Geveci, B.; Law, C. Paraview: An end-user tool for large data visualization. Vis. Handb. 2005, 717. [Google Scholar] [CrossRef]
- Ayachit, U. The ParaView Guide: A Parallel Visualization Application; Kitware, Inc.: Saratoga County, NY, USA, 2015; ISBN 1930934300. [Google Scholar]
- Abramowitz, M. Handbook of Mathematical Functions, With Formulas, Graphs, and Mathematical Tables; Dover Publications, Inc.: New York, NY, USA, 1974; ISBN 0486612724. [Google Scholar]
- Sundholm, D.; Fliegl, H.; Berger, R.J.F. Calculations of magnetically induced current densities: Theory and applications. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2016, 6, 639–678. [Google Scholar] [CrossRef]
- Bohmann, J.A.; Weinhold, F.; Farrar, T.C. Natural chemical shielding analysis of nuclear magnetic resonance shielding tensors from gauge-including atomic orbital calculations. J. Chem. Phys. 1997, 107, 1173–1184. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Landis, C.R.; Weinhold, F. Natural Bond Orbital Analysis Program: NBO 6.0; Theoretical Chemistry Institute and Department of Chemistry, University of Wisconsin: Madison, WI, USA, 2013. [Google Scholar]
- Childs, H.; Brugger, E.; Whitlock, B.; Meredith, J.; Ahern, S.; Pugmire, D.; Biagas, K.; Miller, M.; Harrison, C.; Weber, G.H.; et al. VisIt: An End-User Tool For Visualizing and Analyzing Very Large Data. In High Performance Visualization—Enabling Extreme-Scale Scientific Insight; CRC Press: Boca Raton, FL, USA, 2012; pp. 357–372. [Google Scholar]
- Llusar, R.; Beltrán, A.; Andrés, J.; Noury, S.; Silvi, B. Topological analysis of electron density in depleted homopolar chemical bonds. J. Comput. Chem. 1999, 20, 1517–1526. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Hückel, E. Quantentheoretische Beiträge zum Benzolproblem. Zeitschrift Phys. 1931, 70, 204–286. [Google Scholar] [CrossRef]
- Hückel, E. Quanstentheoretische Beiträge zum Benzolproblem. Zeitschrift Phys. 1931, 72, 310–337. [Google Scholar] [CrossRef]
- Hückel, E. Zur Quantentheorie der Doppelbindung. Zeitschrift Phys. 1930, 60, 423–456. [Google Scholar] [CrossRef]
- Lazzeretti, P. Ring currents. Prog. Nucl. Magn. Reson. Spectrosc. 2000, 36, 1–88. [Google Scholar] [CrossRef]
- Juse, J.; Sundholm, D. Ab initio determination of the induced ring current in aromatic molecules. Phys. Chem. Chem. Phys. 1999, 1, 3429–3435. [Google Scholar]
- Inostroza, D.; García, V.; Yañez, O.; Torres-Vega, J.J.; Vásquez-Espinal, A.; Pino-Rios, R.; Báez-Grez, R.; Tiznado, W. On the NICS limitations to predict local and global current pathways in polycyclic systems. New J. Chem. 2021, 45, 8345–8351. [Google Scholar] [CrossRef]
- Aihara, J. Circuit resonance energy: A key quantity that links energetic and magnetic criteria of aromaticity. J. Am. Chem. Soc. 2006, 128, 2873–2879. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torres-Vega, J.J.; Alcoba, D.R.; Oña, O.B.; Vásquez-Espinal, A.; Báez-Grez, R.; Lain, L.; Torre, A.; García, V.; Tiznado, W. Analysis of Local and Global Aromaticity in Si3C5 and Si4C8 Clusters. Aromatic Species Containing Planar Tetracoordinate Carbon. Chemistry 2021, 3, 1101-1112. https://doi.org/10.3390/chemistry3040080
Torres-Vega JJ, Alcoba DR, Oña OB, Vásquez-Espinal A, Báez-Grez R, Lain L, Torre A, García V, Tiznado W. Analysis of Local and Global Aromaticity in Si3C5 and Si4C8 Clusters. Aromatic Species Containing Planar Tetracoordinate Carbon. Chemistry. 2021; 3(4):1101-1112. https://doi.org/10.3390/chemistry3040080
Chicago/Turabian StyleTorres-Vega, Juan J., Diego R. Alcoba, Ofelia B. Oña, Alejandro Vásquez-Espinal, Rodrigo Báez-Grez, Luis Lain, Alicia Torre, Víctor García, and William Tiznado. 2021. "Analysis of Local and Global Aromaticity in Si3C5 and Si4C8 Clusters. Aromatic Species Containing Planar Tetracoordinate Carbon" Chemistry 3, no. 4: 1101-1112. https://doi.org/10.3390/chemistry3040080
APA StyleTorres-Vega, J. J., Alcoba, D. R., Oña, O. B., Vásquez-Espinal, A., Báez-Grez, R., Lain, L., Torre, A., García, V., & Tiznado, W. (2021). Analysis of Local and Global Aromaticity in Si3C5 and Si4C8 Clusters. Aromatic Species Containing Planar Tetracoordinate Carbon. Chemistry, 3(4), 1101-1112. https://doi.org/10.3390/chemistry3040080