
Fully Biobased Reactive Extrusion of Biocomposites Based on PLA Blends and Hazelnut Shell Powders (HSP)

 ,
,  ,
,  and
and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
3. Results
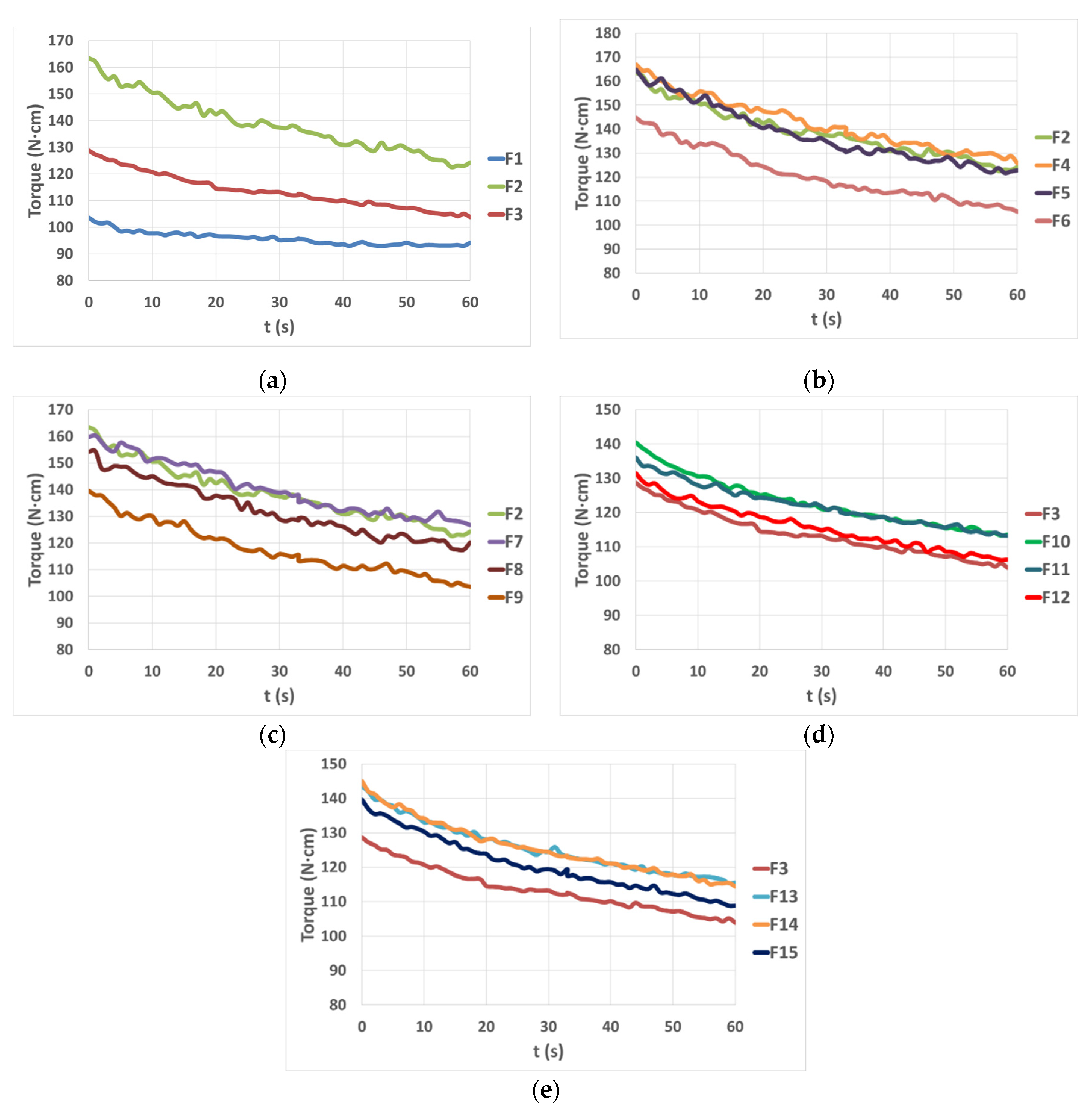
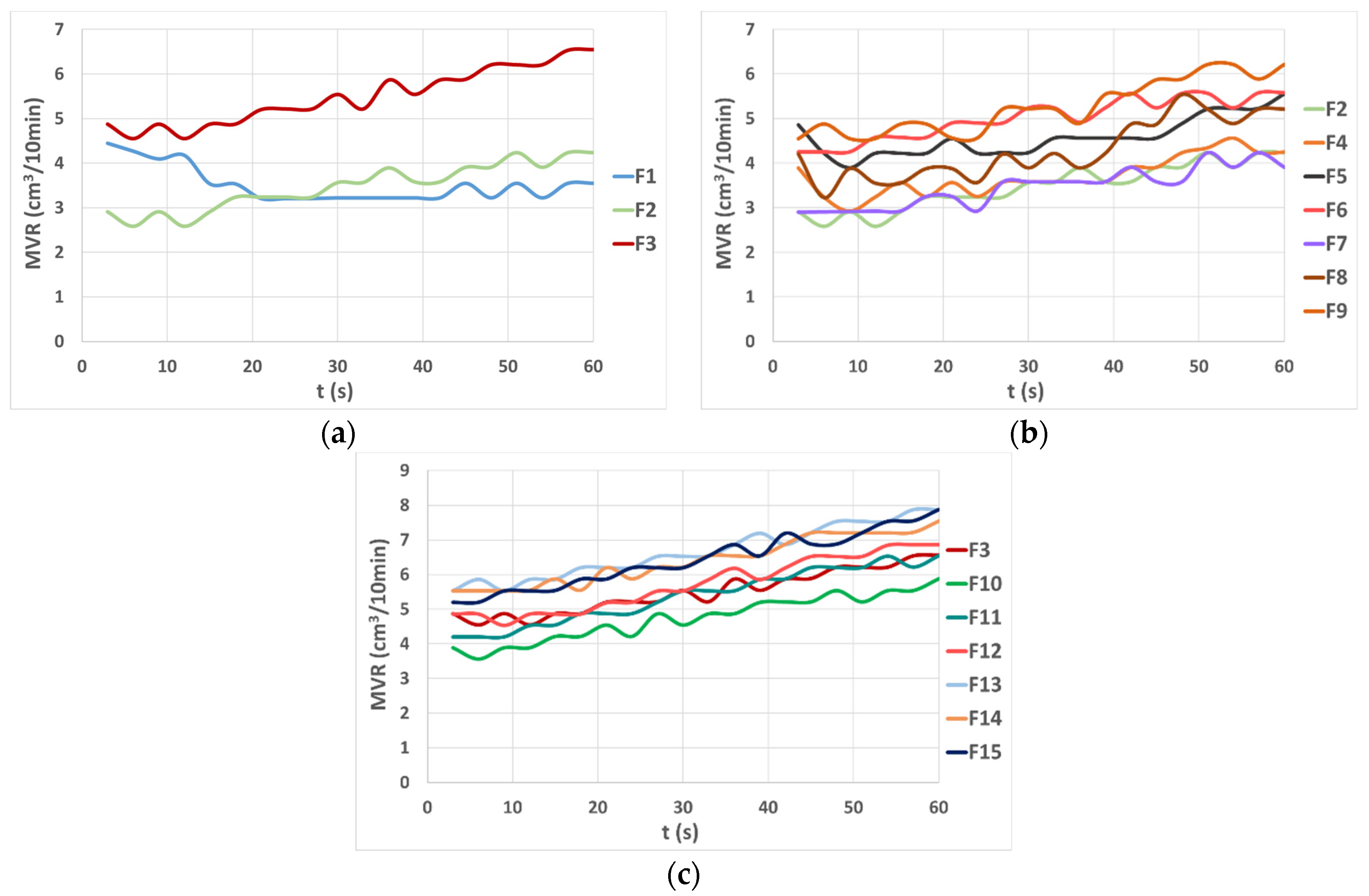
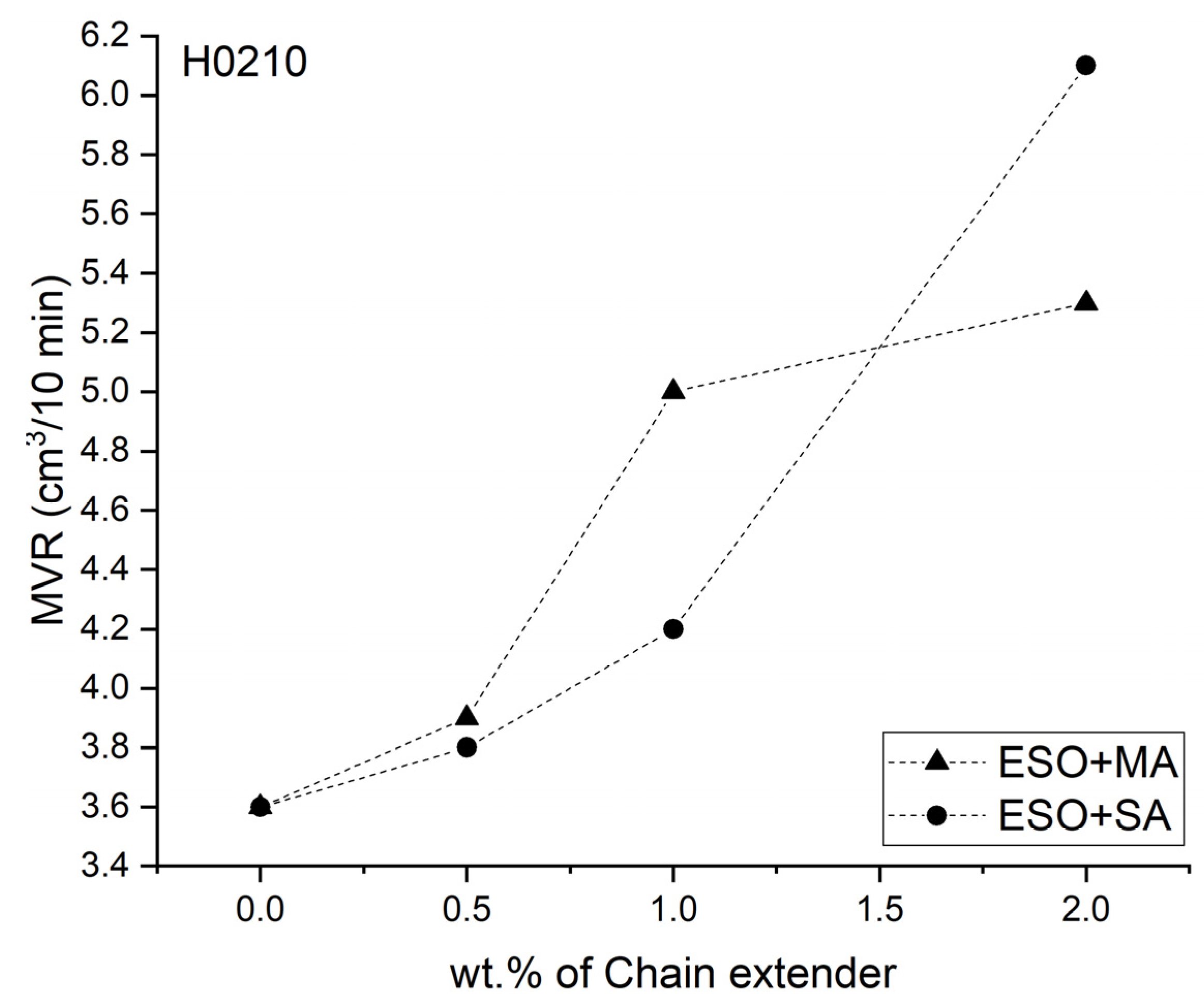
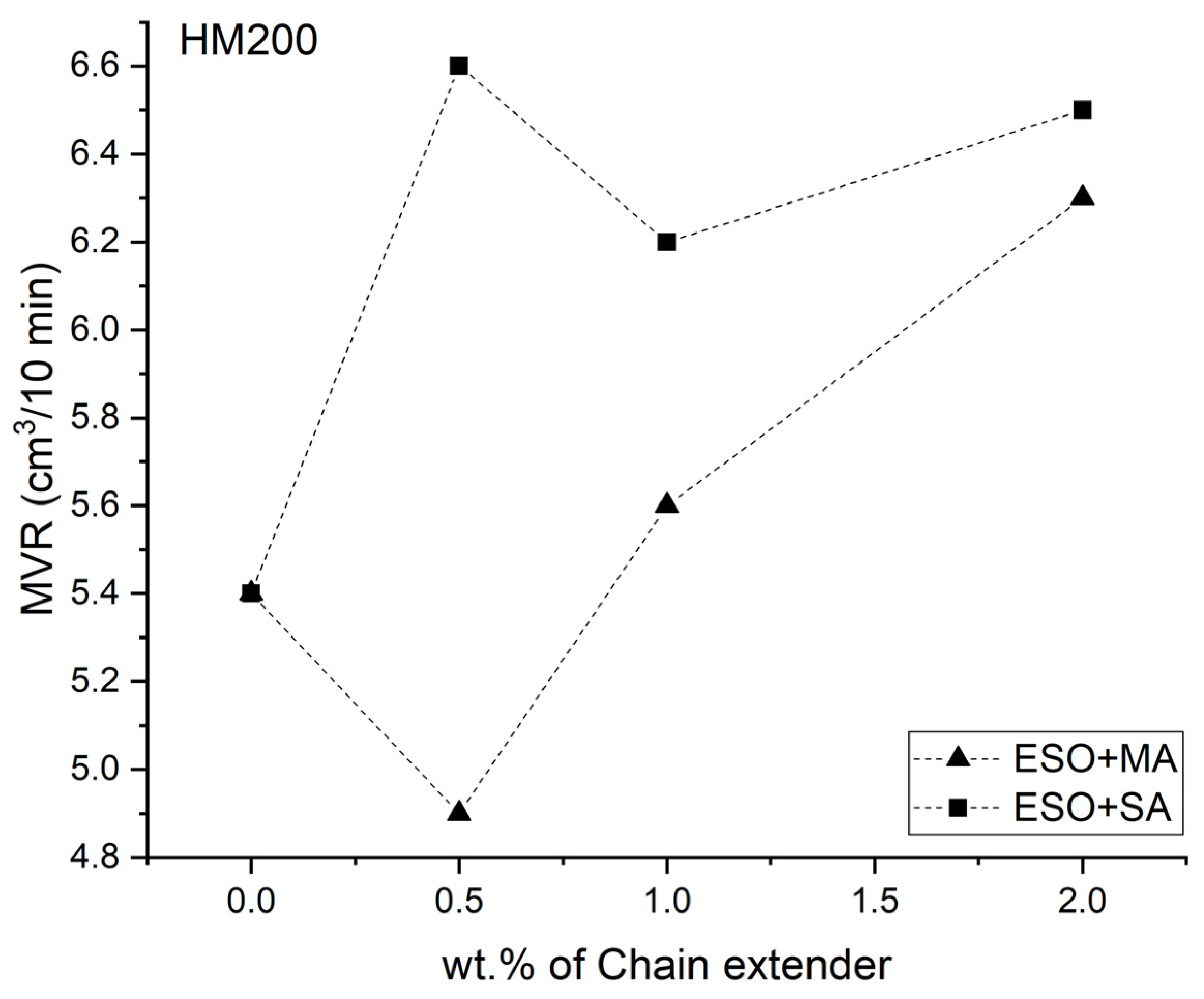
3.1. Melt Flow Analysis
3.2. Thermal Characterization
3.3. Mechanical Characterization
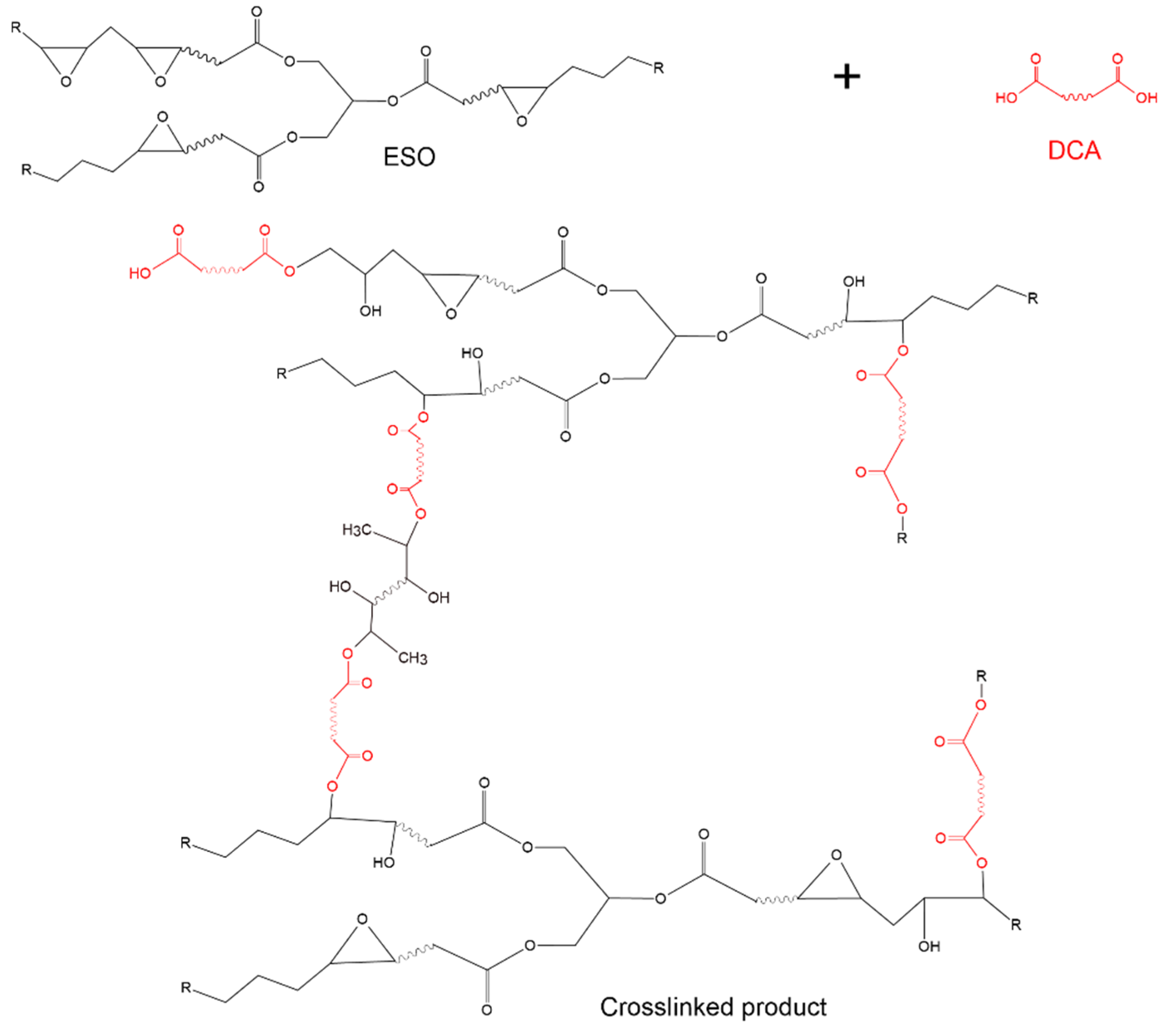
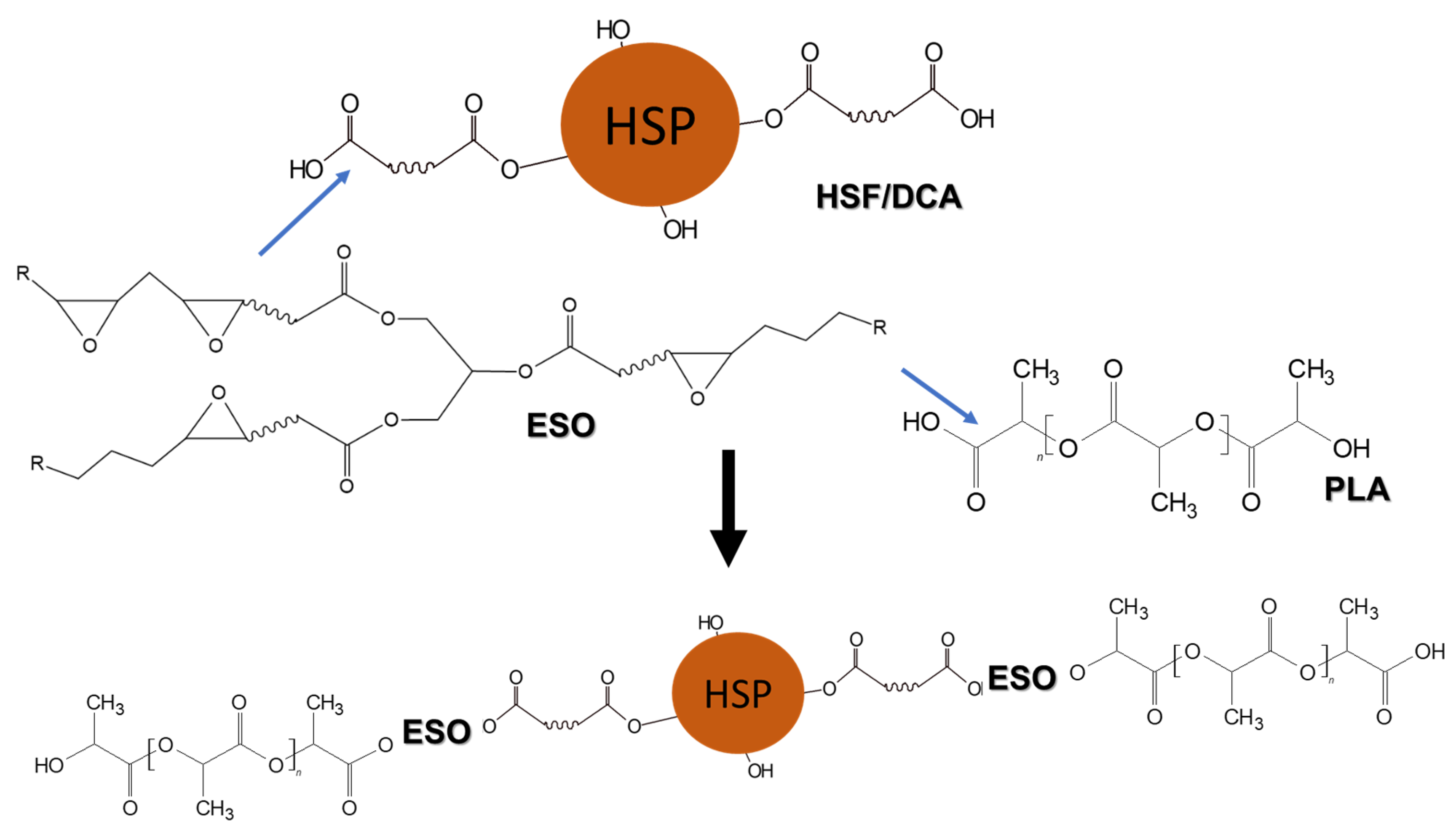
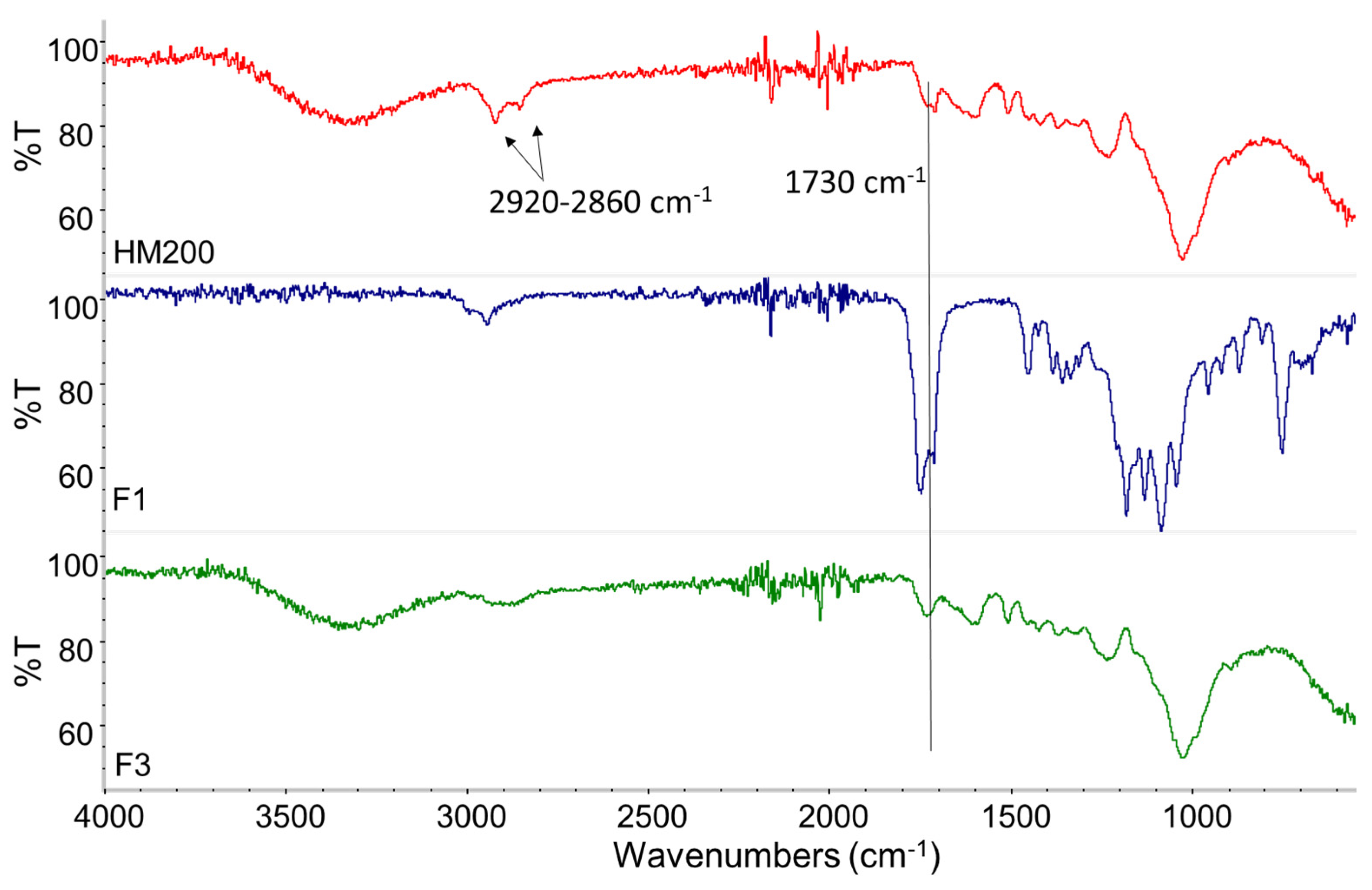
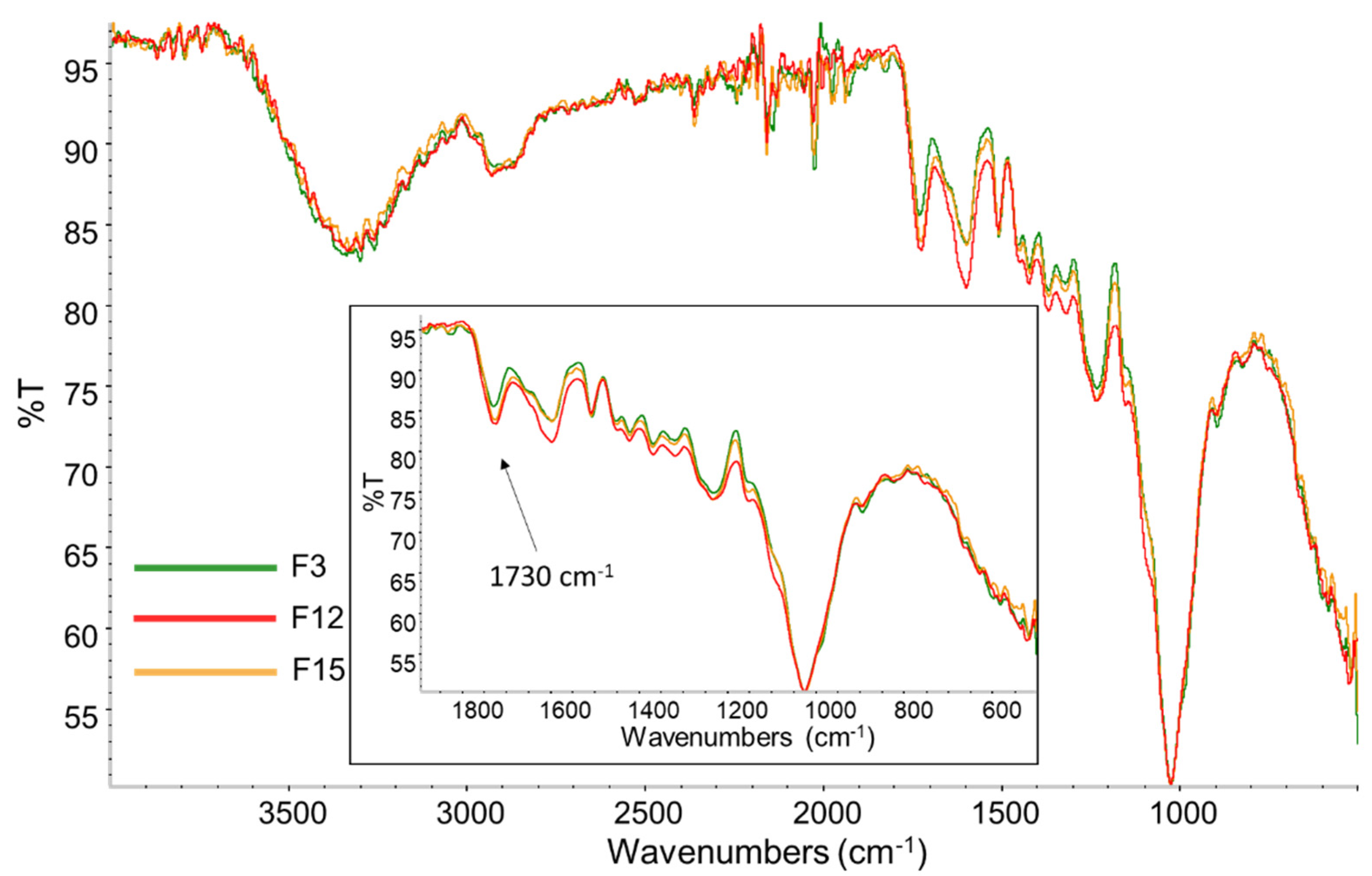
3.4. Reactivity Study
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Muhammad Shamsuddin, I. Bioplastics as Better Alternative to Petroplastics and Their Role in National Sustainability: A Review. Adv. Biosci. Bioeng. 2017, 5, 63. [Google Scholar] [CrossRef] [Green Version]
- Hottle, T.A.; Bilec, M.M.; Landis, A.E. Biopolymer production and end of life comparisons using life cycle assessment. Resour. Conserv. Recycl. 2017, 122, 295–306. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, M.; John, B. Synthetic biopolymer nanocomposites for tissue engineering scaffolds. Prog. Polym. Sci. 2013, 38, 1487–1503. [Google Scholar] [CrossRef]
- Azimi, B.; Maleki, H.; Zavagna, L.; la Ossa, J.G.; Linari, S.; Lazzeri, A.; Danti, S. Bio-Based Electrospun Fibers for Wound Healing. J. Funct. Biomater. 2020, 11, 67. [Google Scholar] [CrossRef] [PubMed]
- Coltelli, M.-B.; Danti, S.; De Clerck, K.; Lazzeri, A.; Morganti, P. Pullulan for Advanced Sustainable Body- and Skin-Contact Applications. J. Funct. Biomater. 2020, 11, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristallini, C.; Danti, S.; Azimi, B.; Tempesti, V.; Ricci, C.; Ventrelli, L.; Cinelli, P.; Barbani, N.; Lazzeri, A. Multifunctional Coatings for Robotic Implanted Device. Int. J. Mol. Sci. 2019, 20, 5126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, J.; Winkeljann, B.; Lieleg, O. Biopolymer-Based Coatings: Promising Strategies to Improve the Biocompatibility and Functionality of Materials Used in Biomedical Engineering. Adv. Mater. Interfaces 2020, 7, 2000850. [Google Scholar] [CrossRef]
- Rusa, C.C.; Shuai, X.; Shin, I.D.; Bullions, T.A.; Wei, M.; Porbeni, F.E.; Lu, J.; Huang, L.; Fox, J.; Tonelli, A.E. Controlling the Behaviors of Biodegradable/Bioabsorbable Polymers with Cyclodextrins. J. Polym. Environ. 2004, 12, 157–163. [Google Scholar] [CrossRef]
- Kaplan, D.L. Introduction to Biopolymers from Renewable Resources. In Biopolymers from Renewable Resources; Kaplan, D.L., Ed.; Springer: Berlin/Heidelberg, Germany, 1998; pp. 1–29. ISBN 978-3-662-03680-8. [Google Scholar]
- Lezak, E.; Kulinski, Z.; Masirek, R.; Piorkowska, E.; Pracella, M.; Gadzinowska, K. Mechanical and Thermal Properties of Green Polylactide Composites with Natural Fillers. Macromol. Biosci. 2008, 8, 1190–1200. [Google Scholar] [CrossRef]
- Jappes, J.T.W.; Senthil Muthu Kumar, T.; Rajini, N.; Muthulaksmi, L.; Jawaid, M.; Varadharajulu, A. Preparation and Properties of Cellulose/Tamarind Nut Powder Green Composites. J. Nat. Fibers 2018, 15, 11–20. [Google Scholar] [CrossRef]
- Badia, J.D.; Strömberg, E.; Kittikorn, T.; Ek, M.; Karlsson, S.; Ribes-Greus, A. Relevant factors for the eco-design of polylactide/sisal biocomposites to control biodegradation in soil in an end-of-life scenario. Polym. Degrad. Stab. 2017, 143, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Brebu, M. Environmental Degradation of Plastic Composites with Natural Fillers—A Review. Polymers 2020, 12, 166. [Google Scholar] [CrossRef] [Green Version]
- Shah, A.A.; Hasan, F.; Hameed, A.; Ahmed, S. Biological degradation of plastics: A comprehensive review. Biotechnol. Adv. 2008, 26, 246–265. [Google Scholar] [CrossRef]
- Ledbetter, C.A. Shell cracking strength in almond (Prunus dulcis [Mill.] D.A. Webb.) and its implication in uses as a value-added product. Bioresour. Technol. 2008, 99, 5567–5573. [Google Scholar] [CrossRef]
- Lin, C.S.K.; Pfaltzgraff, L.A.; Herrero-Davila, L.; Mubofu, E.B.; Abderrahim, S.; Clark, J.H.; Koutinas, A.A.; Kopsahelis, N.; Stamatelatou, K.; Dickson, F.; et al. Food waste as a valuable resource for the production of chemicals, materials and fuels. Current situation and global perspective. Energy Environ. Sci. 2013, 6, 426–464. [Google Scholar] [CrossRef]
- Essabir, H.; Achaby, M.E.I.; Hilali, E.I.M.; Bouhfid, R.; Qaiss, A. Morphological, Structural, Thermal and Tensile Properties of High Density Polyethylene Composites Reinforced with Treated Argan Nut Shell Particles. J. Bionic Eng. 2015, 12, 129–141. [Google Scholar] [CrossRef]
- Jannat, N.; Latif Al-Mufti, R.; Hussien, A.; Abdullah, B.; Cotgrave, A. Utilisation of nut shell wastes in brick, mortar and concrete: A review. Constr. Build. Mater. 2021, 293, 123546. [Google Scholar] [CrossRef]
- Ebikade, E.O.; Sadula, S.; Gupta, Y.; Vlachos, D.G. A review of thermal and thermocatalytic valorization of food waste. Green Chem. 2021, 23, 2806–2833. [Google Scholar] [CrossRef]
- Rahib, Y.; Elorf, A.; Sarh, B.; Ezahri, M.; Rahib, Y.; Bonnamy, S. Experimental Analysis on Thermal Characteristics of Argan Nut Shell (ANS) Biomass as a Green Energy Resource. Int. J. Renew. Energy Res. 2019, 9, 1606–1615. [Google Scholar]
- Rahib, Y.; Boushaki, T.; Sarh, B.; Chaoufi, J. Combustion and pollutant emission characteristics of argan nut shell (ANS) biomass. Fuel Process. Technol. 2021, 213, 106665. [Google Scholar] [CrossRef]
- Rawlins, C.H.; Sadeghi, F. Experimental Study on Oil Removal in Nutshell Filters for Produced-Water Treatment. SPE Prod. Oper. 2017, 33, 145–153. [Google Scholar] [CrossRef]
- Moccia, F.; Agustin-Salazar, S.; Berg, A.-L.; Setaro, B.; Micillo, R.; Pizzo, E.; Weber, F.; Gamez-Meza, N.; Schieber, A.; Cerruti, P.; et al. Pecan (Carya illinoinensis (Wagenh.) K. Koch) Nut Shell as an Accessible Polyphenol Source for Active Packaging and Food Colorant Stabilization. ACS Sustain. Chem. Eng. 2020, 8, 6700–6712. [Google Scholar] [CrossRef]
- Haykiri-Acma, H.; Yaman, S.; Kucukbayrak, S. Comparison of the thermal reactivities of isolated lignin and holocellulose during pyrolysis. Fuel Process. Technol. 2010, 91, 759–764. [Google Scholar] [CrossRef]
- Esposito, T.; Sansone, F.; Franceschelli, S.; Del Gaudio, P.; Picerno, P.; Aquino, R.P.; Mencherini, T. Hazelnut (Corylus avellana L.) Shells Extract: Phenolic Composition, Antioxidant Effect and Cytotoxic Activity on Human Cancer Cell Lines. Int. J. Mol. Sci. 2017, 18, 392. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Sismour, E.N.; Parry, J.; Hanna, M.A.; Li, H. Nutritional composition and antioxidant activity in hazelnut shells from US-grown cultivars. Int. J. Food Sci. Technol. 2012, 47, 940–946. [Google Scholar] [CrossRef]
- Balart, J.F.; García-Sanoguera, D.; Balart, R.; Boronat, T.; Sánchez-Nacher, L. Manufacturing and properties of biobased thermoplastic composites from poly(lactid acid) and hazelnut shell wastes. Polym. Compos. 2018, 39, 848–857. [Google Scholar] [CrossRef]
- Teuber, L.; Osburg, V.-S.; Toporowski, W.; Militz, H.; Krause, A. Wood polymer composites and their contribution to cascading utilisation. J. Clean. Prod. 2016, 110, 9–15. [Google Scholar] [CrossRef]
- Keener, T.J.; Stuart, R.K.; Brown, T.K. Maleated coupling agents for natural fibre composites. Compos. Part A Appl. Sci. Manuf. 2004, 35, 357–362. [Google Scholar] [CrossRef]
- Maldas, D.; Kokta, B.V.; Daneault, C. Influence of coupling agents and treatments on the mechanical properties of cellulose fiber–polystyrene composites. J. Appl. Polym. Sci. 1989, 37, 751–775. [Google Scholar] [CrossRef]
- Aliotta, L.; Lazzeri, A. A proposal to modify the Kelly-Tyson equation to calculate the interfacial shear strength (IFSS) of composites with low aspect ratio fibers. Compos. Sci. Technol. 2020, 186, 107920. [Google Scholar] [CrossRef]
- Tillet, G.; Boutevin, B.; Ameduri, B. Chemical reactions of polymer crosslinking and post-crosslinking at room and medium temperature. Prog. Polym. Sci. 2011, 36, 191–217. [Google Scholar] [CrossRef]
- Lee, T.; Lee, C.; Cho, S.; Lee, D.; Yoon, K.-B. Enhancement of physical properties of thermoplastic polyether-ester elastomer by reactive extrusion with chain extender. Polym. Bull. 2011, 66, 979–990. [Google Scholar] [CrossRef]
- Frenz, V.; Scherzer, D.; Villalobos, M.; Awojulu, A.; Edison, M.; Meer, R. Multifunctional Polymers as Chain Extenders and Compatibilizers for Polycondensates and Biopolymers. In Proceedings of the Technical Papers, Regional Technical Conference-Society of Plastics Engineers, Wyandotte, MI, USA, 4–8 May 2008; Volume 3. [Google Scholar]
- Kahraman, Y.; Özdemir, B.; Kılıç, V.; Goksu, Y.A.; Nofar, M. Super toughened and highly ductile PLA/TPU blend systems by in situ reactive interfacial compatibilization using multifunctional epoxy-based chain extender. J. Appl. Polym. Sci. 2021, 138, 50457. [Google Scholar] [CrossRef]
- de CD Nunes, E.; de Souza, A.G.; dos S Rosa, D. Use of a chain extender as a dispersing agent of the CaCO3 into PBAT matrix. J. Compos. Mater. 2020, 54, 1373–1382. [Google Scholar] [CrossRef]
- Liu, C.; Jia, Y.; He, A. Preparation of Higher Molecular Weight Poly (L-lactic Acid) by Chain Extension. Int. J. Polym. Sci. 2013, 2013, 315917. [Google Scholar] [CrossRef] [Green Version]
- Beltrán, F.R.; Infante, C.; de la Orden, M.U.; Martínez Urreaga, J. Mechanical recycling of poly(lactic acid): Evaluation of a chain extender and a peroxide as additives for upgrading the recycled plastic. J. Clean. Prod. 2019, 219, 46–56. [Google Scholar] [CrossRef]
- Pilla, S.; Kramschuster, A.; Yang, L.; Lee, J.; Gong, S.; Turng, L.-S. Microcellular injection-molding of polylactide with chain-extender. Mater. Sci. Eng. C 2009, 29, 1258–1265. [Google Scholar] [CrossRef]
- Arruda, L.C.; Magaton, M.; Bretas, R.E.S.; Ueki, M.M. Influence of chain extender on mechanical, thermal and morphological properties of blown films of PLA/PBAT blends. Polym. Test. 2015, 43, 27–37. [Google Scholar] [CrossRef]
- Najafi, N.; Heuzey, M.C.; Carreau, P.J. Polylactide (PLA)-clay nanocomposites prepared by melt compounding in the presence of a chain extender. Compos. Sci. Technol. 2012, 72, 608–615. [Google Scholar] [CrossRef]
- Li, X.; Ai, X.; Pan, H.; Yang, J.; Gao, G.; Zhang, H.; Yang, H.; Dong, L. The morphological, mechanical, rheological, and thermal properties of PLA/PBAT blown films with chain extender. Polym. Adv. Technol. 2018, 29, 1706–1717. [Google Scholar] [CrossRef]
- Anakabe, J.; Zaldua Huici, A.M.; Eceiza, A.; Arbelaiz, A. The effect of the addition of poly(styrene-co-glycidyl methacrylate) copolymer on the properties of polylactide/poly(methyl methacrylate) blend. J. Appl. Polym. Sci. 2016, 133. [Google Scholar] [CrossRef]
- Chaiwutthinan, P.; Leejarkpai, T.; Kashima, D.P.; CHUAYJULJIT, S. Poly(Lactic Acid)/Poly(Butylene Succinate) Blends Filled with Epoxy Functionalised Polymeric Chain Extender. Adv. Mater. Res. 2013, 664, 644–648. [Google Scholar] [CrossRef]
- Cao, Z.; Lu, Y.; Zhang, C.; Zhang, Q.; Zhou, A.; Hu, Y.; Wu, D.; Tao, G.; Gong, F.; Ma, W.; et al. Effects of the chain-extender content on the structure and performance of poly(lactic acid)–poly(butylene succinate)–microcrystalline cellulose composites. J. Appl. Polym. Sci. 2017, 134. [Google Scholar] [CrossRef]
- Bobade, S.K.; Paluvai, N.R.; Mohanty, S.; Nayak, S.K. Bio-Based Thermosetting Resins for Future Generation: A Review. Polym. Plast. Technol. Eng. 2016, 55, 1863–1896. [Google Scholar] [CrossRef]
- Coltelli, M.-B.; Bertolini, A.; Aliotta, L.; Gigante, V.; Vannozzi, A.; Lazzeri, A. Chain Extension of Poly(Lactic Acid) (PLA)–Based Blends and Composites Containing Bran with Biobased Compounds for Controlling Their Processability and Recyclability. Polymers 2021, 13, 3050. [Google Scholar] [CrossRef]
- Sharma, B.K.; Adhvaryu, A.; Liu, Z.; Erhan, S.Z. Chemical modification of vegetable oils for lubricant applications. J. Am. Oil Chem. Soc. 2006, 83, 129–136. [Google Scholar] [CrossRef]
- Sharmin, E.; Zafar, F.; Akram, D.; Alam, M.; Ahmad, S. Recent advances in vegetable oils based environment friendly coatings: A review. Ind. Crops Prod. 2015, 76, 215–229. [Google Scholar] [CrossRef]
- Alam, M.; Akram, D.; Sharmin, E.; Zafar, F.; Ahmad, S. Vegetable oil based eco-friendly coating materials: A review article. Arab. J. Chem. 2014, 7, 469–479. [Google Scholar] [CrossRef]
- No, S.-Y. Inedible vegetable oils and their derivatives for alternative diesel fuels in CI engines: A review. Renew. Sustain. Energy Rev. 2011, 15, 131–149. [Google Scholar] [CrossRef]
- Chieng, B.W.; Ibrahim, N.A.; Then, Y.Y.; Loo, Y.Y. Epoxidized Vegetable Oils Plasticized Poly(lactic acid) Biocomposites: Mechanical, Thermal and Morphology Properties. Molecules 2014, 19, 16024–16038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamage, P.K.; O’Brien, M.; Karunanayake, L. Epoxidation of some vegetable oils and their hydrolysed products with peroxyformic acid-Optimised to industrial scale. J. Natl. Sci. Found. Sri Lanka 2009, 37, 229–240. [Google Scholar] [CrossRef]
- Ferri, J.M.; Garcia-Garcia, D.; Montanes, N.; Fenollar, O.; Balart, R. The effect of maleinized linseed oil as biobased plasticizer in poly(lactic acid)-based formulations. Polym. Int. 2017, 66, 882–891. [Google Scholar] [CrossRef]
- Garcia-Garcia, D.; Carbonell-Verdu, A.; Arrieta, M.P.; López-Martínez, J.; Samper, M.D. Improvement of PLA film ductility by plasticization with epoxidized karanja oil. Polym. Degrad. Stab. 2020, 179, 109259. [Google Scholar] [CrossRef]
- Auvergne, R.; Caillol, S.; David, G.; Boutevin, B.; Pascault, J.-P. Biobased Thermosetting Epoxy: Present and Future. Chem. Rev. 2014, 114, 1082–1115. [Google Scholar] [CrossRef]
- Xia, Y.; Quirino Rafael, L.; Larock, R.C. Biobased Thermosetting Polymers from Vegetable Oils. J. Renew. Mater. 2013, 1, 3–27. [Google Scholar] [CrossRef]
- Aliotta, L.; Gigante, V.; Coltelli, M.-B.; Lazzeri, A. Volume Change during Creep and Micromechanical Deformation Processes in PLA–PBSA Binary Blends. Polymers 2021, 13, 2379. [Google Scholar] [CrossRef]
- Aliotta, L.; Vannozzi, A.; Bonacchi, D.; Coltelli, M.-B.; Lazzeri, A. Analysis, Development, and Scaling-Up of Poly(lactic acid) (PLA) Biocomposites with Hazelnuts Shell Powder (HSP). Polymers 2021, 13, 4080. [Google Scholar] [CrossRef]
- Aliotta, L.; Vannozzi, A.; Canesi, I.; Cinelli, P.; Coltelli, M.-B.; Lazzeri, A. Poly(lactic acid) (PLA)/Poly(butylene succinate-co-adipate) (PBSA) Compatibilized Binary Biobased Blends: Melt Fluidity, Morphological, Thermo-Mechanical and Micromechanical Analysis. Polymers 2021, 13, 218. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.-T.; Wu, Y.; Li, Y.-D.; Wang, M.; Zeng, J.-B. Curing behavior of epoxidized soybean oil with biobased dicarboxylic acids. Polym. Test. 2017, 57, 281–287. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Li, K. Pressure-Sensitive Adhesives Based on Epoxidized Soybean Oil and Dicarboxylic Acids. ACS Sustain. Chem. Eng. 2014, 2, 2090–2096. [Google Scholar] [CrossRef]
- Zhao, T.-H.; Wu, Y.; Li, Y.-D.; Wang, M.; Zeng, J.-B. High Performance and Thermal Processable Dicarboxylic Acid Cured Epoxidized Plant Oil Resins through Dynamic Vulcanization with Poly(lactic acid). ACS Sustain. Chem. Eng. 2017, 5, 1938–1947. [Google Scholar] [CrossRef]
- Garea, S.-A.; Corbu, A.-C.; Deleanu, C.; Iovu, H. Determination of the epoxide equivalent weight (EEW) of epoxy resins with different chemical structure and functionality using GPC and 1H-NMR. Polym. Test. 2006, 25, 107–113. [Google Scholar] [CrossRef]
- Yussuf, A.A.; Massoumi, I.; Hassan, A. Comparison of Polylactic Acid/Kenaf and Polylactic Acid/Rise Husk Composites: The Influence of the Natural Fibers on the Mechanical, Thermal and Biodegradability Properties. J. Polym. Environ. 2010, 18, 422–429. [Google Scholar] [CrossRef]
- Sanivada, U.K.; Mármol, G.; Brito, F.P.; Fangueiro, R. PLA Composites Reinforced with Flax and Jute Fibers—A Review of Recent Trends, Processing Parameters and Mechanical Properties. Polymers 2020, 12, 2373. [Google Scholar] [CrossRef] [PubMed]
- Sykacek, E.; Schlager, W.; Mundigler, N. Compatibility of softwood flour and commercial biopolymers in injection molding. Polym. Compos. 2010, 31, 443–451. [Google Scholar] [CrossRef]
- Aliotta, L.; Gigante, V.; Coltelli, M.-B.; Cinelli, P.; Lazzeri, A.; Seggiani, M. Thermo-Mechanical Properties of PLA/Short Flax Fiber Biocomposites. Appl. Sci. 2019, 9, 3797. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Xu, H.; Odelius, K.; Hakkarainen, M. Poly(lactide)-g-poly(butylene succinate-co-adipate) with High Crystallization Capacity and Migration Resistance. Materials 2016, 9, 313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patrício, T.; Bártolo, P. Thermal Stability of PCL/PLA Blends Produced by Physical Blending Process. Procedia Eng. 2013, 59, 292–297. [Google Scholar] [CrossRef]
- Li, M.-X.; Kim, S.-H.; Choi, S.-W.; Goda, K.; Lee, W.-I. Effect of reinforcing particles on hydrolytic degradation behavior of poly (lactic acid) composites. Compos. Part B Eng. 2016, 96, 248–254. [Google Scholar] [CrossRef]
- Elsawy, M.A.; Kim, K.-H.; Park, J.-W.; Deep, A. Hydrolytic degradation of polylactic acid (PLA) and its composites. Renew. Sustain. Energy Rev. 2017, 79, 1346–1352. [Google Scholar] [CrossRef]
- Agustin-Salazar, S.; Cerruti, P.; Medina-Juárez, L.Á.; Scarinzi, G.; Malinconico, M.; Soto-Valdez, H.; Gamez-Meza, N. Lignin and holocellulose from pecan nutshell as reinforcing fillers in poly (lactic acid) biocomposites. Int. J. Biol. Macromol. 2018, 115, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Al-Mulla, E.A.J.; Yunus, W.M.Z.W.; Ibrahim, N.A.B.; Rahman, M.Z.A. Properties of epoxidized palm oil plasticized polytlactic acid. J. Mater. Sci. 2010, 45, 1942–1946. [Google Scholar] [CrossRef]
- Coltelli, M.-B.; Aliotta, L.; Vannozzi, A.; Morganti, P.; Panariello, L.; Danti, S.; Neri, S.; Fernandez-Avila, C.; Fusco, A.; Donnarumma, G.; et al. Properties and Skin Compatibility of Films Based on Poly(Lactic Acid) (PLA) Bionanocomposites Incorporating Chitin Nanofibrils (CN). J. Funct. Biomater. 2020, 11, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thielemans, W.; Can, E.; Morye, S.S.; Wool, R.P. Novel applications of lignin in composite materials. J. Appl. Polym. Sci. 2002, 83, 323–331. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | PLA/PBSA (wt.%) | H0210 (wt.%) | HM200 (wt.%) | ESO+MA (wt.%) | ESO+SA (wt.%) |
|---|---|---|---|---|---|
| F1 | 100 | - | - | - | - |
| F2 | 80 | 20 | - | - | - |
| F3 | 80 | - | 20 | - | - |
| F4 | 80 | 19.5 | - | 0.5 | - |
| F5 | 80 | 19 | - | 1 | - |
| F6 | 80 | 18 | - | 2 | - |
| F7 | 80 | 19.5 | - | - | 0.5 |
| F8 | 80 | 19 | - | - | 1 |
| F9 | 80 | 18 | - | - | 2 |
| F10 | 80 | - | 19.5 | 0.5 | - |
| F11 | 80 | - | 19 | 1 | - |
| F12 | 80 | - | 18 | 2 | - |
| F13 | 80 | - | 19.5 | - | 0.5 |
| F14 | 80 | - | 19 | - | 1 |
| F15 | 80 | - | 18 | - | 2 |
| Sample | Torque (N·cm) | MFR (g/10 min) | MVR (cm3/10 min) | Melt Density (g/cm3) |
|---|---|---|---|---|
| F1 | 64 ± 4 | 3.3 ± 0.5 | 2.8 ± 0.7 | 1.16 |
| F2 | 124 ± 8 | 4.5 ± 0.1 | 3.6 ± 0.2 | 1.25 |
| F3 | 105 ± 17 | 6.5 ± 0.1 | 5.4 ± 0.1 | 1.20 |
| F4 | 126 ± 6 | 4.7 ± 0.2 | 3.9 ± 0.3 | 1.23 |
| F5 | 123 ± 6 | 5.4 ± 0.1 | 5.0 ± 0.6 | 1.09 |
| F6 | 106 ± 3 | 6.1 ± 0.2 | 5.3 ± 0.4 | 1.15 |
| F7 | 127 ± 5 | 4.5 ± 0.2 | 3.8 ± 0.4 | 1.18 |
| F8 | 120 ± 11 | 5.2 ± 0.2 | 4.2 ± 0.2 | 1.22 |
| F9 | 104 ± 8 | 7.0 ± 1.1 | 6.1 ± 1.3 | 1.15 |
| F10 | 114 ± 12 | 6.0 ± 0.4 | 4.9 ± 0.4 | 1.22 |
| F11 | 113 ± 4 | 6.8 ± 0.3 | 5.6 ± 0.3 | 1.22 |
| F12 | 106 ± 2 | 7.6 ± 0.7 | 6.3 ± 0.7 | 1.20 |
| F13 | 116 ± 3 | 7.9 ± 0.3 | 6.6 ± 0.2 | 1.19 |
| F14 | 114 ± 3 | 7.6 ± 0.3 | 6.2 ± 0.3 | 1.22 |
| F15 | 109 ± 4 | 7.8 ± 0.1 | 6.5 ± 0.2 | 1.19 |
| Sample | Torque Difference (T0-Tf) (N·cm) |
|---|---|
| F1 | 9.43 |
| F2 | 39.14 |
| F3 | 29.00 |
| F4 | 41.29 |
| F5 | 41.86 |
| F6 | 39.00 |
| F7 | 33.00 |
| F8 | 34.14 |
| F9 | 36.00 |
| F10 | 26.86 |
| F11 | 20.00 |
| F12 | 25.14 |
| F13 | 28.00 |
| F14 | 30.57 |
| F15 | 30.71 |
| Sample | PLA Tg (°C) | PBSA Tm (°C) | PLA Tcc (°C) | PLA Tm (°C) | XC,PLA (%) |
|---|---|---|---|---|---|
| F1 | 57.64 | 82.62 | 117.83 | 150.01 | 4.2 |
| F2 | 57.06 | 86.19 | 117.27 | 150.00 | 4.8 |
| F3 | 55.68 | 85.86 | 114.22 | 149.15 | 4.3 |
| F4 | 57.43 | 86.46 | 119.09 | 150.67 | 2.2 |
| F5 | 56.97 | 85.99 | 117.21 | 149.83 | 3.1 |
| F6 | 56.62 | 86.26 | 117.29 | 149.99 | 4.6 |
| F7 | 57.62 | 86.26 | 118.47 | 150.43 | 4.4 |
| F8 | 57.32 | 86.77 | 118.24 | 150.74 | 3.6 |
| F9 | 56.48 | 86.24 | 117.14 | 149.89 | 7.3 |
| F10 | 56.67 | 86.31 | 113.40 | 149.50 | 6.3 |
| F11 | 55.61 | 86.11 | 111.45 | 148.66 | 6.5 |
| F12 | 55.62 | 86.05 | 112.37 | 148.76 | 6.7 |
| F13 | 56.10 | 85.80 | 112.89 | 148.90 | 6.4 |
| F14 | 55.50 | 85.91 | 112.83 | 148.97 | 5.4 |
| F15 | 55.71 | 85.78 | 111.87 | 148.45 | 7.2 |
| Sample | σy (MPa) | εy (%) | σb (MPa) | εb (%) |
|---|---|---|---|---|
| F1 | 28.8 ± 0.7 | 4.5 ± 0.4 | 25.2 ± 1.3 | 170.1 ± 56.4 |
| F2 | 29.8 ± 2.6 | 3.7 ± 0.6 | 25.4 ± 4.6 | 7.2 ± 1.9 |
| F3 | 23.8 ± 0.5 | 5.0 ± 0.4 | 19.7 ± 0.7 | 22.6 ± 6.4 |
| F4 | 30.7 ± 1.9 | 3.9 ± 0.5 | 28.2 ± 2.3 | 7.0 ± 3.5 |
| F5 | 27.4 ± 7.8 | 3.8 ± 0.4 | 27.3 ± 1 | 7.0 ± 1.8 |
| F6 | 29.6 ± 1.5 | 4.0 ± 0.5 | 26.7 ± 2 | 9.3 ± 4.8 |
| F7 | 30.0 ± 1.9 | 3.8 ± 0.3 | 25.3 ± 7.5 | 6.2 ± 1.2 |
| F8 | 28.0 ± 1.8 | 3.7 ± 0.4 | 25.6 ± 1.3 | 6.0 ± 1.5 |
| F9 | 27.6 ± 1.9 | 3.7 ± 0.3 | 24.2 ± 1.8 | 6.9 ± 1.6 |
| F10 | 23.2 ± 0.9 | 4.5 ± 0.3 | 18.5 ± 0.7 | 21.2 ± 6.9 |
| F11 | 23.0 ± 0.7 | 4.2 ± 0.2 | 18.3 ± 0.9 | 23.4 ± 6.5 |
| F12 | 21.8 ± 0.5 | 4.2 ± 0.6 | 17.1 ± 0.6 | 29.5 ± 5.5 |
| F13 | 22.8 ± 0.6 | 4.4 ± 0.3 | 18.2 ± 0.7 | 22.9 ± 7.3 |
| F14 | 22.4 ± 0.6 | 4.3 ± 0.3 | 17.9 ± 0.8 | 21.5 ± 7.4 |
| F15 | 21.7 ± 1.1 | 4.0 ± 0.5 | 17.5 ± 0.7 | 28.6 ± 10.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panariello, L.; Coltelli, M.-B.; Vannozzi, A.; Bonacchi, D.; Aliotta, L.; Lazzeri, A. Fully Biobased Reactive Extrusion of Biocomposites Based on PLA Blends and Hazelnut Shell Powders (HSP). Chemistry 2021, 3, 1464-1480. https://doi.org/10.3390/chemistry3040104
Panariello L, Coltelli M-B, Vannozzi A, Bonacchi D, Aliotta L, Lazzeri A. Fully Biobased Reactive Extrusion of Biocomposites Based on PLA Blends and Hazelnut Shell Powders (HSP). Chemistry. 2021; 3(4):1464-1480. https://doi.org/10.3390/chemistry3040104
Chicago/Turabian StylePanariello, Luca, Maria-Beatrice Coltelli, Alessandro Vannozzi, Daniele Bonacchi, Laura Aliotta, and Andrea Lazzeri. 2021. "Fully Biobased Reactive Extrusion of Biocomposites Based on PLA Blends and Hazelnut Shell Powders (HSP)" Chemistry 3, no. 4: 1464-1480. https://doi.org/10.3390/chemistry3040104
APA StylePanariello, L., Coltelli, M.-B., Vannozzi, A., Bonacchi, D., Aliotta, L., & Lazzeri, A. (2021). Fully Biobased Reactive Extrusion of Biocomposites Based on PLA Blends and Hazelnut Shell Powders (HSP). Chemistry, 3(4), 1464-1480. https://doi.org/10.3390/chemistry3040104