
Regioselective Bond-Forming and Hydrolysis Reactions of Doubly Charged Vanadium Oxide Anions in the Gas Phase


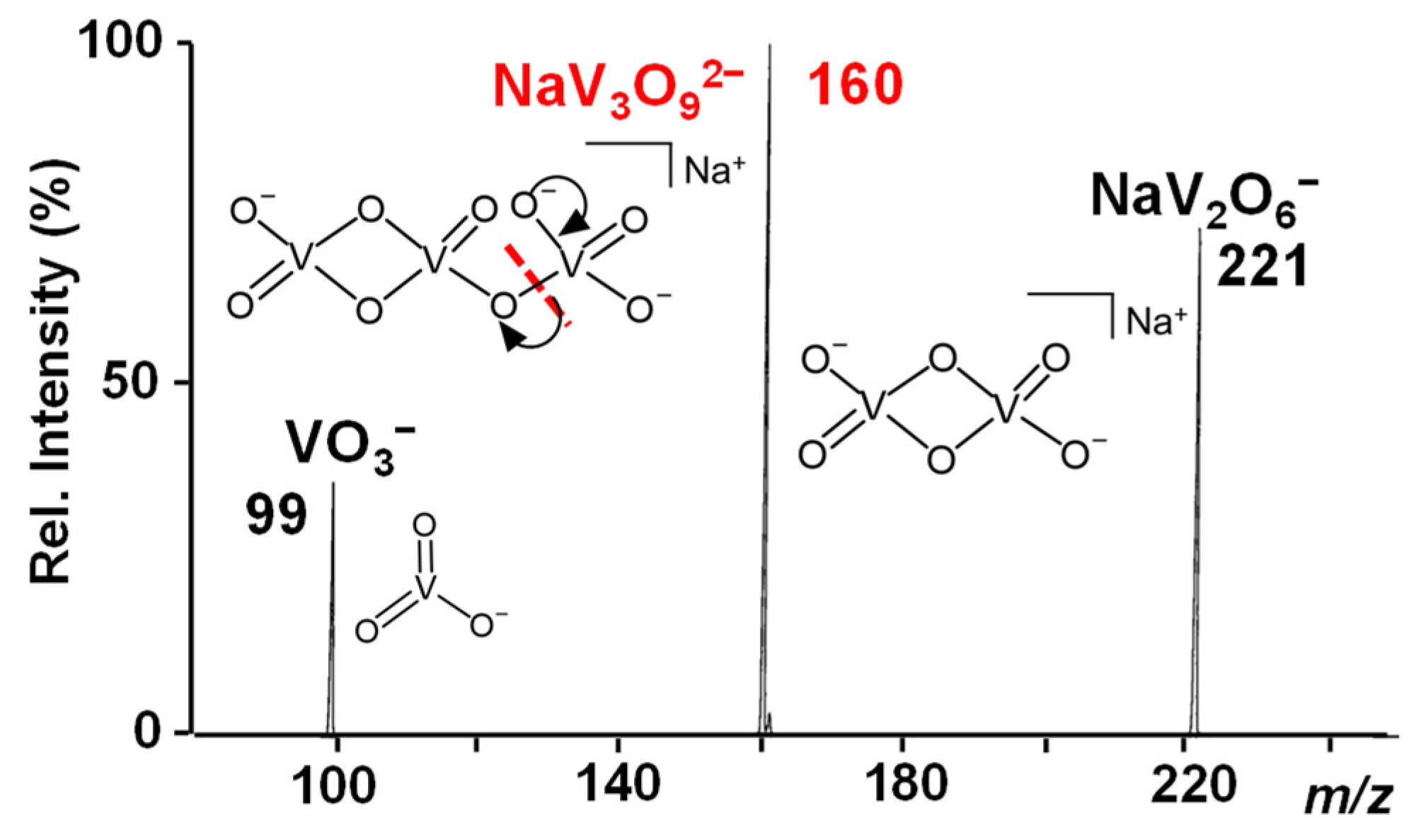{kind=link}
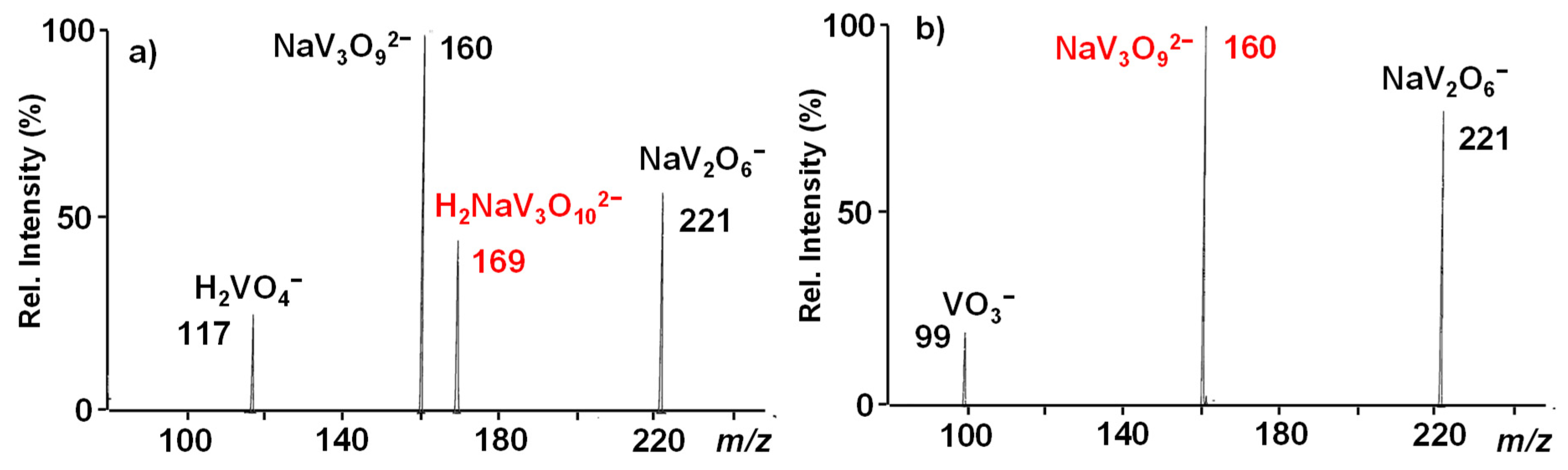{kind=link}
{kind=link}
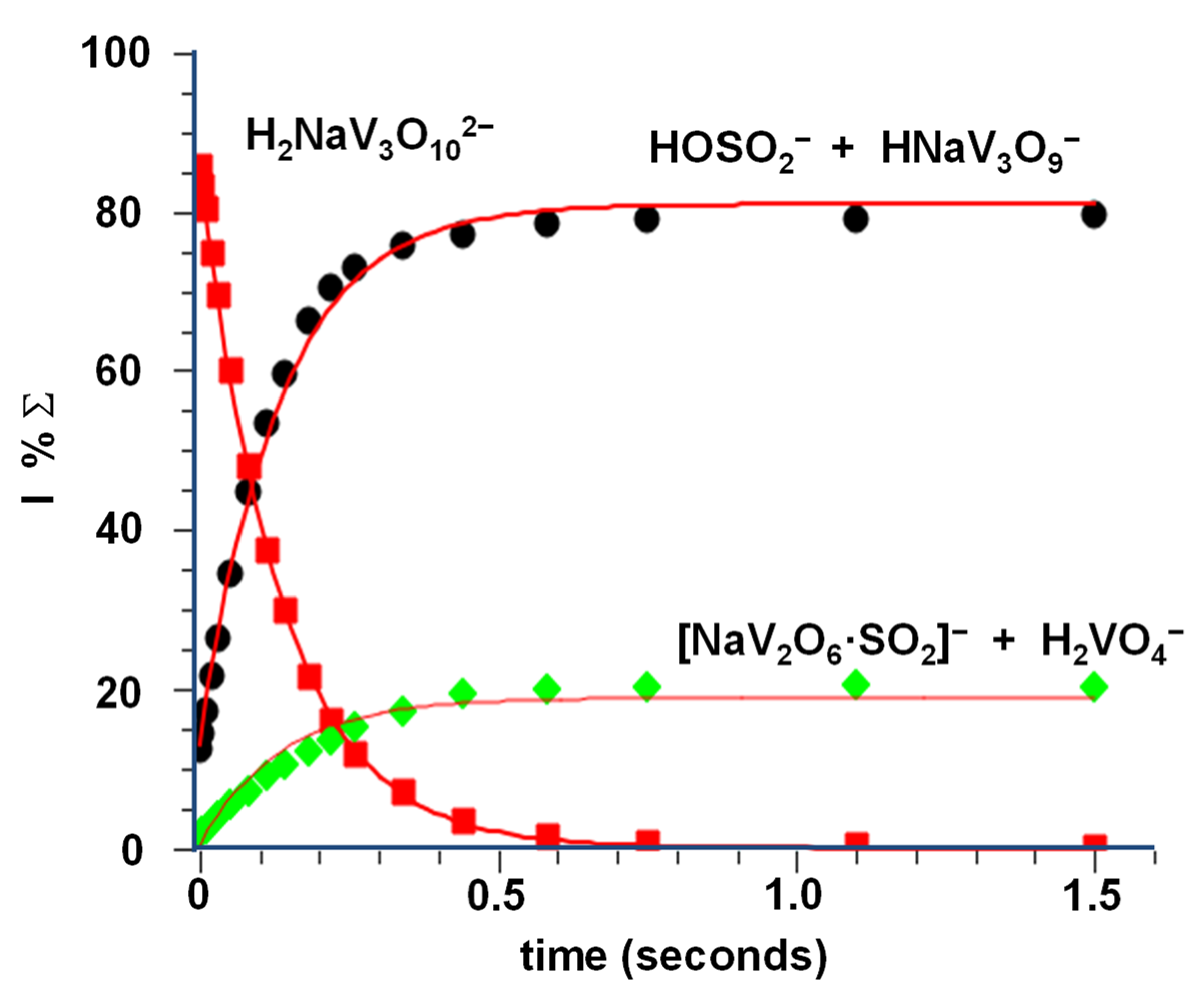{kind=link}
{kind=link}
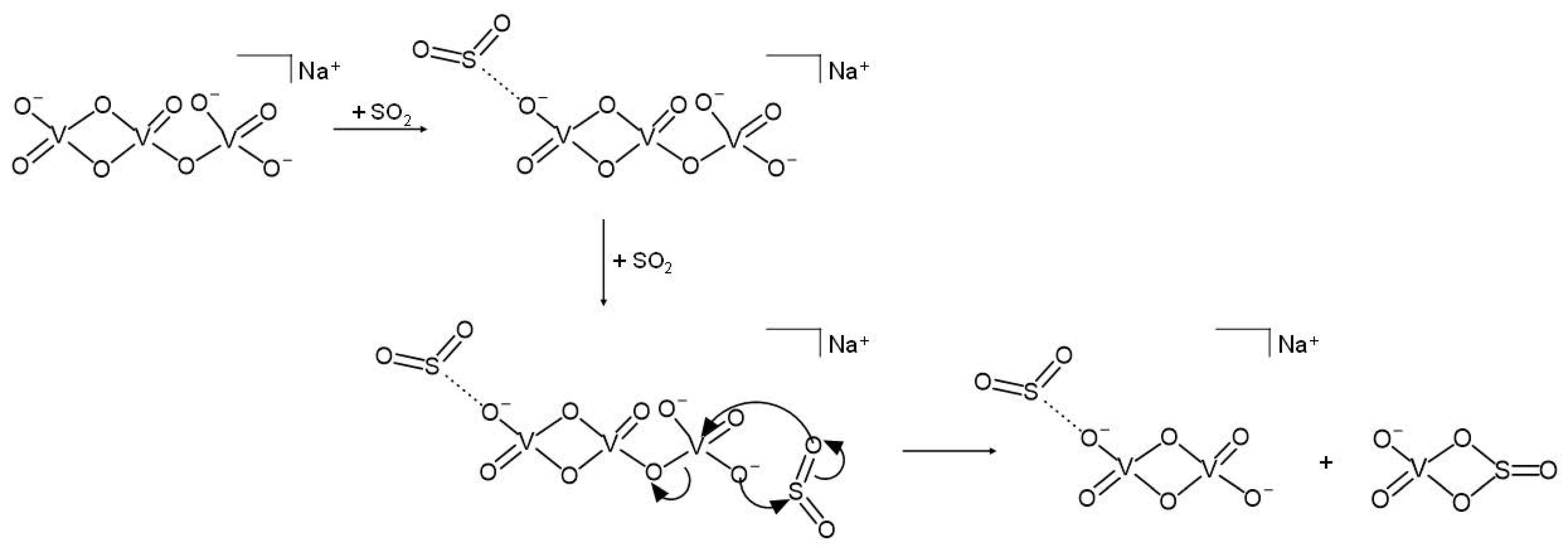{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Mass Spectrometry
3. Results and Discussion
3.1. The NaV3O92− and H2NaV3O102− Reactant Dianions
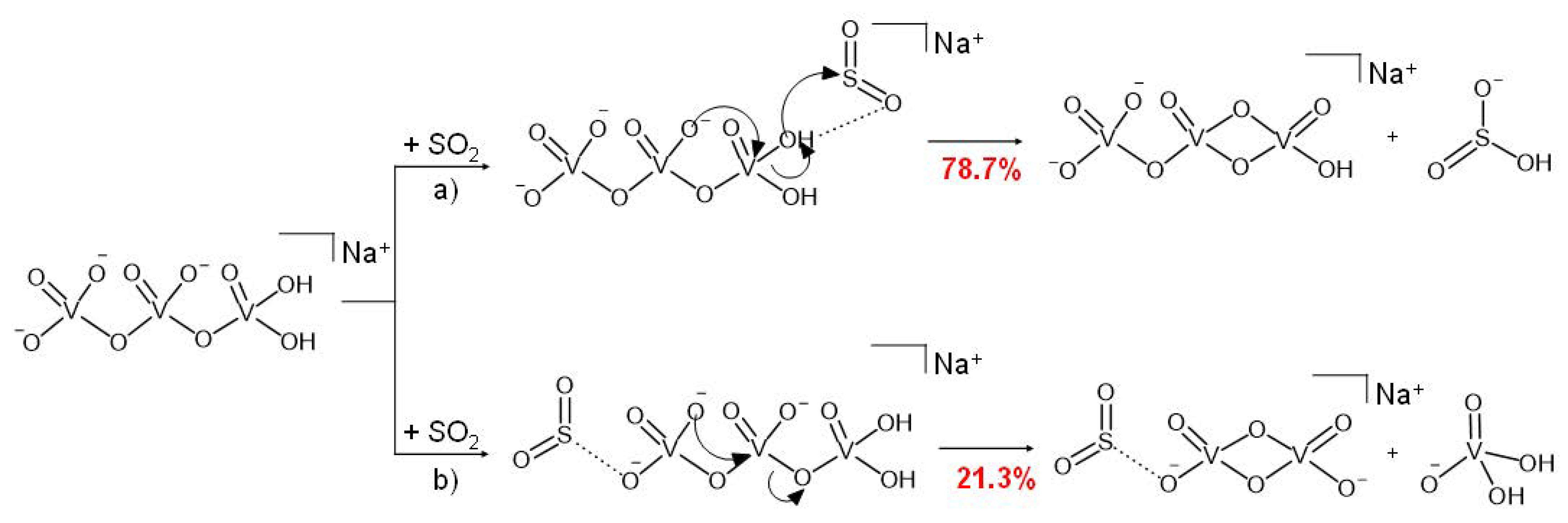
3.2. Reactivity of NaV3O92− and H2NaV3O102− Dianions towards SO2
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Weckhuysen, B.M.; Keller, D.E. Chemistry, Spectroscopy and the role of supported vanadium oxides. Catal. Today 2003, 78, 25–46. [Google Scholar] [CrossRef]
- Chenier, J.P. Survey of Industrial Chemistry; John Wiley & Sons: New York, NY, USA, 1987; pp. 45–57. [Google Scholar]
- Whittingham, M.S. Lithium batteries and cathode materials. Chem. Rev. 2004, 104, 4271–4302. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Song, H.; Zhang, C.; Liu, C.; Liu, Y.; Cao, G. Interface reduction synthesis of H2V2O3 nanobelts-graphene for high-rate Li-ion batteries. J. Phys. Chem. C 2015, 119, 11399. [Google Scholar]
- Xu, X.; Yan, M.; Tian, X.; Yang, C.; Shi, M.; Wei, Q.; Xu, L.; Mai, L. In situ investigation of Li and Na transport with single nanowire electrochemical devices. Nano Lett. 2015, 15, 3879–3884. [Google Scholar] [CrossRef]
- Anjass, M.; Lowe, G.A.; Streb, C. Molecular vanadium oxides for energy conversion and energy storage: Current trends and emerging opportunities. Angew. Chem. Int. Ed. 2021, 60, 7522–7532. [Google Scholar] [CrossRef]
- Johnson, G.; Tyo, C.C.; Castleman, A.W. Cluster reactivity experiments: Employing mass spectrometry to investigate the molecular level details of catalytic oxidation reactions. Proc. Natl. Acad. Sci. USA 2008, 105, 18108–18113. [Google Scholar] [CrossRef]
- Johnson, G.E.; Mitrić, R.; Bonačić-Koutecký, V.; Castleman, A. Clusters as model systems for investigating nanoscale oxidation catalysis. Chem. Phys. Lett. 2009, 475, 1–9. [Google Scholar] [CrossRef]
- Schlangen, M.; Schwarz, H. Effects of ligands, cluster size, and charge state in gas-phase catalysis: A happy marriage of experimental and computational studies. Catal. Lett. 2012, 142, 1265–1278. [Google Scholar] [CrossRef]
- Dietl, D.-C.N.; Troiani, A.; Schlangen, M.; Ursini, O.; Angelini, G.; Apeloig, Y.; de Petris, G.; Schwarz, H. Mechanistic aspects of gas-phase hydrogen-atom transfer from methane to [CO]+ and [SiO]+: Why do they differ? Chem. Eur. J. 2013, 19, 6662–6669. [Google Scholar] [CrossRef]
- Schlangen, M.; Schwarz, H. Probing elementary steps of nickel-mediated bond activation in gas-phase reactions: Ligand- and cluster-size effects. J. Catal. 2011, 284, 126–137. [Google Scholar] [CrossRef]
- Vikse, K.L.; McIndoe, J.S. Mechanistic insights from mass spectrometry: Examination of the elementary steps of catalytic reactions in the gas phase. Pure Appl. Chem. 2015, 87, 361–377. [Google Scholar] [CrossRef]
- Vikse, K.L.; Ahmadi, Z.; McIndoe, J.S. The application of electrospray ionization mass spectrometry to homogeneous catalysis. Coord. Chem. Rev. 2014, 279, 96–114. [Google Scholar] [CrossRef]
- Bell, R.C.; Zemski, K.A.; Kerns, K.P.; Deng, H.T.; Castleman, A.W. Reactivities and collision-induced dissociation of Vanadium oxide clusters cations. J. Phys. Chem. A 1998, 102, 1733–1742. [Google Scholar] [CrossRef]
- Zemski, K.A.; Justes, D.R.; Castleman, A.W. Reactions of group V transition metal oxide cluster ions with ethane and ethylene. J. Phys. Chem. A 2001, 105, 10237–10245. [Google Scholar] [CrossRef]
- Feyel, S.; Schröder, D.; Rozanska, X.; Sauer, J.; Schwarz, H. Gas-phase oxidation of propane and 1-butene with [V3O7]+: Experiment and theory in concert. Angew. Chem. Int. Ed. 2006, 45, 4677–4681. [Google Scholar] [CrossRef] [PubMed]
- Feyel, S.; Döbler, J.; Schröder, D.; Sauer, J.; Schwarz, H. Thermal activation of methane by tetranuclear [V4O10]+. Angew. Chem. Int. Ed. 2006, 45, 4681–4685. [Google Scholar] [CrossRef] [PubMed]
- Dietl, N.; Engeser, M.; Schwarz, H. Competitive hydrogen-atom abstraction versus oxygen-atom and electron transfers in gas-phase reactions of [X4O10]+ (X = P, V) with C2H4. Chem. Eur. J. 2010, 16, 4452–4456. [Google Scholar] [CrossRef]
- Zhou, S.; Li, J.; Schlangen, M.; Schwarz, H. Differences and commonalities in the gas-phase reactions of closed-shell metal dioxide clusters [MO2]+ (M = V, Nb, and Ta) with methane. Chem. Eur. J. 2016, 22, 7225–7228. [Google Scholar] [CrossRef]
- Wang, Z.-C.; Wu, X.N.; Zhao, Y.X.; Ma, J.B.; Ding, X.L.; He, S.G. Room-temperature methane activation by a bimetallic oxide cluster AlVO4+. Chem. Phys. Lett. 2010, 489, 25–29. [Google Scholar] [CrossRef]
- Dietl, N.; Hockendorf, R.F.; Schlangen, M.; Lerch, M.; Beyer, M.K.; Schwarz, H. Generation, reactivity towards hydrocarbons, and electronic structure of heteronuclear vanadium phosphorous oxygen cluster ions. Angew. Chem. Int. Ed. 2011, 50, 1430–1434. [Google Scholar] [CrossRef]
- Wang, Z.-C.; Liu, J.-W.; Schlangen, M.; Weiske, T.; Schröder, D.; Sauer, J.; Schwarz, H. Thermal methane activation by a binary V-Nb transition-metal oxide cluster cation: A further example for the crucial role of oxygen-centered radical. Chem. Eur. J. 2013, 19, 11496–11501. [Google Scholar] [CrossRef] [PubMed]
- Dietl, N.; Wende, T.; Chen, K.; Jiang, L.; Schlangen, M.; Zhang, X.; Asmis, K.R.; Schwarz, H. Structure and chemistry of the heteronuclear oxo-cluster [VPO4]·+: A model system for the gas-phase oxidation of small hydrocarbons. J. Am. Chem. Soc. 2013, 135, 3711–3721. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Li, H.F.; Zhao, Y.X.; He, S.G. Gold(III) mediated activation and transformation of methane on Au1-doped vanadium oxide cluster cations AuV2O6+. J. Am. Chem. Soc. 2016, 138, 9437–9443. [Google Scholar] [CrossRef]
- Bell, R.C.; Castleman, A.W. Reactions of vanadium oxide cluster ions with 1,3-butadiene and isomers of butene. J. Phys. Chem. A 2002, 106, 9893–9899. [Google Scholar] [CrossRef]
- Li, X.-N.; Xu, B.; Ding, X.-L.; He, S.-G. Interaction of vanadium oxide cluster anions with water: An experimental and theoretical study on the reactivity and mechanism. Dalton Trans. 2012, 41, 5562–5570. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Mirabal, A.; Demuth, J.; Wöste, L.; Siebert, T. A complete reactant-product analysis of the oxygen transfer reaction in [V4O11·C3H6]−: A cluster complex for modelling surface activation and reactivity. J. Am. Chem. Soc. 2008, 130, 16832–16833. [Google Scholar] [CrossRef]
- Zhao, Y.-X.; Wu, X.-N.; Ma, J.-B.; He, S.-G.; Ding, X.-L. Experimental and theoretical study of the reactions between vanadium-silicon heteronuclear oxide cluster anions with n-butane. J. Phys. Chem. C 2010, 114, 12271–12279. [Google Scholar] [CrossRef]
- Janssens, E.; Lang, S.M.; Brümmer, M.; Niedziela, A.; Santambrogio, G.; Asmis, K.R.; Sauer, J. Kinetic study of the reaction of vanadium and vanadium-titanium oxide cluster anions with SO2. Phys. Chem. Chem. Phys. 2012, 14, 14344–14353. [Google Scholar] [CrossRef]
- Dinca, A.; Davis, T.P.; Fisher, K.J.; Smith, D.R.; Willett, G.D. Vanadium oxide anion cluster reactions with methyl isobutyrate and methyl methacrylate monomer and dimer: A study by FT/ICR mass spectrometry. Int. J. Mass Spectrom. 1999, 182–183, 73–84. [Google Scholar] [CrossRef]
- Yuan, Z.; Li, Z.-Y.; Zhou, Z.-X.; Liu, Q.-Y.; He, S.-G. Thermal reactions of (V2O5)nO− (n = 1–3) cluster anions with ethylene and propylene: Oxygen atom transfer versus molecular association. J. Phys. Chem. C 2014, 118, 14967–14976. [Google Scholar] [CrossRef]
- Jakubikova, E.; Bernestein, E.R. Reactions of sulfur dioxide with neutral vanadium oxide clusters in the gas phase. I. Density functional theory study. J. Phys. Chem. A 2007, 111, 13339–13346. [Google Scholar] [CrossRef] [PubMed]
- He, S.-G.; Xie, Y.; Dong, F.; Heinbuch, S.; Jakubikova, E.; Rocca, J.J.; Bernstein, E.R. Reactions of sulfur dioxide with neutral vanadium oxide clusters in the gas phase. II. Experimental study employing single-photon ionization. J. Phys. Chem. A 2008, 112, 11067–11077. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Coelho, F.; Eberlin, M.N. The bridge connecting gas-phase and solution chemistries. Angew. Chem. Int. Ed. 2011, 50, 5261–5263. [Google Scholar] [CrossRef] [PubMed]
- Schröder, D.; Engeser, M.; Schwarz, H.; Harvey, J.N. Energetics of the ligated vanadium dications VO2+, VOH2+, and [V,O,H2]2+. ChemPhysChem 2002, 3, 584–591. [Google Scholar] [CrossRef]
- Warzok, U.; Mahnke, L.K.; Bensch, W. Soluble hetero-polyoxovanadates and their solution chemistry analysed by electrospray ionization mass spectrometry. Chem. Eur. J. 2019, 25, 1405–1419. [Google Scholar] [CrossRef]
- Ranasinghe, Y.A.; MacMahon, T.J.; Freiser, B.S. Gas-phase reactions of lanthanum dication with small alkanes and the photodissociation of LaC2H4n+ and LaC3H6n+ (n = 1 and 2). J. Am. Chem. Soc. 1992, 114, 9112–9118. [Google Scholar] [CrossRef]
- Petrie, S.; Javahery, G.; Wang, J.; Bohme, D.K. Derivatization of the fullerene dications C602+ and C702+ by ion-molecule reactions in the gas phase. J. Am. Chem. Soc. 1992, 114, 9177–9181. [Google Scholar] [CrossRef]
- Price, S.D.; Manning, M.; Leone, S.R. Bond-forming reactions of gas-phase molecular dications. J. Am. Chem. Soc. 1994, 116, 8673–8680. [Google Scholar] [CrossRef]
- Roithová, J.; Herman, Z.; Schröder, D.; Schwarz, H. Competition of proton and electron transfers in gas-phase reactions of hydrogen-containing dications CHX2+ (X = F, Cl, Br, I) with atoms, nonpolar and polar molecules. Chem. Eur. J. 2006, 12, 2465–2471. [Google Scholar] [CrossRef]
- Roithová, J.; Schröder, D. Bond-forming reactions of molecular dications as a new route to polyaromatic hydrocarbons. J. Am. Chem. Soc. 2006, 128, 4208–4209. [Google Scholar] [CrossRef]
- Roithová, J.; Žabka, J.; Herman, Z.; Thissen, R.; Schröder, D.; Schwarz, H. Reactivity of the CHBr2+ dication toward molecular hydrogen. J. Phys. Chem. A 2006, 110, 6447–6453. [Google Scholar] [CrossRef]
- Roithová, J.; Schröder, D. Bimolecular reactions of molecular dications: Reactivity paradigms and bond-forming processes. Phys. Chem. Chem. Phys. 2007, 9, 2341–2349. [Google Scholar] [CrossRef] [PubMed]
- Roithová, J.; Schröder, D.; Schwarz, H. The SiF32+ dication: Chemistry counts! Chem. Eur. J. 2009, 15, 9995–9999. [Google Scholar] [CrossRef] [PubMed]
- Blades, A.; Peschke, M.; Verkerk, U.; Kebarle, P. Rates of proton transfer from carboxylic acids to dianions, CO2(CH2)pCO22−, and their significance to observed negative charge states of proteins in the gas phase. J. Phys. Chem. A 2002, 106, 10037–10042. [Google Scholar] [CrossRef]
- Flores, A.E.; Gronert, S. The gas-phase reactions of dianions with alkyl bromides: Direct identification of SN2 and E2 products. J. Am. Chem. Soc. 1999, 121, 2627–2628. [Google Scholar] [CrossRef]
- Gronert, S. Gas phase studies of the competition between substitution and elimination reactions. Acc. Chem. Res. 2003, 36, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Troiani, A.; Rosi, M.; Garzoli, S.; Salvitti, C.; de Petris, G. Vanadium hydroxyde cluster ions in the gas-phase: Bond-forming reactions of doubly-charged negative ions by SO2-promoted V-O activation. Chem. Eur. J. 2017, 23, 11752–11756. [Google Scholar] [CrossRef] [PubMed]
- Troiani, A.; Rosi, M.; Garzoli, S.; Salvitti, C.; de Petris, G. Sulphur dioxide cooperation in hydrolysis of vanadium oxide and hydroxide cluster dianions. New J. Chem. 2018, 42, 4008–4016. [Google Scholar] [CrossRef]
- de Petris, G.; Cartoni, A.; Troiani, A.; Angelini, G.; Ursini, O. Water activation by SO2+ ions: An effective source of OH radicals. PhysChemChemPhys 2009, 11, 9976–9978. [Google Scholar] [CrossRef]
- de Petris, G.; Troiani, A.; Rosi, M.; Angelini, G.; Ursini, O. Selective Activation of C-Cl and C-F Bonds by SO+ Radical Cations: An Experimental and Computational Study. ChemPlusChem 2013, 78, 1065–1072. [Google Scholar] [CrossRef]
- Troiani, A.; Rosi, M.; Salvitti, C.; de Petris, G. The oxidation of sulfur dioxide by single and double oxygen transfer paths. ChemPhysChem 2014, 15, 2723–2731. [Google Scholar] [CrossRef] [PubMed]
- Troiani, A.; Salvitti, C.; de Petris, G. Gas-phase reactivity of carbonate ions with sulfur dioxide: An experimental study of cluster reactions. J. Am. Chem. Soc. Mass Spectrom. 2019, 30, 1964–1972. [Google Scholar] [CrossRef] [PubMed]
- Troiani, A.; Rosi, M.; Garzoli, S.; Salvitti, C.; de Petris, G. Effective redox reactions by chromium oxide anions: Sulfur dioxide oxidation in the gas phase. Int. J. Mass Spectrom. 2019, 436, 18–22. [Google Scholar] [CrossRef]
- Salvitti, C.; Rosi, M.; Pepi, F.; Troiani, A.; de Petris, G. Reactivity of transition metal dioxide anions MO2− (M = Co, Ni, Cu, Zn) with sulfur dioxide in the gas phase: An experimental and theoretical study. Chem. Phys. Lett. 2021, 776, 138555. [Google Scholar] [CrossRef]
- Salvitti, C.; Pepi, F.; Troiani, A.; de Petris, G. Intracluster sulphur dioxide oxidation by sodium chlorite anions: A mass spectrometric study. Molecules 2021, 26, 7114. [Google Scholar] [CrossRef] [PubMed]
- Troiani, A.; Rosi, M.; Garzoli, S.; Salvitti, C.; de Petris, G. Iron-Promoted C-C Bond Formation in the Gas Phase. Angew. Chem. Int. Ed. 2015, 54, 14359–14362. [Google Scholar] [CrossRef] [PubMed]
- Leavitt, C.M.; Bryantsev, V.S.; de Jong, W.A.; Diallo, M.S.; Goddard, W.A., III; Groenewold, G.S.; Van Stipdonk, M.J. Addition of H2O and O2 to acetone and dimethylsulfoxide ligated Uranyl(V) dioxocations. J. Phys. Chem. A 2009, 113, 2350–2358. [Google Scholar] [CrossRef]
- Bartmess, J.E.; Georgiadis, R.M. Empirical methods for determination of ionization gauge relative sensitivities for different gases. Vacuum 1983, 33, 149. [Google Scholar] [CrossRef]
- Kuzmic, P. Program DYNAFIT for the analysis of enzyme kinetic data: Application to HIV proteinase. Anal. Biochem. 1996, 237, 260–273. [Google Scholar] [CrossRef]
- Bowers, M.T.; Su, T. Interactions between Ions and Molecules; Plenum Press: New York, NY, USA, 1975. [Google Scholar]
- Habayeb, M.A.; Hileman, O.E., Jr. 51V- FT-nmr investigations of metavanadate ions in aqueous solutions. Can. J. Chem. 1980, 58, 2255–2261. [Google Scholar] [CrossRef]
- Aureliano, M.; Cran, D.C. Decavanadate (V10O286−) and oxovanadates: Oxometalates with many biological activities. J. Inorg. Biochem. 2009, 103, 536–546. [Google Scholar] [CrossRef] [PubMed]
- Walanda, D.K.; Burns, R.C.; Lawrance, G.A.; Von Nagy-Felsobuki, E.I. New isopolyoxovanadate ions identified by electrospray mass spectrometry. Inorg. Chem. Commun. 1999, 2, 487–489. [Google Scholar] [CrossRef]
- Walanda, D.K.; Burns, R.C.; Lawrance, G.A.; Von Nagy-Felsobuki, E.I. Unknown isopolyoxovanadate species detected by electrospray mass spectrometry. Inorg. Chim. Acta 2000, 305, 118–126. [Google Scholar] [CrossRef]
- Al Hasan, N.M.; Johnson, G.E.; Laskin, J. Gas-phase synthesis of singly and multiply charged polyoxovanadate anions employing electrospray ionization and collision induced dissociation. J. Am. Soc. Mass Spectrom. 2013, 24, 1385–1395. [Google Scholar] [CrossRef] [PubMed]
- Mutlu, E.; Cristy, T.; Graves, S.W.; Hooth, M.J.; Waidyanatha, S. Characterization of aqueous formulations of tetra- and pentavalent forms of vanadium in support of test article selection in toxicology studies. Environ. Sci. Pollut. Res. 2017, 24, 405–416. [Google Scholar] [CrossRef]
- O’Hair, R.A.J.; Freitas, M.A.; Gronert, S.; Schmidt, J.A.R.; Williams, T.D. Concerning the regioselectivity of gas phase reactions of glycine with electrophiles. The cases of the dimethylchlorinium ion and the methoxymethal cation. J. Org. Chem. 1995, 60, 1990–1998. [Google Scholar] [CrossRef]
- Van der Wel, H.; Nibbering, N.M.M. A gas phase study of the regioselective BH4− reduction of some 2-substituted maleic anhydrides. Can. J. Chem. 1998, 66, 2587–2594. [Google Scholar]
- Wernik, M.; Hartmann, P.E.; Sipos, G.; Darvas, F.; Boese, A.D.; Dallinger, D.; Kappe, C.O. On the regioselectivity of the Gould-Jacobs reaction: Gas-phase versus solution-phase thermolysis. Eur. J. Org. Chem. 2020, 2020, 7051–7061. [Google Scholar] [CrossRef]
- Avdeed, V.I.; Tapilin, V.M. Water effect on the electronic structure of active sites of supported Vanadium oxide catalyst VOx/TiO2(001). J. Phys. Chem. C 2010, 114, 3609–3613. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salvitti, C.; Pepi, F.; Troiani, A.; de Petris, G. Regioselective Bond-Forming and Hydrolysis Reactions of Doubly Charged Vanadium Oxide Anions in the Gas Phase. Reactions 2022, 3, 254-264. https://doi.org/10.3390/reactions3020019
Salvitti C, Pepi F, Troiani A, de Petris G. Regioselective Bond-Forming and Hydrolysis Reactions of Doubly Charged Vanadium Oxide Anions in the Gas Phase. Reactions. 2022; 3(2):254-264. https://doi.org/10.3390/reactions3020019
Chicago/Turabian StyleSalvitti, Chiara, Federico Pepi, Anna Troiani, and Giulia de Petris. 2022. "Regioselective Bond-Forming and Hydrolysis Reactions of Doubly Charged Vanadium Oxide Anions in the Gas Phase" Reactions 3, no. 2: 254-264. https://doi.org/10.3390/reactions3020019
APA StyleSalvitti, C., Pepi, F., Troiani, A., & de Petris, G. (2022). Regioselective Bond-Forming and Hydrolysis Reactions of Doubly Charged Vanadium Oxide Anions in the Gas Phase. Reactions, 3(2), 254-264. https://doi.org/10.3390/reactions3020019