

Whole-Exome Sequencing Identified Molecular Variants Linked to the Progression of Gastric Precancerous Lesions in Patients from Southwestern Colombia—An Exploratory Approach

 , and
, and
Abstract
1. Introduction
2. Results
2.1. Molecular Variants Associated with the Progression of Gastric Precancerous Lesions
2.2. Functional Impact Prediction of Nonsynonymous Variants
2.3. Enriched Biological Pathways Linked to the Progression of Gastric Precancerous Lesions
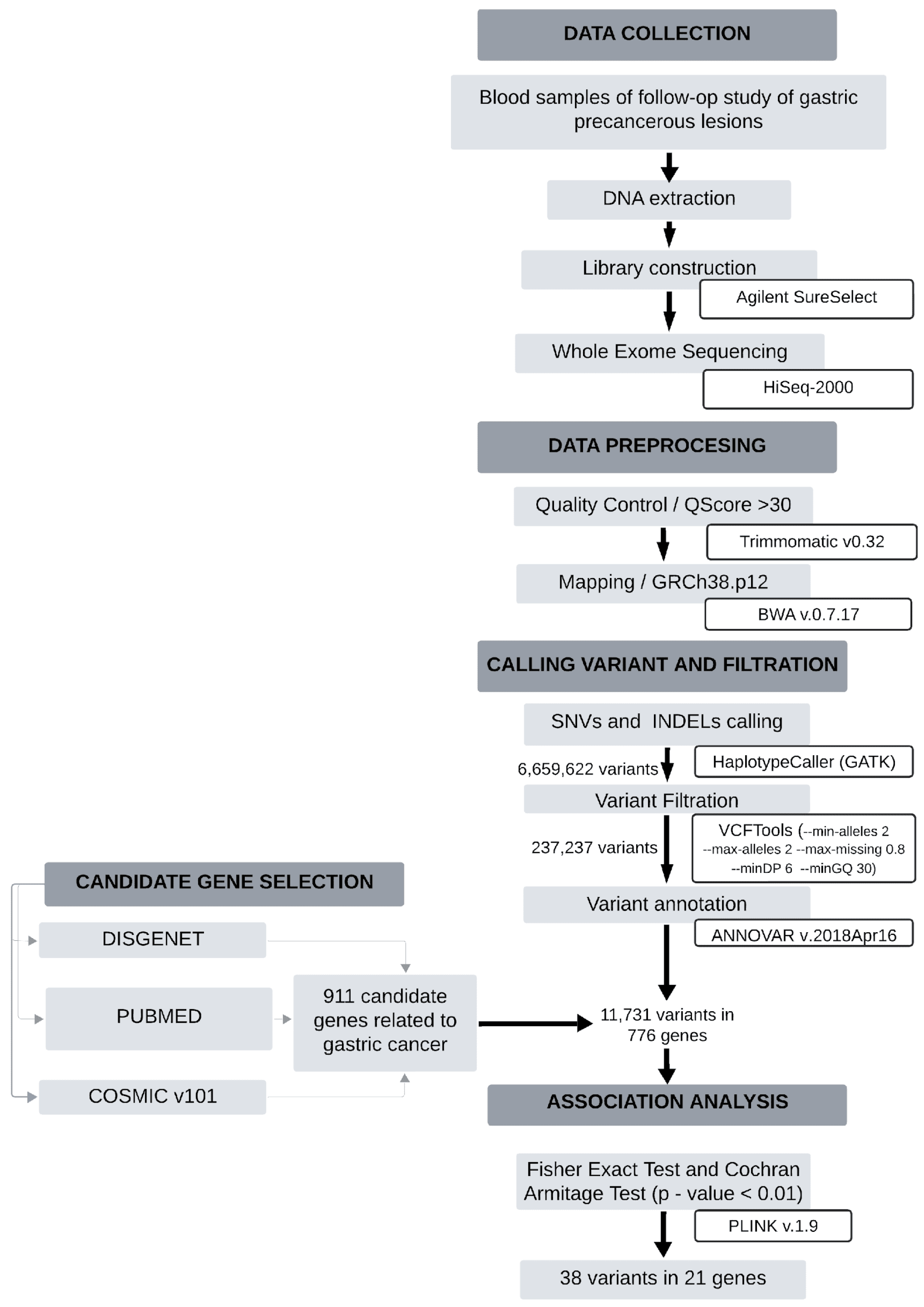
3. Materials and Methods
3.1. Patients and Study Samples
3.2. Sample Preparation and Exome Sequencing
3.3. Bioinformatics Analysis
3.4. Selection of Candidate Genes
3.5. Annotation and Genotyping
3.6. Statistical Analysis
3.7. Enrichment Analysis and Functional Impact
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| MAG | Multifocal Atrophic Gastritis |
| IM | Intestinal Metaplasia |
| GC | Gastric Cancer |
| WES | Whole Exome Sequencing |
References
- IARC GLOBOCAN 2022: Estimated Cancer Incidence, Mortality and Prevalence Worldwide in 2022. Available online: https://gco.iarc.who.int (accessed on 21 February 2024).
- Correa, P.; Piazuelo, M.B. The Gastric Precancerous Cascade. J. Dig. Dis. 2012, 13, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Marin, A.M.; Zambrano, R.; Uribe, P.T.; Arturo, B.L.; Jaramillo, M.d.S.; Lopez, P.A.; Perez, J.M. Asociación Clínica, Patológica y Microbiológica de Helicobacter Pylori En Biopsias Gástricas En El Departamento de Caldas-Colombia. Rev. Gastroenterol. México 2018, 38, 144–150. [Google Scholar]
- Cervantes, E. Helicobacter Pylori: Mecanismos de Patogenicidad. Rev. Latinoam. Patol. Clín. 2016, 63, 100–109. [Google Scholar]
- Correa, P.; Cuello, C.; Duque, E.; Burbano, L.; García, F.; Bolaños, O.; Brown, C.; Haenszel, W. Gastric Cancer in Colombia. III. Natural History of Precursor Lesions. J. Natl. Cancer Inst. 1976, 57, 1027–1035. [Google Scholar] [CrossRef]
- Pardo, C.; Cendales, R. Incidencia, Mortalidad y Prevalencia de Cáncer en Colombia 2007–2011, 1st ed.; Instituto Nacional de Cancerologia: Bogotá, Colombia, 2015; ISBN 9789585883253. [Google Scholar]
- Bedoya-Urresta, Á.; Yépez, Y.; Calvache, D.; Cifuentes, Y.; Lucero, N.; González, P.; Bedoya, G.Á.; Manosalva, E.; Martínez, T.; Peñalosa, A.; et al. Proyecto Urkunina 5000-Investigación de La Prevalencia de Lesiones Precursoras y Del Efecto de La Erradicación de Helicobacter Pylori Como Prevención Primaria Del Cáncer Gástrico En El Departamento de Nariño. Rev. Colomb. Cirugía 2018, 33, 345–352. [Google Scholar] [CrossRef]
- Chiurillo, M.A. Role of Gene Polymorphisms in Gastric Cancer and Its Precursor Lesions: Current Knowledge and Perspectives in Latin American Countries. World J. Gastroenterol. 2014, 20, 4503–4515. [Google Scholar] [CrossRef]
- Correa, P.; Fontham, E.; Bravo, J.; Bravo, L.; Ruiz, B.; Zarama, G.; Realpe, J.; Malcom, G.; Mera, R. Chemoprevention of Gastric Dysplasia: Randomized Trial of Antioxidant Supplements and Anti-Helicobacter Pylori Therapy. J. Natl. Cancer Inst. 2000, 92, 1881–1888. [Google Scholar] [CrossRef]
- Mera, R.; Fontham, E.T.H.; Bravo, L.E.; Bravo, J.C.; Piazuelo, M.B.; Camargo, M.C.; Correa, P. Long Term Follow up of Patients Treated for Helicobacter Pylori Infection. Gut 2005, 54, 1536–1540. [Google Scholar] [CrossRef]
- Mera, R.; Bravo, L.E.; Camargo, C.; Bravo, J.; Delgado, A.; Romero, J.; Yepez, M.; Realpe, J.; Schneider, B.; Morgan, D.; et al. Dynamics of Helicobacter Pylori Infection as a Determinant of Progression of Gastric Precancerous Lesions: 16-Year Follow-up of an Eradication Trial. Gut 2018, 67, 1239–1246. [Google Scholar] [CrossRef]
- Piazuelo, M.B.; Bravo, L.E.; Mera, R.M.; Camargo, M.C.; Bravo, J.C.; Delgado, A.G.; Washington, M.K.; Rosero, A.; Garcia, L.S.; Realpe, J.L.; et al. The Colombian Chemoprevention Trial. Twenty-Year Follow-up of a Cohort of Patients with Gastric Precancerous Lesions. Gastroenterology 2020, 160, 1106–1117. [Google Scholar] [CrossRef]
- Companioni, O.; Bonet, C.; García, N.; Ramírez-Lázaro, M.J.; Lario, S.; Mendoza, J.; Adrados, M.M.; Poves, E.; Espinosa, L.; Pozo-Kreilinger, J.J.; et al. Genetic Variation Analysis in a Follow-up Study of Gastric Cancer Precursor Lesions Confirms the Association of MUC2 Variants with the Evolution of the Lesions and Identifies a Significant Association with NFKB1 and CD14. Int. J. Cancer 2018, 143, 2777–2786. [Google Scholar] [CrossRef]
- Díaz, P.; Valderrama, M.V.; Bravo, J.; Quest, A.F.G. Helicobacter Pylori and Gastric Cancer: Adaptive Cellular Mechanisms Involved in Disease Progression. Front. Microbiol. 2018, 9, 5. [Google Scholar] [CrossRef]
- Rosero-Rojas, S.C.; Chaleal-Cultid, J.A.; Pazos-Moncayo, Á.J.; Rosero-Galindo, C.Y. Polimorfismos IL1B-511 y TNF-A-308 En Una Población Infectada Con Helicobacter Pylori de Una Zona de Bajo Riesgo de Cáncer Gástrico En Nariño-Colombia. Infectio 2020, 24, 81. [Google Scholar] [CrossRef]
- Wu, L.; Schaid, D.; Sicotte, H.; Wieben, E.; Li, H.; Petersen, G. Case-Only Exome Sequencing and Complex Disease Susceptibility Gene Discovery: Study Design Considerations Lang. J. Med. Genet. 2015, 52, 10–16. [Google Scholar] [CrossRef]
- Desai, A.N.; Jere, A. Next-Generation Sequencing for Cancer Biomarker Discovery. In Next Generation Sequencing in Cancer Research; Wu, W., Choudhry, H., Eds.; Springer International Publishing: Cham, Switzerland, 2015; pp. 103–125. [Google Scholar]
- Li, W.Q.; Zhang, L.; Ma, J.L.; Zhang, Y.; Li, J.Y.; Pan, K.F.; You, W.C. Association between Genetic Polymorphisms of DNA Base Excision Repair Genes and Evolution of Precancerous Gastric Lesions in a Chinese Population. Carcinogenesis 2009, 30, 500–505. [Google Scholar] [CrossRef]
- Li, Z.W.; Wu, Y.; Sun, Y.; Liu, L.Y.; Tian, M.M.; Feng, G.S.; You, W.C.; Li, J.Y. Inflammatory Cytokine Gene Polymorphisms Increase the Risk of Atrophic Gastritis and Intestinal Metaplasia. World J. Gastroenterol. 2010, 16, 1788–1794. [Google Scholar] [CrossRef]
- Marín, F.; Bonet, C.; Muñoz, X.; García, N.; Pardo, M.L.; Ruiz-Liso, J.M.; Alonso, P.; Capellà, G.; Sanz-Anquela, J.M.; González, C.A.; et al. Genetic Variation in MUC1, MUC2 and MUC6 Genes and Evolution of Gastric Cancer Precursor Lesions in a Long-Term Follow-up in a High-Risk Area in Spain. Carcinogenesis 2012, 33, 1072–1080. [Google Scholar] [CrossRef]
- Meyerson, M.; Gabriel, S.; Getz, G. Advances in Understanding Cancer Genomes through Second-Generation Sequencing. Nat. Rev. Genet. 2010, 11, 685–696. [Google Scholar] [CrossRef]
- Chang, V. Whole Exome Sequencing of Pediatric Gastric Adenocarcinoma: A Germline and Somatic Mutation Analysis. Master’s Thesis, University of California, Los Angeles, CA, USA, 2012. [Google Scholar]
- Valdés-Mas, R.; Bea, S.; Puente, D.A.; López-Otín, C.; Puente, X.S. Estimation of Copy Number Alterations from Exome Sequencing Data. PLoS ONE 2012, 7, e51422. [Google Scholar] [CrossRef]
- Kim, Y.C.; Cui, J.; Luo, J.; Xiao, F.; Downs, B.; Wang, S.M. Exome-Based Variant Detection in Core Promoters. Sci. Rep. 2016, 6, 30716. [Google Scholar] [CrossRef]
- Wang, K.; Kan, J.; Yuen, S.T.; Shi, S.T.; Chu, K.M.; Law, S.; Chan, T.L.; Kan, Z.; Chan, A.S.Y.; Tsui, W.Y.; et al. Exome Sequencing Identifies Frequent Mutation of ARID1A in Molecular Subtypes of Gastric Cancer. Nat. Genet. 2011, 43, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Zang, Z.J.; Cutcutache, I.; Poon, S.L.; Zhang, S.L.; Mcpherson, J.R.; Tao, J.; Rajasegaran, V.; Heng, H.L.; Deng, N.; Gan, A.; et al. Exome Sequencing of Gastric Adenocarcinoma Identifies Recurrent Somatic Mutations in Cell Adhesion and Chromatin Remodeling Genes. Nat. Genet. 2012, 44, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler Transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map (SAM) Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- DePristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Sivachenko, A.Y.; Cibulskis, K.; Gabriel, S.B.; et al. A Framework for Variation Discovery and Genotyping Using Next- Generation DNA Sequencing Data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET Knowledge Platform for Disease Genomics: 2019 Update. Nucleic Acids Res. 2019, 48, D845–D855. [Google Scholar] [CrossRef]
- Park, J.; Yoo, H.M.; Jang, W.; Shin, S.; Kim, M.; Kim, Y.; Lee, S.W.; Kim, J.G.; Gurzu, S. Distribution of Somatic Mutations of Cancerrelated Genes According to Microsatellite Instability Status in Korean Gastric Cancer. Medicine 2017, 96, e7224. [Google Scholar] [CrossRef]
- Slavin, T.; Neuhausen, S.; Rybak, C.; Solomon, I.; Nehoray, B.; Blazer, K. Genetic Gastric Cancer Susceptibility in the International Clinical Cancer Genomics Community Research Network. Cancer Genet. 2017, 216–217, 111–119. [Google Scholar] [CrossRef]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.N.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-Genome Sequencing and Comprehensive Molecular Profiling Identify New Driver Mutations in Gastric Cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef]
- Abbas, M.; Faggian, A.; Sintali, D.N.; Khan, G.J.; Naeem, S.; Shi, M.; Dingding, C. Current and Future Biomarkers in Gastric Cancer. Biomed. Pharmacother. 2018, 103, 1688–1700. [Google Scholar] [CrossRef]
- Rosero, G.C.Y.; Mejía, O.L.; Corredor, M. A Review of Polymorphisms in Genes Involved in the Development of Gastric Cancer. Rev. Colomb. Gastroenterol. 2016, 31, 391–402. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Lee, J.; Lee, S.; Jang, J.-Y.; Park, T. Exact Association Test for Small Size Sequencing Data. BMC Med. Genom. 2018, 11, 30. [Google Scholar] [CrossRef]
- Ghodsi, M.; Amiri, S.; Hassani, H.; Ghodsi, Z. An Enhanced Version of Cochran-Armitage Trend Test for Genome-Wide Association Studies. Meta Gene 2016, 9, 225–229. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Manning, S.E.; Ku, H.-C.; Dluzen, D.F.; Xing, C.; Zhou, Z. A Nonparametric Alternative to the Cochran-Armitage Trend Test in Genetic Case-Control Association Studies: The Jonckheere-Terpstra Trend Test. PLoS ONE 2023, 18, e0280809. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting Functional Effect of Human Missense Mutations Using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 76, 7–20. [Google Scholar] [CrossRef]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the Effects of Coding Non-Synonymous Variants on Protein Function Using the SIFT Algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN Web Server: A Tool to Predict the Functional Effect of Amino Acid Substitutions and Indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef]
- Bravo, L.E.; Van Doorn, L.J.; Realpe, J.L.; Correa, P. Virulence-Associated Genotypes of Helicobacter Pylori: Do They Explain the African Enigma? Am. J. Gastroenterol. 2002, 97, 2839–2842. [Google Scholar] [CrossRef]
- Cuello, C.; Correa, P.; Haenszel, W.; Gordillo, G.; Brown, C.; Archer, M. Gastric Cancer in Colombia. I. Cancer Risk and Suspect Enviromental Agents. J. Natl. Cancer Inst. 1976, 37, 1015–1020. [Google Scholar]
- Chatrath, A.; Ratan, A.; Dutta, A. Germline Variants That Affect Tumor Progression. Trends Genet. 2021, 37, 433–443. [Google Scholar] [CrossRef]
- Baker, S.J.; Poulikakos, P.I.; Irie, H.Y.; Parekh, S.; Reddy, E.P. CDK4: A Master Regulator of the Cell Cycle and Its Role in Cancer. Genes Cancer 2022, 13, 21–45. [Google Scholar] [CrossRef]
- Fassl, A.; Geng, Y.; Sicinski, P. CDK4 and CDK6 Kinases: From Basic Science to Cancer Therapy. Science 2022, 375, eabc1495. [Google Scholar] [CrossRef]
- Javed, A.; Yarmohammadi, M.; Korkmaz, K.S.; Rubio-Tomás, T. The Regulation of Cyclins and Cyclin-Dependent Kinases in the Development of Gastric Cancer. Int. J. Mol. Sci. 2023, 24, 2848. [Google Scholar] [CrossRef]
- Ullah Shah, A.; Mahjabeen, I.; Kayani, M.A. Genetic Polymorphisms in Cell Cycle Regulatory Genes CCND1 and CDK4 Are Associated with Susceptibility to Breast Cancer. J. BUON 2015, 20, 985–993. [Google Scholar]
- Roa, S.J.C.; Vo, Q.; Araya, O.J.C.; Villaseca, H.M.; Guzmán, G.P.; Ibacache, S.G.; de Aretxabala, U.X.; Roa, E.I. Inactivación Del Gen CDKN2A (P16) En Cáncer de La Vesícula Biliar. Rev. Médica Chile 2004, 132, 11. [Google Scholar] [CrossRef]
- Royds, J.A.; Pilbrow, A.P.; Ahn, A.; Morrin, H.R.; Frampton, C.; Russell, I.A.; Moravec, C.S.; Sweet, W.E.; Tang, W.H.W.; Currie, M.J.; et al. The Rs11515 Polymorphism Is More Frequent and Associated With Aggressive Breast Tumors with Increased ANRIL and Decreased P16INK4a Expression. Front. Oncol. 2016, 5, 306. [Google Scholar] [CrossRef]
- Danishevich, A.; Bilyalov, A.; Nikolaev, S.; Khalikov, N.; Isaeva, D.; Levina, Y.; Makarova, M.; Nemtsova, M.; Chernevskiy, D.; Sagaydak, O.; et al. CDKN2A Gene Mutations: Implications for Hereditary Cancer Syndromes. Biomedicines 2023, 11, 3343. [Google Scholar] [CrossRef]
- Steri, M.; Idda, M.L.; Whalen, M.B.; Orrù, V. Genetic Variants in MRNA Untranslated Regions. WIREs RNA 2018, 9, e1474. [Google Scholar] [CrossRef]
- Sauroja, I.; Smeds, J.; Vlaykova, T.; Kumar, R.; Talve, L.; Hahka-Kemppinen, M.; Punnonen, K.; Jansèn, C.T.; Hemminki, K.; Pyrhönen, S. Analysis of G1/S Checkpoint Regulators in Metastatic Melanoma. Genes Chromosomes Cancer 2000, 28, 404–414. [Google Scholar] [CrossRef]
- Geddert, H.; Kiel, S.; Zotz, R.B.; Zhang, J.; Willers, R.; Gabbert, H.E.; Sarbia, M. Polymorphism of P16 INK4A and Cyclin D1 in Adenocarcinomas of the Upper Gastrointestinal Tract. J. Cancer Res. Clin. Oncol. 2005, 131, 803–808. [Google Scholar] [CrossRef]
- Moreira, A.M.; Pereira, J.; Melo, S.; Fernandes, M.S.; Carneiro, P.; Seruca, R.; Figueiredo, J. The Extracellular Matrix: An Accomplice in Gastric Cancer Development and Progression. Cells 2020, 9, 394. [Google Scholar] [CrossRef]
- Xu, F.; Chang, K.; Ma, J.; Qu, Y.; Xie, H.; Dai, B.; Gan, H.; Zhang, H.; Shi, G.; Zhu, Y.; et al. The Oncogenic Role of COL23A1 in Clear Cell Renal Cell Carcinoma. Sci. Rep. 2017, 7, 9846. [Google Scholar] [CrossRef]
- Packer, D.; Martin, P.T. Micro-Laminin Gene Therapy Can Function as an Inhibitor of Muscle Disease in the DyW Mouse Model of MDC1A. Mol. Ther. Methods Clin. Dev. 2021, 21, 274–287. [Google Scholar] [CrossRef]
- Wang, R.-Q.; Lan, Y.-L.; Lou, J.-C.; Lyu, Y.-Z.; Hao, Y.-C.; Su, Q.-F.; Ma, B.-B.; Yuan, Z.-B.; Yu, Z.-K.; Zhang, H.-Q.; et al. Expression and Methylation Status of LAMA2 Are Associated with the Invasiveness of Nonfunctioning PitNET. Ther. Adv. Endocrinol. Metab. 2019, 10, 2042018818821296. [Google Scholar] [CrossRef]
- Shibuya, M. Involvement of Flt-1 (VEGF Receptor-1) in Cancer and Preeclampsia. Proc. Jpn. Acad. Ser. B 2011, 87, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Malespín-Bendaña, W.; Alpízar-Alpízar, W.; Figueroa-Protti, L.; Reyes, L.; Molina-Castro, S.; Une, C.; Ramírez-Mayorga, V. Helicobacter Pylori Infection Induces Gastric Precancerous Lesions and Persistent Expression of Angpt2, Vegf-A and Tnf-A in a Mouse Model. Front. Oncol. 2023, 13, 1072802. [Google Scholar] [CrossRef]
- Ma, Y.; Wang, W.; Liu, L.; Liu, Y.; Bi, W. Co-Expression of VEGF-B and FLT-1 Correlates with Malignancy and Prognosis of Gastric Cancer. Biomark. Med. 2021, 15, 481–488. [Google Scholar] [CrossRef]
- Qian, B.-Z.; Zhang, H.; Li, J.; He, T.; Yeo, E.-J.; Soong, D.Y.H.; Carragher, N.O.; Munro, A.; Chang, A.; Bresnick, A.R.; et al. FLT1 Signaling in Metastasis-Associated Macrophages Activates an Inflammatory Signature That Promotes Breast Cancer Metastasis. J. Exp. Med. 2015, 212, 1433–1448. [Google Scholar] [CrossRef] [PubMed]
- Jaroenlapnopparat, A.; Bhatia, K.; Coban, S. Inflammation and Gastric Cancer. Diseases 2022, 10, 35. [Google Scholar] [CrossRef] [PubMed]
- Ida, S.; Watanabe, M.; Baba, H. Chronic Inflammation and Gastrointestinal Cancer. J. Cancer Metastasis Treat. 2015, 1, 138. [Google Scholar] [CrossRef]
- Cho, N.; Seong, S. Apolipoproteins Inhibit the Innate Immunity Activated by Necrotic Cells or Bacterial Endotoxin. Immunology 2009, 128, e479–e486. [Google Scholar] [CrossRef]
- Ren, L.; Yi, J.; Li, W.; Zheng, X.; Liu, J.; Wang, J.; Du, G. Apolipoproteins and Cancer. Cancer Med. 2019, 8, 7032–7043. [Google Scholar] [CrossRef]
- Pih, G.Y.; Gong, E.J.; Choi, J.Y.; Kim, M.-J.; Ahn, J.Y.; Choe, J.; Bae, S.E.; Chang, H.-S.; Na, H.K.; Lee, J.H.; et al. Associations of Serum Lipid Level with Gastric Cancer Risk, Pathology, and Prognosis. Cancer Res. Treat. 2021, 53, 445–456. [Google Scholar] [CrossRef]
- Aceves-Ramírez, M.; Valle, Y.; Casillas-Muñoz, F.; Martínez-Fernández, D.E.; Parra-Reyna, B.; López-Moreno, V.A.; Flores-Salinas, H.E.; Valdés-Alvarado, E.; Muñoz-Valle, J.F.; García-Garduño, T.; et al. Analysis of the APOB Gene and Apolipoprotein B Serum Levels in a Mexican Population with Acute Coronary Syndrome: Association with the Single Nucleotide Variants Rs1469513, Rs673548, Rs676210, and Rs1042034. Genet. Res. 2022, 2022, 4901090. [Google Scholar] [CrossRef]
- Liu, C.; Yang, J.; Han, W.; Zhang, Q.; Shang, X.; Li, X.; Lu, F.; Liu, X. Polymorphisms in ApoB Gene Are Associated with Risk of Myocardial Infarction and Serum ApoB Levels in a Chinese Population. Int. J. Clin. Exp. Med. 2015, 8, 16571–16577. [Google Scholar]
- Falanga, A.; Marchetti, M.; Vignoli, A. Coagulation and Cancer: Biological and Clinical Aspects. J. Thromb. Haemost. 2013, 11, 223–233. [Google Scholar] [CrossRef]
- Moik, F.; Ay, C. Hemostasis and Cancer: Impact of Haemostatic Biomarkers for the Prediction of Clinical Outcomes in Patients with Cancer. J. Thromb. Haemost. 2022, 20, 2733–2745. [Google Scholar] [CrossRef]
- Liu, Y.; Liao, X.-W.; Qin, Y.-Z.; Mo, X.-W.; Luo, S.-S. Identification of F5 as a Prognostic Biomarker in Patients with Gastric Cancer. BioMed Res. Int. 2020, 2020, 9280841. [Google Scholar] [CrossRef] [PubMed]
- Tinholt, M.; Sandset, P.M.; Iversen, N. Polymorphisms of the Coagulation System and Risk of Cancer. Thromb. Res. 2016, 140, S49–S54. [Google Scholar] [CrossRef] [PubMed]
- Tinholt, M.; Garred, Ø.; Borgen, E.; Beraki, E.; Schlichting, E.; Kristensen, V.; Sahlberg, K.K.; Iversen, N. Subtype-specific Clinical and Prognostic Relevance of Tumor-expressed F5 and Regulatory F5 Variants in Breast Cancer: The CoCaV Study. J. Thromb. Haemost. 2018, 16, 1347–1356. [Google Scholar] [CrossRef]
- Jorgensen, A.L.; Al-Zubiedi, S.; Zhang, J.E.; Keniry, A.; Hanson, A.; Hughes, D.A.; Eker, D.V.; Stevens, L.; Hawkins, K.; Toh, C.H.; et al. Genetic and Environmental Factors Determining Clinical Outcomes and Cost of Warfarin Therapy: A Prospective Study. Pharmacogenet. Genom. 2009, 19, 800–812. [Google Scholar] [CrossRef]
- GeneCards Gene-Fibrinogen Alpha Chain. Available online: https://www.genecards.org/cgi-bin/carddisp.pl?gene=FGA (accessed on 3 November 2024).
- Shi, F.; Wu, H.; Qu, K.; Sun, Q.; Li, F.; Shi, C.; Li, Y.; Xiong, X.; Qin, Q.; Yu, T.; et al. Identification of Serum Proteins AHSG, FGA and APOA-I as Diagnostic Biomarkers for Gastric Cancer. Clin. Proteom. 2018, 15, 18. [Google Scholar] [CrossRef]

{kind=link}
| GRCh38 Chr: Position | Gene | Ref/Alt Alleles 1 | Variant Type | SNP ID | Fisher Exact Test | Cochran–Armitage Test | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| MAF 2 Progression (N = 21) | MAF 2 Regression (N = 16) | p-Value | Alt/Ref Progression (N = 21) | Alt/Ref Regression (N = 16) | p-Value | |||||
| 13:28390061 | FLT1 | C/T | Exonic | rs17537350 | 0.67 | 0.28 | 0.002 | 28/14 | 9/23 | 0.001 |
| 13:28494902 | FLT1 | G/A | UTR5 | rs55927955 | 0.62 | 0.25 | 0.002 | 26/16 | 8/24 | 0.001 |
| 13:28322842 | FLT1 | C/T | Exonic | rs56314249 | 0.60 | 0.25 | 0.004 | 25/17 | 8/24 | 0.003 |
| 3:195870036 | TNK2 | T/C | Exonic | rs2278034 | 0.52 | 0.22 | 0.009 | 22/20 | 7/25 | 0.003 |
| 7:84129375 | SEMA3A | C/T | Exonic | rs7789342 | 0.79 | 0.47 | 0.007 | 33/9 | 15/17 | 0.003 |
| 2:21008652 | APOB | G/A | Exonic | rs676210 | 0.50 | 0.19 | 0.007 | 21/21 | 6/26 | 0.003 |
| 2:21014672 | APOB | G/A | Intronic | rs673548 | 0.50 | 0.19 | 0.007 | 21/21 | 6/26 | 0.003 |
| 6:129349403 | LAMA2 | C/T | Intronic | rs17057158 | 0.33 | 0.06 | 0.009 | 14/28 | 2/30 | 0.006 |
| 6:129369987 | LAMA2 | C/G | Exonic | rs17057184 | 0.33 | 0.06 | 0.009 | 14/28 | 2/30 | 0.006 |
| 3:195868079 | TNK2 | G/A | Exonic | rs56260729 | 0.31 | 0.06 | 0.010 | 13/29 | 2/30 | 0.006 |
| 18:27993437 | CDH2 | T/C | Intronic | rs11564295 | 0.31 | 0.06 | 0.010 | 13/29 | 2/30 | 0.012 |
| 2:137275858 | THSD7B | G/A | Intronic | rs67227746 | 0.31 | 0.06 | 0.010 | 13/29 | 2/30 | 0.028 |
| 3:195885163 | TNK2 | T/C | Intronic | rs3747672 | 0.68 | 0.39 | 0.040 | 23/11 | 11/17 | 0.003 |
| 7:84134993 | SEMA3A | C/G | Intronic | rs17241389 | 0.79 | 0.50 | 0.014 | 33/9 | 16/16 | 0.005 |
| 9:21968200 | CDKN2A | C/G | UTR3 | rs11515 | 1.00 | 0.84 | 0.013 | 42/0 | 27/5 | 0.006 |
| 1:175355515 | TNR | A/G | Exonic | rs2228359 | 0.88 | 0.66 | 0.025 | 37/5 | 21/11 | 0.006 |
| 7:6402057 | RAC1 | T/C | Intronic | rs2303364 | 0.24 | 0.03 | 0.019 | 10/32 | 1/31 | 0.006 |
| 9:8527144 | PTPRD | AC/A | Intronic | rs113334891 | 0.24 | 0.03 | 0.019 | 10/32 | 1/31 | 0.006 |
| 17:82081605 | FASN | G/A | Exonic | rs1140616 | 0.21 | 0.47 | 0.026 | 9/33 | 15/17 | 0.008 |
| 4:125415548 | FAT4 | T/C | Exonic | rs17009618 | 0.45 | 0.19 | 0.025 | 19/23 | 6/26 | 0.010 |
| 7:84110699 | SEMA3A | T/C | Intronic | rs1990045 | 0.76 | 0.47 | 0.014 | 32/10 | 15/17 | 0.010 |
| 3:195888505 | TNK2 | A/G | Exonic | rs3747669 | 0.55 | 0.28 | 0.033 | 23/19 | 9/23 | 0.010 |
| 16:85657272 | GSE1 | C/CA | Intronic | rs34805959 | 0.30 | 0.06 | 0.016 | 12/28 | 2/30 | 0.004 |
| 3:105704000 | CBLB | C/T | Exonic | rs2305037 | 0.86 | 0.53 | 0.004 | 36/6 | 17/15 | 0.004 |
| 12:57750882 | CDK4 | C/T | Intronic | rs2069502 | 0.36 | 0.09 | 0.013 | 15/27 | 3/29 | 0.008 |
| 12:51975516 | ACVR1B | C/T | Intronic | rs2242106 | 0.79 | 0.50 | 0.014 | 33/9 | 16/16 | 0.009 |
| 12:51976172 | ACVR1B | G/GA | Intronic | rs3214829 | 0.79 | 0.50 | 0.014 | 33/9 | 16/16 | 0.009 |
| 1:169514323 | F5 | T/C | Exonic | rs6027 | 0.29 | 0.03 | 0.005 | 12/30 | 1/31 | 0.004 |
| 1:169541110 | F5 | T/C | Exonic | rs1800595 | 0.29 | 0.03 | 0.005 | 12/30 | 1/31 | 0.004 |
| 1:169542640 | F5 | T/G | Exonic | rs6018 | 0.29 | 0.03 | 0.005 | 12/30 | 1/31 | 0.004 |
| 1:169542801 | F5 | T/C | Exonic | rs6024 | 0.29 | 0.03 | 0.005 | 12/30 | 1/31 | 0.004 |
| 1:169541286 | F5 | A/G | Exonic | rs1800594 | 0.76 | 0.44 | 0.007 | 32/10 | 14/18 | 0.009 |
| 1:169529737 | F5 | T/C | Exonic | rs6030 | 0.74 | 0.44 | 0.016 | 31/11 | 14/18 | 0.010 |
| 1:223766378 | CAPN2 | A/C | Exonic | rs17599 | 0.43 | 0.13 | 0.005 | 18/24 | 4/28 | 0.002 |
| 5:178256904 | COL23A1 | G/A | Exonic | rs61739424 | 0.19 | 0.00 | 0.009 | 8/34 | 0/32 | 0.005 |
| 5:178256458 | COL23A1 | G/A | Intronic | rs11948620 | 0.31 | 0.00 | 0.001 | 11/25 | 0/30 | 0.001 |
| 5:178259790 | COL23A1 | T/C | Intronic | rs2973748 | 0.26 | 0.00 | 0.002 | 11/31 | 0/32 | 0.002 |
| 4:154586438 | FGA | T/C | Exonic | rs6050 | 0.50 | 0.19 | 0.007 | 21/21 | 6/26 | 0.003 |
| Gene | SNP ID | Amino Acid Change | Prediction | ||
|---|---|---|---|---|---|
| SIFT | POLYPHEN | PROVEAN | |||
| F5 | rs6027 | D2222G | D | Probably damaging | D |
| F5 | rs1800595 | H1327R | T | Benign | N |
| F5 | rs6018 | N817T | D | Benign | D |
| F5 | rs6030 | M1764V | T | Benign | N |
| FGA | rs6050 | T331A | T | Benign | N |
| TNK2 | rs56260729 | P757L | T | Benign | N |
| CAPN2 | rs17599 | K490Q | D | Benign | D |
| COL23A1 | rs61739424 | R267W | D | - | D |
| APOB | rs676210 | P2739L | D | Probably damaging | D |
| Pathway ID | Description | p-Value | Genes |
|---|---|---|---|
| R-HSA-8957275 | Post-translational protein phosphorylation | 0.000025 | APOB, CDH2, F5, FGA |
| R-HSA-1474244 | Extracellular matrix organization | 0.000105 | CAPN2, COL23A1, FGA, LAMA2, TNR |
| R-HSA-3000178 | ECM proteoglycans | 0.006982 | LAMA2, TNR |
| R-HSA-1474228 | Degradation of the extracellular matrix | 0.022382 | CAPN2, COL23A1 |
| R-HSA-5686938 | Regulation of TLR by endogenous ligand | 0.000443 | APOB, FGA |
| R-HSA-168898 | Toll-like Receptor Cascades | 0.026409 | APOB, FGA |
| R-HSA-9006927 | Signaling by Non-Receptor Tyrosine Kinases | 0.003583 | CDK4, RAC1 |
| R-HSA-194138 | Signaling by VEGF | 0.013237 | FLT1, RAC1 |
| R-HSA-69231 | Cyclin D-associated events in G1 | 0.002392 | CDK4, CDKN2A |
| R-HSA-69236 | G1 Phase | 0.002392 | CDK4, CDKN2A |
| R-HSA-109582 | Hemostasis | 0.023201 | F5, FGA |
| R-HSA-140877 | Formation of Fibrin Clot (Clotting Cascade) | 0.001883 | F5, FGA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mejia-Ortiz, L.; Zabaleta, J.; Garai, J.; Bravo, L.E.; Castillo, A. Whole-Exome Sequencing Identified Molecular Variants Linked to the Progression of Gastric Precancerous Lesions in Patients from Southwestern Colombia—An Exploratory Approach. Gastrointest. Disord. 2025, 7, 30. https://doi.org/10.3390/gidisord7020030
Mejia-Ortiz L, Zabaleta J, Garai J, Bravo LE, Castillo A. Whole-Exome Sequencing Identified Molecular Variants Linked to the Progression of Gastric Precancerous Lesions in Patients from Southwestern Colombia—An Exploratory Approach. Gastrointestinal Disorders. 2025; 7(2):30. https://doi.org/10.3390/gidisord7020030
Chicago/Turabian StyleMejia-Ortiz, Lizeth, Jovanny Zabaleta, Jone Garai, Luis Eduardo Bravo, and Andres Castillo. 2025. "Whole-Exome Sequencing Identified Molecular Variants Linked to the Progression of Gastric Precancerous Lesions in Patients from Southwestern Colombia—An Exploratory Approach" Gastrointestinal Disorders 7, no. 2: 30. https://doi.org/10.3390/gidisord7020030
APA StyleMejia-Ortiz, L., Zabaleta, J., Garai, J., Bravo, L. E., & Castillo, A. (2025). Whole-Exome Sequencing Identified Molecular Variants Linked to the Progression of Gastric Precancerous Lesions in Patients from Southwestern Colombia—An Exploratory Approach. Gastrointestinal Disorders, 7(2), 30. https://doi.org/10.3390/gidisord7020030