Abstract
Background: Gastrointestinal Stromal Tumors (GISTs) are characterized as round, well–defined mass lesions in the submucosal layer of the gastrointestinal (GI) tract. GISTs often present histological diversity and mutations in c-KIT and PDGFRA genes. Symptoms usually appear as abdominal pain, often accompanied by gastrointestinal bleeding or abdominal mass. The prognosis relies on tumor size, mitotic index, and different mutations, such as KIT mutations. There are a variety of diagnostic measures in the case of GISTs. However, it is important to note that ultrasound is the most common and reliable method for diagnosing gastric GISTs. The treatment methods followed vary from preoperative systemic therapy to surgical interventions. Depending on the type of GIST, professionals decide upon the best treatment plan for the patient. Objective: This review aims to inform the scientific community about the intricacies of gastric and small intestine GISTs to enhance understanding and improve patient management, with a particular focus on the importance of understanding and interpreting the unique microscopic histopathological findings of GISTs.
1. The Context of the Epidemic Rise in GISTs
The research findings on Gastrointestinal Stromal Tumor (GIST) incidences are estimated to be 7–15 cases per 1 million people annually [1,2,3,4]. GISTs are rare and account for only about 1–2% of all primary malignant tumors that originate in the gastrointestinal (GI) tract [5,6]. The two main events in GIST pathogenesis are KIT and PDGFRA gene mutations. According to molecular epidemiology, it is suggested that GISTs with KIT mutations have an incidence of close to 8 cases per 106 individuals per year in most regions. In comparison, GISTs with PDGFRA mutations have an incidence of fewer than 3 cases per 106 individuals per year. Other forms of GISTs have an incidence of less than 1 case per 106 individuals per year [7,8].
While GISTs can develop anywhere along the GI tract, they are predominantly found in the stomach (60%) or small intestine (20–30%). However, they can occasionally be found in the momentum, mesentery, and peritoneum [9,10], and thus, they are of the utmost importance for medical professionals. A recently published, descriptive, population-based cohort study by Alvarez et al. [11] concluded that the incidence of GISTs in major organ sites has increased among various population groups in the past two decades. More precisely, by using the data from the National Cancer Institute Surveillance, Epidemiology, and End Results (SEER) Program, including the SEER-22 and SEER-17 registries, this study underlined that for 23,001 patients, the age-adjusted incidence rates for common digestive GISTs (i.e., gastric, small intestine, and colon GISTs) increased between 2% and 7% during the period from 2000 and 2019, primarily for early-stage tumors [11]. Regarding the risk factors associated with the high prevalence of GISTs, the authors observed that male sex, Black race, older age at diagnosis, advanced stage at diagnosis, tumor size greater than 5 cm, poorly and undifferentiated grade, and early year of diagnosis are associated with worse GIST-specific survival [11]. These results were similar to the results of the study by Khan et al. [6], who, by retrieving data from 10,833 patients using the SEER database for the period 2000–2018, found that an age over 60 years old, male gender, race, tumor size, and tumor stage were associated with worse overall survival, consistent also with a previous SEER database study covering data from 1998 to 2011 [12].
However, even though remarkable improvements in diagnostic imaging tools have been made, the National Comprehensive Cancer Network (NCCN) and the European Society for Medical Oncology clinical practice guidelines for GIST diagnosis have remained almost unchanged in the past two decades [13,14]. This suggests that while better diagnostic methods are available, there are still challenges in their implementation, and they may be unrelated to the high prevalence of GISTs. In the same context are environmental and lifestyle exposures [11].
2. Types, Classification, and Histologic Spectrum of GISTs
GISTs are characterized as round, well-defined mass lesions in the submucosal layer of the GI tract, which express the KIT (CD117) protein [15]. The KIT protein is a classical oncogene that encodes a receptor tyrosine kinase that responds to stem cell factor [16]. The types of cells that consist of GISTs are spindle, epithelioid, or pleomorphic mesenchymal cells. GISTs can be histologically classified into eight distinct subgroups based on their size, differentiation, and probability of metastasis based on their location. To be more precise, the subgroups, based on the National Institute of Health NIH classification [17] and related articles, are defined as follows [18,19,20]:
- Tumors equal to or less than 2 cm with five or fewer mitoses per 50 high-power fields (HPFs) have a 0% probability of giving metastasis in the stomach and small intestine.
- Tumors equal to or less than 2 cm with over five mitoses per 50 HPFs have a 0% probability of giving metastasis in the stomach. However, these tumors have a high probability (50%) of giving metastasis in the duodenum of the small intestine.
- Tumors with a size between 3 and 5 cm and five or fewer mitoses per 50 HPFs have a 1.9%, 4.3%, and 8.3% probability of giving metastasis in the stomach, duodenum, and jejunum–ileum, respectively.
- Tumors with a size between 3 and 5 cm and over five mitoses per 50 HPFs have a 16%, 73%, and 50% probability of giving metastasis in the stomach, duodenum, and jejunum–ileum, respectively.
- Tumors with a size between 6 and 10 cm and five or fewer mitoses per 50 HPFs have a 3.6%, 24%, and 34% probability of giving metastasis in the stomach, duodenum, and jejunum–ileum, respectively.
- Tumors with a size between 6 and 10 cm and over five mitoses per 50 HPFs have a 55%, 85%, and 86% probability of giving metastasis in the stomach, duodenum, and jejunum–ileum, respectively.
- Tumors over 11 cm with five or fewer mitoses per 50 HPFs have a 12%, 52%, and 34% probability of giving metastasis in the stomach, duodenum, and jejunum–ileum, respectively.
- Tumors over 11 cm with over five mitoses per 50 HPFs have a 86%, 90%, and 86% probability of giving metastasis in the stomach, duodenum, and jejunum–ileum, respectively.
However, apart from the above GIST subgroups, the Armed Forces Institute of Pathology (AFIP) [21] has also proposed a GIST classification since primary tumor location is a significant prognostic risk factor for patients with GISTs. Goh et al. [22] compared and validated the performance of the NIH and AFIP risk criteria for GISTs. They modified the NIH and AFIP risk criteria for GISTs and applied them to several surgically treated localized primary GISTs at one institution. The goal was to determine the most influential risk stratification system for GIST. The study found that the proposed modified AFIP criteria demonstrated the best independent predictive value of RFS compared to other systems. The researchers concluded that the NIH, modified NIH, and AFIP criteria are all helpful in prognosticating GIST. The AFIP risk criteria provided the best prognostication for primary localized GIST among the three systems.
These subtypes form a continuum based on their biological potential, and their specific identification is crucial for understanding these tumors’ behavior and treatment response. GIST differentiation in the GI tract is mainly contributed by three markers: (1) the KIT (CD117) protein, the DOG-1 protein, and the protein kinase C theta (PKC-theta) [23,24]. However, although 95% of GISTs express CD117, a high percentage (70–90%) of GISTs also express the human progenitor cell antigen (CD34), contributing to these tumors’ differentiation throughout the GI tract [25,26].
GIST classification can also be performed based on microscopic findings. Microscopically, GISTs may exhibit moderate or high cellularity and can be categorized into three main types: (1) spindle cell type (70%), (2) epithelioid type (20%), and (3) mixed type (10%) [27,28,29]. Regarding the gastric GISTs, up to 70% of them can be histologically subclassified into eight subtypes: four describe spindle cell tumors and four describe epithelioid tumors [30]. Unlike gastric GISTs, small intestine GISTs do not form distinctive histologic subtypes. Most small intestinal GISTs comprise spindle cells, and around 40–50% contain distinct, round, oval, or elongated eosinophilic and periodic acid–Schiff-positive aggregates of extracellular collagen fibers [30,31]. In addition, most GISTs of sites other than stomach and small intestine are spindle cell tumors [32].
3. Mutations and Pathogenesis of GISTs
3.1. KIT Mutations
The groundbreaking discovery of activating mutations of c-KIT in GISTs was reported in 1998 [33]. This discovery has not only significantly advanced our understanding of GISTs but also opened up new avenues for treatment. It is now well established that 75% of GISTs harbor KIT mutations. The KIT gene, which encodes the c-KIT receptor tyrosine kinase, a type III tyrosine kinase receptor, plays a crucial role in the development of GISTs. This receptor binds with the stem cell factor (SCF) ligand, leading to c-KIT dimerization and activation of the receptor. This activation triggers downstream signaling through PI3K/AKT and RAS pathways [34]. The mutations in the KIT gene result in uncontrolled activation of the tyrosine kinase site and increased cellular growth. These mutations can be categorized as primary or secondary, with secondary mutations emerging during tyrosine kinase inhibitor treatment, leading to secondary drug resistance [33]. Recent studies have revealed that in addition to the more common sporadic GISTs with somatic KIT mutation, there are rare cases of familial GISTs linked to heritable germline mutations of KIT. To date, more than 31 families and six individuals have been identified with germline mutations in KIT in exons 8, 9, 11, 13, and 17 [35,36,37].
From the KIT mutations mentioned above, the most popular primary mutations in the KIT gene have been found in exon 11 coding for intracellular juxta membrane (66–71%) [38]. In this mutation, a duplication in the 3′ region of the KIT exon 11 causes the gastric GIST, and the deletions in the exon 11 can result in a higher level of aggressiveness compared to point mutations [33]. Next in line are mutations in exon 9 coding for the extracellular domain (13%) [38], often located in the small intestine, colon, or rectum [9,39,40]. In this mutation, a duplication of A502–Y503 codons in exon 9 emerges. However, the frequency of exon 11 and exon 9 mutations varies across different anatomical sites. The prevalence of KIT exon 11 mutations in the gastric is between 54 and 60%; in the small intestine, it is between 43 and 50%; and in the rectum, it is between 70% and 80%. Respectively, the prevalence of KIT exon 9 mutations in the gastric is <5%; in the small intestine, it is between 20% and 25%; and in the rectum, it is between 10% and 15% [9,40,41,42,43]. Regarding other mutations, it is important to note that primary missense mutations in KIT exon 13 and exon 17 are rare occurrences. Specifically, the most frequently observed primary mutation in exon 13 is a K642E substitution, while the most primary and secondary KIT exon 17 mutations are missense mutations found in codons 820, 822, and 823 [44,45].
3.2. PDGFRA Mutations
Mutations in PDGFRA are primarily located in exon 18, a region where they are most commonly found. Less common occurrences are in exon 12 or 14. Specifically, PDGFRA mutations in exon 12 occur in the juxta membrane regulatory domain, and in exon 14, they occur in the tyrosine kinase domains-ATP-binding region. It is important to note that most PDGFRA-mutated GISTs have a mostly epithelioid histology and are mainly located in the stomach (15–18%), followed by the small intestine (5–7%) [16,36,38,46,47].
3.3. Other Mutations
GISTs without KIT or PDGFRA mutations can be divided into SSDH-deficient and SDH-competent GISTs. SDH-deficient GISTs include those with mutations in genes encoding SDH subunits (A, B, C, and D) [48,49,50]. SDH deficiency leads to the buildup of intracellular succinate, which in turn inhibits a wide range of enzymes, including prolyl hydroxylases, JmjC domain-containing histone demethylases (KDMs), and the TET family of dioxygenases [51,52,53]. In addition, it is noteworthy that SDH-deficient GISTs can arise from genetic mutations (SDHA, SDHB, SDHC, and SDHD) or epigenetic changes (SDHC promoter methylation). These tumors can be sporadic, caused by somatic mutations within the cancer, or familial, related to germline mutations. Currently, most cases are characterized as familial [49,53].
However, studies have shown that SDH-deficient GISTs that develop in children and young patients can be associated with Carney–Stratakis syndrome (CSS) and Carney triad (CT). Carney–Stratakis syndrome is a rare hereditary condition characterized by multifocal paragangliomas and GISTs. Considering the molecular pathogenesis of this autosomal-dominant syndrome, most CSS patients have confirmed constitutional inactivating mutations of SDHB, SDHC, or SDHD. On the other hand, CT, which was first described in 1977 as a triad of gastric leiomyosarcoma (the GIST we know today), paragangliomas, and pulmonary chondromas, is a rare non-hereditary disease caused by the association of the above elements [54,55,56,57]. In contrast, SDH components include those with mutations in the RAS–MEK–MAPK pathway genes, those with translocations involving NTRK or FGFR genes, and others with rare mutations [45,48,49,50,58,59].
Moreover, GISTs have been associated with Neurofibromatosis type 1 (NF-1), an autosomal dominant tumor syndrome caused by a mutation of the NF1 gene located on chromosome 17. GISTs related to NF-1 are typically small in size, have a low mitotic rate, are located in the small intestine, are often multifocal spindle cells, and have a good prognosis. Based on the literature, around 7% of patients will develop a GIST at some point in their lifetime [60,61].
4. The Challenging Road to Diagnosis of GISTs
Ultrasound, magnetic resonance imaging (MRI), computed tomography (CT), PET-CT imaging, endoscopy, and endoscopic-guided fine needle aspiration (FNA) or core biopsy are some of the various diagnostic modalities used for GISTs.
4.1. Endoscopy and Endoscopic Ultrasound
The most preferred method for GIST diagnosis is endoscopic ultrasound [62], as it distinguishes the gastrointestinal system’s layers and facilitates needle or core biopsy. To be more specific, when it comes to gastric GISTs, the primary diagnostic methods are endoscopy and CT [20]. Endoscopy is a powerful tool for detecting intramural tumors and is critical in collecting cytological and histological samples. This step is crucial for a definitive diagnosis. Unlike histology, cytology does not provide information on the mitotic rate. In emergencies requiring immediate surgery, the suspected diagnosis of GIST is confirmed post-surgery through a critical step-histological examination of the resected tumor specimen [63].
4.2. Computed Tomography
Contrast-enhanced CT, such as GISTs, is the preferred imaging method for identifying and describing neoplasms. It is also effective in assessing the extent and presence of metastatic disease and gathering additional information about the relationship of GIST to the gastrointestinal wall and the surrounding organs [64]. Regarding GIST size, CT can provide indirect hints regarding tumor size, growth pattern, dignity, and prognostic outcome. Those with size <5 cm are typically benign and appear as sharply margined, smooth-walled, homogeneous, soft-tissue masses on CT. They usually involve the stomach or small intestine wall and may have an intraluminal component [65]. In addition, they typically appear similar in contrast-enhanced CT images. However, some focal areas of low attenuation on CT may be due to hypocellular tumor, hemorrhage, necrosis, cystic degeneration, and ulcer fluid. There is no proven relationship between these findings and malignant potential [66,67].
Larger tumors, with a size over 6 cm, often have central areas of necrosis or hemorrhage. Mural calcifications are rare and typically have a unique ‘dumbbell-like’ appearance. This distinctive shape, with the tumor protruding into the bowel wall’s lumen and growing outward from the serosa, is crucial in identifying large GISTs. Large GISTs may also appear as well-demarcated extraluminal masses, a characteristic that can make it difficult to appreciate the natural origin of the tumor. However, the use of multiplanar reconstructions on multidetector-row CT can be instrumental in identifying the organ of origin, underscoring the importance of this technique in the diagnosis of large GISTs [66,68,69,70,71,72].
4.3. Magnetic Resonance Imaging
MRI provides high soft-tissue contrast, enabling the visualization of tumor extent, central necrosis, and hemorrhage [73]. MRI findings confirm that small GISTs tend to be round with substantial, homogeneous arterial enhancement. In contrast, large GISTs tend to have a lobulated appearance and typically exhibit mild, heterogeneous, gradual enhancement with intratumoral cystic changes [74]. Even though MRI’s diagnostic performance is comparable to CT, CT scans remain the preferred initial imaging method for staging the disease. However, MRI may be a better imaging option in some cases (i.e., large-in-size GISTs, GISTs metastases, and rectum GISTs) [75,76].
4.4. Immunohistochemical Methods
Immunohistochemical methods are considered alternative strategies for diagnosing GISTs, which are hard to diagnose with just hematoxylin–eosin staining—a technique commonly used in histology to visualize the cells and the tissue’s morphology and structure. Less than 5% of GIST cases do not express CD117 (KIT-KIT proto-oncogene receptor tyrosine kinase) and DOG-1 mutations. Approximately 80% of the cases express PDGFRA (platelet-derived growth factor receptor α), and 70 to 80% express CD34 [77,78,79].
4.5. Diagnostic Challenges
GIST diagnosis is challenging due to the unexpected nature of their discovery. Asymptomatic mini-GISTs smaller than 2 cm are incidentally found during endoscopy, and micro-GISTs smaller than 1 cm are incidentally discovered during pathological examinations of resected specimens [3,38,79]. During random diagnostic endoscopy, micro-GISTs are found in the stomachs and rectums of middle-aged adults in 10–35% and 0.1–0.2% of cases, respectively [79,80,81,82]. In the same context, endoscopy may also detect small neoplastic submucosal tumors in 0.15% of middle-aged adults, half of which are considered GISTs [83,84].
Small intestine GISTs, once often overlooked and delayed in diagnosis, have recently seen a ray of hope. This is due to the scarcity and difficulty in detecting small intestine neoplasms in early images, presenting a continuous clinical challenge characterized by low morbidity, common symptoms, diverse imaging manifestations, pleomorphic bowel, and overlapping intestinal loops [85]. However, the incidence of GISTs in the small intestine is currently considered more invasive than GISTs of the same size in the gastric. This increase in occurrence is a phenomenon that some scholars attribute to advances in radiology and endoscopy techniques and improved physician awareness [86,87]. Original microscopic copyright data from the Laboratory of Autopsy Pathology, Forensic Service of Thessaloniki, Ministry of Justice, Greece, revealed that an intestinal histotome, which was taken after the death of a male person and was an incidental autopsy finding, presented unique features, with elements of autolysis, areas showing inflammatory cells, and vessels variably congested. The tumor was categorized as low-grade malignant, with 2–3 mitoses per 10 HPFs. The microscopically described nodules were characterized in some places by abundant interwoven bundles of fibrillar cells oriented in various directions with eosinophilic and focal lightly colored cytoplasm and elongated nuclei with relative hyperchromasia. The layer showed several variably sized, sometimes hyperemic vessels and focal sparse chronic inflammatory changes. Overall, the presence of nodules corresponded to a rare, benign positive to CD117 GIST (Figure 1). Figure 2 presents GIST cells that express the KIT (CD117) protein.
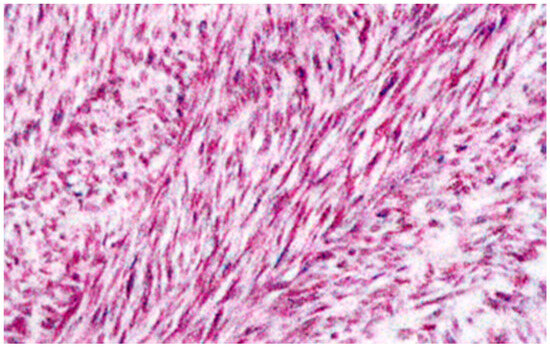
Figure 1.
Pathology findings: Microscopic examination of the intestinal histotome revealed benign GISTs (×100 magnification, hematoxylin–eosin stain). Original copyright data from the Laboratory of Autopsy Pathology, Forensic Service of Thessaloniki, Ministry of Justice, Greece.
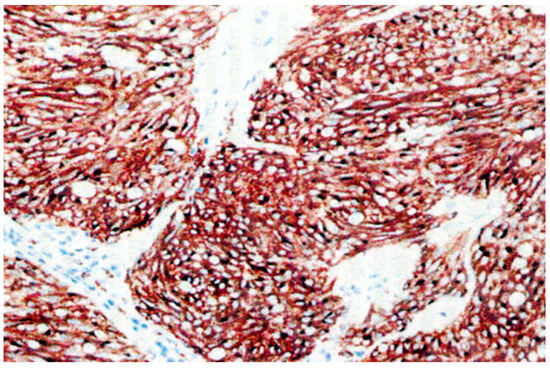
Figure 2.
Pathology findings: Microscopic examination of the intestinal histotome revealed GIST cells that express the KIT (CD117) protein (×100 magnification). Original copyright data from the Laboratory of Autopsy Pathology, Forensic Service of Thessaloniki, Ministry of Justice, Greece.
5. Symptoms and Clinical Signs of GISTs
Regarding the GIST clinical signs, the most frequent symptom of GIST tumors is abdominal pain. This pain can be accompanied by GI bleeding, a common symptom of GISTs, especially in gastric GISTs. Gastric GIST bleeding is usually associated with the growing submucosal tumor’s ulceration of the mucosal surface [88]. Symptomatic patients, except for the clinical signs of abdominal pain, may present nonspecific symptoms, such as nausea, vomiting, abdominal distension, and early satiety [62]. Moreover, an abdominal mass may also be present in some cases. It Is important to note that not all GIST tumors cause symptoms, and some people may be asymptomatic [89].
6. GIST Prognosis
The mitotic rate, tumor size, and tumor site are widely recognized prognostic factors, according to the latest clinical guidelines, including NCCN, ESMO/EURACAN, and French Intergroup Clinical Practice guidelines [90]. Additionally, the prognosis of GISTs is significantly influenced by the presence of KIT mutations and the nature of exon mutation, even though mutational status is not currently used to assess the malignant potential of a localized GIST. For instance, patient mutations in codons 557–558 often lead to a more challenging prognosis, as they are associated with the malignant behavior of tumors, highlighting the critical role of these mutations in disease progression [91,92,93]. A better prognosis is associated with C-KIT exon 11 point mutation and insertion mutation, while C-KIT exon 11 deletion mutation and exon nine mutation are linked with a worse prognosis [94]. KIT exon nine mutations predominantly manifest outside the stomach, leading to a more unfavorable clinical prognosis and necessitating a higher dosage of imatinib for adjuvant therapy. However, the poor prognosis related to KIT exon nine mutations is attributed to the tumor location rather than the inherently invasive biological nature of the mutation [95]. While regarding mutations in KIT exon 13 and exon 17, a multicenter study by Lasota et al. [96] showed that these mutations are more commonly found in the small intestine than other mutation types. However, the study did not find any significant difference in clinical prognosis.
Moreover, in a long-term follow-up study of 1765 patients with gastric GIST, it was observed that only 2–3% of the tumors smaller than 10 cm in size and having less than five mitoses per 50 HPFs presented with metastatic relapse. However, 86% of tumors over 10 cm and having more than five mitoses per 50 HPFs showed metastatic relapse [21]. In the same study, KIT exon 11 mutations were revealed in 119 patients, and interestingly, those with point mutations fared better than those with deletions [21]. Similarly, in a study by Miettinen et al. [30], which included 906 cases of small intestinal GIST, tumors larger than 10 cm with a mitotic index of 5 or less per 50 HPFs and those smaller than 5 cm with a mitotic index greater than 5 per 50 HPFs, had a relapse rate of over 50%. This rate was higher than that of gastric GIST with similar tumor parameters. In the EORTC phase II trial, researchers found that KIT exon nine mutations were the most significant adverse prognostic factor for response to imatinib. This increased the relative risk of progression by 171% and the relative risk of death by 190% when compared with KIT exon 11 mutants. They also noticed that the relative risk of progression was increased by 108% and the relative risk of death by 76% in patients without detectable KIT or PDGFRA mutations. However, in patients whose tumors expressed an exon 9 KIT oncoprotein, treatment with the high-dose regimen led to significantly better progression-free survival, reducing the risk by 61%. Thus, the study concludes that tumor genotypes are of high prognostic importance for assessing the prognosis and predicting how efficient the use of imatinib is for advanced GISTs [41].
7. Treatment
7.1. Preoperative Systemic Therapy
Neoadjuvant treatment, a type of preoperative systemic therapy, and preoperative tumor downstaging treatment are both included in the preoperative systemic therapy. Neoadjuvant treatment refers to the use of therapeutic interventions before the primary treatment, such as surgery, to reduce the size of the tumor or make it easier to remove. As for the tumor downstaging treatment, it is considered an innovative treatment for unresectable malignant tumors. In the case of advanced tumors, the combination of both imatinib—a medicine that hinders the growth of specific cancer cells—and surgical resection is a common path to follow when it comes to the treatment plan. Neoadjuvant imatinib treatment can be considered practical despite the limitations regarding clinical trial evidence. The NCCN’s guidelines suggest that this treatment method reduces the size of preoperative tumors, especially the large ones or those that require surgery.
Moreover, the preoperative use of imatinib for high-risk patients or those requiring extensive surgery has shown promising results, bolstering confidence in its efficacy. The recommendation to administer imatinib 6 to 12 months before surgery is based on the understanding that a treatment duration of two or three months may not significantly reduce tumor size. This underscores the need to revisit the evidence on the optimal treatment period for advanced GISTs. Nevertheless, prolonged imatinib treatment can be associated with some disadvantages, like tumor necrosis, a condition where the tumor cells die, cystic changes, fluid-filled sacs that can form within the tumor, rupture, and hemorrhage. Moreover, this treatment method can be used for high karyokinesis exponents or tumors of significant size so that the hazard of intraoperative complications, like tumor crack, decreases [97].
The surgical method should be operated on within a specific timeline, so the patient does not resist imatinib. The surgical treatment is vital for the management of the disease during the imatinib treatment, even though organ function loss may be caused, and the postoperative quality of life may be dramatically affected because of the wide range of resection [98].
7.1.1. KIT or PDGFRA Mutations Response to Imatinib
Imatinib is effective against the KIT and PDGFRA tyrosine kinases [99]. It is the primary medication for advanced GIST and is also utilized for neoadjuvant and adjuvant therapy in localized GIST. KIT mutations in exon 9 or 8 typically duplicated the insertion of A502–Y503 codons and primarily located in the small intestine, show a varying sensitivity to imatinib, with lower response rates (17%) at 400 mg per day and higher response rates (67%) at 800 mg daily. In addition, the progression-free survival (PFS) achieved with imatinib remedy is between 7 and 12.6 months for doses equal to 400 mg per day and between 16.7 and 18.0 months for doses equal to 800 mg per day for KIT mutations in exon 9. In contrast, KIT mutations in exon 11, located at all GI sites, show higher response rates (71.7%) and PFS (24.7–39.4 months) to imatinib, compared to KIT mutations in exon 9. Patients with KIT mutations in exon 13 show ambiguous results. In some cases, there is a response to imatinib therapy, but in others, there is not [38,100]. Overall, KIT mutations in exons 13 and 17 show a significantly lower response to imatinib than exons 11 and 9 [38,100]. Regarding PDGFRA mutations, studies show that PFS achieved with imatinib therapy has a mean of 6.4 months, while the response rates to this remedy range between 18% and 30% [39,40,41,101]. Interestingly, adjuvant imatinib treatment is ineffective in GISTs without KIT or PDGFRA mutations or in GISTs with KIT or PDGFRA mutations that confer imatinib insensitivity. These patients should be observed after surgical resection of localized disease [43,59].
7.1.2. Imatinib-Resistant KIT or PDGFRA GISTs
While imatinib was shown to improve prognosis and survival outcomes as the first-line therapy in 2001 [102,103], it is essential to acknowledge that it is rarely remedial due to the emergence of treatment-resistant cells within the tumor [104]. As a result, many research efforts have been focused on discovering new tyrosine kinase inhibitors that can target both imatinib-sensitive and -resistant mutants at the same time. Sunitinib was the first multi-targeted tyrosine kinase inhibitor approved in 2006 as a second-line therapy for imatinib-resistant metastatic GISTs. Like imatinib, sunitinib targets KIT and PDGFR. However, due to its broader binding profile and affinity, sunitinib may be effective in imatinib-resistant GISTs. Dimitri et al. [105], by performing a randomized controlled trial in patients with advanced GISTs, found that patients receiving sunitinib had a median PFS of 6.8 months, compared to 1.6 months for those on placebo.
Similarly, Heinrich et al. [106], by evaluating the impact of primary and secondary kinase genotype on sunitinib activity in 97 patients with metastatic, imatinib-resistant/intolerant GISTs, noticed that the clinical benefit of sunitinib in those with partial response or stable disease for ≥6 months, was 58% for KIT exon 9, 34% for KIT exon 11, and 56% for wild–type KIT/PDGFRA mutations. The authors concluded that the clinical activity of sunitinib after imatinib failure is significantly influenced by both primary and secondary mutations in the predominant pathogenic kinases [106]. However, there is limited knowledge about the correlation between PDGFRA mutation status and the clinical benefit in patients treated with sunitinib, emphasizing the crucial need for further research. Reichardt et al. [107] performed a non-interventional, retrospective analysis in patients with imatinib-resistant or intolerant GIST who were treated in a worldwide, open-label treatment-use study (Study 1036; NCT00094029) in which sunitinib was administered at a starting dose of 50 mg/day on a 4-week-on, 2-week-off schedule. This study confirmed the effectiveness of sunitinib as a post-imatinib therapy, regardless of mutational status [107]. In addition, a Korean study by Yoon et al. [108] showed that sunitinib was effective for patients with advanced GIST with KIT (exons 9, 11, 13, 17) and PDGFRA (exons 12 and 18) mutations. However, those with KIT exon 9 mutations had numerically better clinical benefits from sunitinib therapy than those with KIT exon 11 mutant GISTs.
Furthermore, regorafenib was introduced in 2012 as a third-line treatment for GISTs that are resistant to sunitinib [105,109]. Regorafenib has the broadest kinase inhibitory activity among approved agents. It is a competitive inhibitor of the ATP-binding site for PDGFR, vascular endothelial growth factor receptor 1–3 (VEGFR1–3), TEK, KIT, RET, RAF1, BRAF, and FGFR [110]. In a multicenter, randomized controlled trial, Demetri et al. [109] revealed that oral regorafenib could significantly improve progression-free survival compared with placebo in patients with metastatic GIST after progression on standard treatments. Meanwhile, in a case report of a patient with PDGFRA-mutated GIST, treatment with regorafenib resulted in a prolonged response. Twenty months after treatment onset, the patient is still under treatment and maintaining a partial response [111].
Next-generation, fourth-line tyrosine kinase inhibitors for GISTs are ripretinib and avapritinib. Ripretinib is a new type II switch-controlled kinase inhibitor that modulates the kinase switch pocket and the activation loop [112]. According to the latest Polish and international guidelines (ESMO 2022 and NCCN 2022), ripretinib is the preferred fourth-line treatment option in patients with inoperable, progressive, or metastatic GIST after treatment with imatinib, sunitinib, and regorafenib. The recommended dosage is 150 mg/day [13]. However, these guidelines also suggest a ripretinib dosage regimen of 150 mg twice per day for patients whose disease has progressed while taking the drug at a dose of 150 mg/day [14,113]. Its dual mechanism of action keeps KIT and PDGFRA in an inactive conformation, inhibiting downstream signaling regardless of the mutation type [114]. Ripretinib, the first of the KIT inhibitors, has been comprehensively tested and effectively inhibits all tested KIT and PDGFRA mutations except for the D842V mutation and wild-type GISTs. In addition, in vitro studies have shown that ripretinib has favorable effects in treating secondary mutations, as it inhibits other kinases, such as PDGFRB, TIE2, VEGFR2, and BRAF [114,115,116].
Unlike early-generation TKIs, avapritinib was designed as a potent and highly selective type I inhibitor of mutations affecting the activation loop (encoded by exon 17 in KIT and exon 18 in PDGFRA). Avapritinib was approved by the FDA in 2020 based on the phase I/II trial results for advanced or metastatic PDGFRA-mutated GIST, including the exon 18 D842V mutation [112,117,118]. During the first-in-human phase I clinical trial, avapritinib had a minimum starting dose of 300 mg/day and a maximum tolerated dose of 400 mg/day [117]. The efficacy results were impressive: the overall response rate was 91% at 300 mg daily, the clinical benefit rate was 98%, and the mean PFS was 34.0 months [119].
Hashimoto et al. [120] conducted a study to evaluate the use of circulating tumor DNA (ctDNA) for making treatment decisions in 133 patients with advanced GIST. The study found that patients who tested positive for ctDNA had significantly shorter progression-free survival than those who tested negative for ctDNA. In addition, when evaluating the effectiveness of different tyrosine kinase inhibitors for 17 patients with KIT or PDGFRA mutations based on their specific mutation, it was found that patients who received tyrosine kinase inhibitors matching their mutation had a much longer progression-free survival (PFS) compared to those who received mismatched inhibitors (median PFS, 8.23 vs. 2.43 months). Their findings confirmed the low maximum variant allele frequency (VAF) of ctDNA without KIT or PDGFRA mutations. This led them to hypothesize that clonal evolution related to acquired resistance mechanisms may provide insight into this phenomenon. Avapritinib is the most promising treatment for advanced GISTs with the mutation PDGFRAD842V. However, patients may develop secondary resistance in some cases, indicating treatment response and the low tumor fraction carrying original alterations. Resistance mutations have been detected in PDGFRA exons 13, 14, and 15, leading to secondary PDGFRA mutations causing V658A, N659K, Y676C, and G680R substitutions, which impede avapritinib binding [121].
7.2. Endoscopic Ultrasonography
In cases of small intestine GISTs when the surgical method (surgical removal) is not feasible, CT-guided radiofrequency is suggested in addition to surgery and targeted therapy. There are thoughts that alcohol septal ablation, which is a non-surgical treatment, can be an alternative option and targeted to the cases where surgery cannot take place [20]. Despite the efficiency of this method, there is still room for enhancement in its practical application. A positive aspect of this method is that it can be used not only in small intestine GISTs but also for the ablation of liver and adrenal or pelvic lymph node metastases.
More information regarding the etiopathogenesis of GISTSs has been issued because of the increasing use of EUS for small intestine GISTs, as it has a significant role in distinguishing small intestine GISTs from other submucosal tumors. CT-guided radiofrequency ablation and EUS alcohol ablation are considerable alternatives for small intestine GIST treatment when surgery is impossible. However, the effectiveness of these methods may differ when it comes to diverse patients, and that is why consultation with healthcare professionals is vital for deciding on a more efficient treatment plan [122].
7.3. Surgical Therapy
Radical resection is the most preferred treatment for small GISTs. Sufficiency is being assessed by borderline status and complete resection without tumor overflow or rupture. However, from an oncological overview, the ideal surgical approach is limited resection of small intestinal GISTs [123]. Furthermore, laparoscopic surgery’s evolution has increased its use in small intestine GISTs. Low tumor crack rate, which refers to the minimal incidence of tumor rupture during surgery, short operative time, early recovery of intestinal function, and less postoperative pain are some of the main advantages of laparoscopic surgery in small intestine GISTs. According to the NCCN’s guidelines, to use laparoscopic surgery in small intestine GISTs, the tumor has to be less than 5 cm. However, some studies suggest increasing the limit to under 10 cm. In the case of a positive resection margin, there are different guidelines regarding its management. While the NCCN suggests not continuing with an operation, the European Society of Medical Oncology recommends reoperation to achieve curative resection (RO resection) and remove the tumor. Despite the positive aspects of using laparoscopic surgery in small intestine GISTs, there are some drawbacks, too.
To start, this method requires the digestive tract to undergo anastomosis. A wound protector must be used to prevent the tumor from becoming infected. This device covers surgical wounds and helps prevent contamination. Thus, the navel incision widens [124]. Using a wound protector not only reduces the operation time but also makes the surgery more accessible for operators, thereby enhancing the adaptability of laparoscopic surgery. This method also decreases the likelihood of adhesion and incisional hernia, demonstrating its versatility [124,125]. Regrettably, tumor pathology remains a significant factor in relapses, which underscores the fact that the surgical method may not always be a definitive solution. It is crucial to be aware of these limitations to make informed decisions.
8. Conclusions
In conclusion, a specific approach is needed to manage gastric and small intestine GISTs. This article presented a review of our current knowledge on GISTs, from their molecular basis to diagnostic measures and treatment strategies. Genetic analysis contributes to the estimation of the prognosis of GISTs, and simultaneously, other diagnostic approaches exist, like imaging and immunochemistry, which inform patients of the most effective treatment for each one. Treatment plans continue to evolve, aiming at the enhancement of patients’ life quality. The cooperation between healthcare providers and individualized patient assessments is vital in choosing the right treatment plan for each patient.
Author Contributions
All authors contributed to the study conception and design. Material preparation and data collection were performed by R.T., A.A., D.T. and D.A. The first draft of the manuscript was written by P.E., V.M., R.T., A.A. and D.T. The second draft of the manuscript was edited by P.E., V.M. and D.A. All authors read and approved of the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author due to ethical restrictions.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Ma, G.L.; Murphy, J.D.; Martinez, M.E.; Sicklick, J.K. Epidemiology of Gastrointestinal Stromal Tumors in the Era of Histology Codes: Results of a Population–Based Study. Cancer Epidemiol. Biomark. Prev. 2015, 24, 298–302. [Google Scholar] [CrossRef]
- van der Graaf, W.T.A.; Tielen, R.; Bonenkamp, J.J.; Lemmens, V.; Verhoeven, R.H.A.; de Wilt, J.H.W. Nationwide trends in the incidence and outcome of patients with gastrointestinal stromal tumour in the imatinib era. Br. J. Surg. 2018, 105, 1020–1027. [Google Scholar] [CrossRef]
- Demetri, G.D.; Von Mehren, M.; Antonescu, C.R.; DeMatteo, R.P.; Ganjoo, K.N.; Maki, R.G.; Pisters, P.W.T.; Raut, C.P.; Riedel, R.F.; Schuetze, S.; et al. NCCN Task Force Report: Update on the Management of Patients with Gastrointestinal Stromal Tumors. J. Natl. Compr. Cancer Netw. 2010, 8 (Suppl. S2), S-1–S-41. [Google Scholar] [CrossRef]
- Søreide, K.; Sandvik, O.M.; Søreide, J.A.; Giljaca, V.; Jureckova, A.; Bulusu, V.R. Global epidemiology of gastrointestinal stromal tumours (GIST): A systematic review of population–based cohort studies. Cancer Epidemiol. 2016, 40, 39–46. [Google Scholar] [CrossRef]
- Miettinen, M.; Lasota, J. Gastrointestinal stromal tumors-definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchows Arch. 2001, 438, 1–12. [Google Scholar] [CrossRef]
- Khan, J.; Ullah, A.; Waheed, A.; Karki, N.R.; Adhikari, N.; Vemavarapu, L.; Belakhlef, S.; Bendjemil, S.M.; Seraj, S.M.; Sidhwa, F.; et al. Gastrointestinal Stromal Tumors (GIST): A Population-Based Study Using the SEER Database, including Management and Recent Advances in Targeted Therapy. Cancers 2022, 14, 3689. [Google Scholar] [CrossRef]
- A Cassier, P.; Ducimetière, F.; Lurkin, A.; Ranchère-Vince, D.; Scoazec, J.-Y.; Bringuier, P.-P.; Decouvelaere, A.-V.; Méeus, P.; Cellier, D.; Blay, J.-Y.; et al. A prospective epidemiological study of new incident GISTs during two consecutive years in Rhône Alpes region: Incidence and molecular distribution of GIST in a European region. Br. J. Cancer 2010, 103, 165–170. [Google Scholar] [CrossRef]
- Verschoor, A.J.; Bovée, J.V.M.G.; Overbeek, L.I.H.; Hogendoorn, P.C.W.; Gelderblom, H. The incidence, mutational status, risk classification and referral pattern of gastro–intestinal stromal tumours in the Netherlands: A nationwide pathology registry (PALGA) study. Virchows Arch. 2018, 472, 221–229. [Google Scholar] [CrossRef]
- Joensuu, H.; Hohenberger, P.; Corless, C.L. Gastrointestinal stromal tumour. Lancet 2013, 382, 973–983. [Google Scholar] [CrossRef]
- Nishida, T.; Blay, J.-Y.; Hirota, S.; Kitagawa, Y.; Kang, Y.-K. The standard diagnosis, treatment, and follow-up of gastrointestinal stromal tumors based on guidelines. Gastric Cancer 2015, 19, 3–14. [Google Scholar] [CrossRef]
- Alvarez, C.S.; Piazuelo, M.B.; Fleitas-Kanonnikoff, T.; Ruhl, J.; Pérez-Fidalgo, J.A.; Camargo, M.C. Incidence and Survival Outcomes of Gastrointestinal Stromal Tumors. JAMA Netw. Open 2024, 7, e2428828. [Google Scholar] [CrossRef]
- Güller, U.; Tarantino, I.; Cerny, T.; Schmied, B.M.; Warschkow, R. Population–based SEER trend analysis of overall and cancer–specific survival in 5138 patients with gastrointestinal stromal tumor. BMC Cancer 2015, 15, 557. [Google Scholar] [CrossRef]
- Casali, P.G.; Blay, J.Y.; Abecassis, N.; Bajpai, J.; Bauer, S.; Biagini, R.; Bielack, S.; Bonvalot, S.; Boukovinas, I.; Bovee, J.V.M.G.; et al. Gastrointestinal stromal tumours: ESMO-EURACAN-GENTURIS Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2022, 33, 20–33. [Google Scholar] [CrossRef]
- von Mehren, M.; Kane, J.M.; Riedel, R.F.; Sicklick, J.K.; Pollack, S.M.; Agulnik, M.; Sundar, H.; Hang, L.E. NCCN Guidelines® Insights: Gastrointestinal Stromal Tumors, Version 2.2022. J. Natl. Compr. Cancer Netw. 2022, 20, 1204–1214. [Google Scholar] [CrossRef]
- Akahoshi, K.; Oya, M.; Koga, T.; Shiratsuchi, Y. Current clinical management of gastrointestinal stromal tumor. World J. Gastroenterol. 2018, 24, 2806–2817. [Google Scholar] [CrossRef]
- Sheikh, E.; Tran, T.; Vranic, S.; Levy, A.; Bonfil, R.D. Role and Significance of c-KIT Receptor Tyrosine Kinase in Cancer: A Review. Bosn. J. Basic Med. Sci. 2022, 22, 683–698. [Google Scholar] [CrossRef]
- Fletcher, C.D.M.; Berman, J.J.; Corless, C.; Gorstein, F.; Lasota, J.; Longley, B.J.; Miettinen, M.; O’Leary, T.J.; Remotti, H.; Rubin, B.P.; et al. Diagnosis of gastrointestinal stromal tumors: A consensus approach. Hum. Pathol. 2002, 33, 459–465. [Google Scholar] [CrossRef]
- Miettinen, M.; Kopczynski, J.; Makhlouf, H.R.; Sarlomo-Rikala, M.; Gyorffy, H.; Burke, A.; Sobin, L.H.; Lasota, J. Gastrointestinal Stromal Tumors, Intramural Leiomyomas, and Leiomyosarcomas in the Duodenum: A Clinicopathologic, Immunohistochemical, and Molecular Genetic Study of 167 Cases. Am. J. Surg. Pathol. 2003, 27, 625–641. [Google Scholar] [CrossRef]
- Benjamin, R.S.; Casali, P.G. Adjuvant Imatinib for GI Stromal Tumors: When and For How Long? J. Clin. Oncol. 2016, 34, 215–218. [Google Scholar] [CrossRef]
- Parab, T.M.; DeRogatis, M.J.; Boaz, A.M.; Grasso, S.A.; Issack, P.S.; Duarte, D.A.; Urayeneza, O.; Vahdat, S.; Qiao, J.-H.; Hinika, G.S. Gastrointestinal stromal tumors: A comprehensive review. J. Gastrointest. Oncol. 2018, 10, 144–154. [Google Scholar] [CrossRef]
- Miettinen, M.; Sobin, L.H.; Lasota, J. Gastrointestinal Stromal Tumors of the Stomach. Am. J. Surg. Pathol. 2005, 29, 52–68. [Google Scholar] [CrossRef]
- Goh, B.K.P.; Chow, P.K.H.; Yap, W.-M.; Kesavan, S.M.; Song, I.-C.; Paul, P.G.; Ooi, B.-S.; Chung, Y.-F.A.; Wong, W.-K. Which Is the Optimal Risk Stratification System for Surgically Treated Localized Primary GIST? Comparison of Three Contemporary Prognostic Criteria in 171 Tumors and a Proposal for a Modified Armed Forces Institute of Pathology Risk Criteria. Ann. Surg. Oncol. 2008, 15, 2153–2163. [Google Scholar] [CrossRef]
- Loughrey, M.B.; Trivett, M.; Beshay, V.; Dobrovic, A.; Kovalenko, S.; Murray, W.; Lade, S.; Turner, H.; McArthur, G.A.; Zalcberg, J.; et al. KIT immunohistochemistry and mutation status in gastrointestinal stromal tumours (GISTs) evaluated for treatment with imatinib. Histopathology 2006, 49, 52–65. [Google Scholar] [CrossRef]
- Rubin, B.P.; Fletcher, J.A.; Fletcher, C.D.M. Molecular Insights into the Histogenesis and Pathogenesis of Gastrointestinal Stromal Tumors. Int. J. Surg. Pathol. 2000, 8, 5–10. [Google Scholar] [CrossRef]
- Miettinen, M.; Sarlomo-Rikala, M.; Sobin, L.H.; Lasota, J. Gastrointestinal Stromal Tumors and Leiomyosarcomas in the Colon. Am. J. Surg. Pathol. 2000, 24, 1339–1352. [Google Scholar] [CrossRef]
- Ueyama, T.; Guo, K.-J.; Hashimoto, H.; Daimaru, Y.; Enjoji, M. A clinicopathologic and immunohistochemical study of gastrointestinal stromal tumors. Cancer 1992, 69, 947–955. [Google Scholar] [CrossRef]
- Gheorghe, G.; Bacalbasa, N.; Ceobanu, G.; Ilie, M.; Enache, V.; Constantinescu, G.; Bungau, S.; Diaconu, C.C. Gastrointestinal Stromal Tumors—A Mini Review. J. Pers. Med. 2021, 11, 694. [Google Scholar] [CrossRef]
- Lasota, J.; Jasinski, M.; Sarlomo-Rikala, M.; Miettinen, M. Mutations in Exon 11 of c-Kit Occur Preferentially in Malignant versus Benign Gastrointestinal Stromal Tumors and Do Not Occur in Leiomyomas or Leiomyosarcomas. Am. J. Pathol. 1999, 154, 53–60. [Google Scholar] [CrossRef]
- Fülöp, E.; Marcu, S.; Milutin, D.; Borda, A. Gastrointestinal stromal tumors: Review on morphology, diagnosis and management. Rom. J. Morphol. Embryol. 2009, 50, 319–326. [Google Scholar]
- Miettinen, M.; Makhlouf, H.; Sobin, L.H.; Lasota, J. Gastrointestinal Stromal Tumors of the Jejunum and Ileum. Am. J. Surg. Pathol. 2006, 30, 477–489. [Google Scholar] [CrossRef]
- Min, K.W.M.D. mall intestinal stromal tumors with skeinoid fibers. Clinicopathological, immunohistochemical, and ultrastructural investigations. Am. J. Surg. Pathol. 1992, 16, 145–155. [Google Scholar] [CrossRef]
- Miettinen, M.; Lasota, J. Gastrointestinal Stromal Tumors: Review on Morphology, Molecular Pathology, Prognosis, and Differential Diagnosis. Arch. Pathol. Lab. Med. 2006, 130, 1466–1478. [Google Scholar] [CrossRef]
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; et al. Gain-of-Function Mutations of c– kit in Human Gastrointestinal Stromal Tumors. Science 1998, 279, 577–580. [Google Scholar] [CrossRef]
- Bauer, S.; Duensing, A.; Demetri, G.D.; Fletcher, J.A. KIT oncogenic signaling mechanisms in imatinib–resistant gastrointestinal stromal tumor: PI3-kinase/AKT is a crucial survival pathway. Oncogene 2007, 26, 7560–7568. [Google Scholar] [CrossRef]
- Gopie, P.; Mei, L.; Faber, A.C.; Grossman, S.R.; Smith, S.C.; Boikos, S.A. Classification of gastrointestinal stromal tumor syndromes. Endocr. Relat. Cancer 2018, 25, R49–R58. [Google Scholar] [CrossRef]
- Ricci, R. Syndromic gastrointestinal stromal tumors. Hered. Cancer Clin. Pr. 2016, 14, 15. [Google Scholar] [CrossRef]
- Forde, P.M.; Cochran, R.L.; Boikos, S.A.; Zabransky, D.J.; Beaver, J.A.; Meyer, C.F.; Thornton, K.A.; Montgomery, E.A.; Lidor, A.O.; Donehower, R.C.; et al. Familial GI Stromal Tumor With Loss of Heterozygosity and Amplification of Mutant KIT. J. Clin. Oncol. 2016, 34, e13–e16. [Google Scholar] [CrossRef]
- Blay, J.Y.; Kang, Y.K.; Nishida, T.; von Mehren, M. Gastrointestinal stromal tumours. Nat. Rev. Dis. Primers 2021, 7, 22. [Google Scholar] [CrossRef]
- Debiec-Rychter, M.; Sciot, R.; Le Cesne, A.; Schlemmer, M.; Hohenberger, P.; van Oosterom, A.T.; Blay, J.-Y.; Leyvraz, S.; Stul, M.; Casali, P.G.; et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur. J. Cancer 2006, 42, 1093–1103. [Google Scholar] [CrossRef]
- Wozniak, A.; Rutkowski, P.; Piskorz, A.; Ciwoniuk, M.; Osuch, C.; Bylina, E.; Sygut, J.; Chosia, M.; Rys, J.; Urbanczyk, K.; et al. Prognostic value of KIT/PDGFRA mutations in gastrointestinal stromal tumours (GIST): Polish Clinical GIST Registry experience. Ann. Oncol. 2012, 23, 353–360. [Google Scholar] [CrossRef]
- Gastrointestinal Stromal Tumor Meta–Analysis Group (MetaGIST). Comparison of Two Doses of Imatinib for the Treatment of Unresectable or Metastatic Gastrointestinal Stromal Tumors: A Meta-Analysis of 1640 Patients. J. Clin. Oncol. 2010, 28, 1247–1253. [Google Scholar] [CrossRef]
- von Mehren, M.; Randall, R.L.; Benjamin, R.S.; Boles, S.; Bui, M.M.; Casper, E.S.; Conrad, E.U., III; DeLaney, T.F.; Ganjoo, K.N.; George, S.; et al. Gastrointestinal Stromal Tumors, Version 2.2014. J. Natl. Compr. Cancer Netw. 2014, 12, 853–862. [Google Scholar] [CrossRef]
- Casali, P.G.; Abecassis, N.; Bauer, S.; Biagini, R.; Bielack, S.; Bonvalot, S.; Boukovinas, I.; Bovee, J.V.M.G.; Brodowicz, T.; Broto, J.M.; et al. Gastrointestinal stromal tumours: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv68–iv78. [Google Scholar] [CrossRef]
- Bachet, J.-B.; Landi, B.; Laurent-Puig, P.; Italiano, A.; Le Cesne, A.; Lévy, P.; Safar, V.; Duffaud, F.; Blay, J.-Y.; Emile, J.-F. Diagnosis, prognosis and treatment of patients with gastrointestinal stromal tumour (GIST) and germline mutation of KIT exon 13. Eur. J. Cancer 2013, 49, 2531–2541. [Google Scholar] [CrossRef]
- Corless, C.L.; Barnett, C.M.; Heinrich, M.C. Gastrointestinal stromal tumours: Origin and molecular oncology. Nat. Rev. Cancer 2011, 11, 865–878. [Google Scholar] [CrossRef]
- Watkins, J.A.; Hatcher, H.; Malhotra, S.; Amen, F.; Bruty, J.; Trotman, J.; Tarpey, P.; Tadross, J.A. Identification of an Activating PDGFRA Deletion in a Novel Sinonasal Soft Tissue Neoplasm. Head Neck Pathol. 2023, 17, 576–580. [Google Scholar] [CrossRef]
- Peng, F.; Liu, Y. Gastrointestinal Stromal Tumors of the Small Intestine: Progress in Diagnosis and Treatment Research. Cancer Manag. Res. 2020, 12, 3877–3889. [Google Scholar] [CrossRef]
- Janeway, K.A.; Kim, S.Y.; Lodish, M.; Nosé, V.; Rustin, P.; Gaal, J.; Dahia, P.L.M.; Liegl, B.; Ball, E.R.; Raygada, M.; et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc. Natl. Acad. Sci. USA 2010, 108, 314–318. [Google Scholar] [CrossRef]
- Boikos, S.A.; Pappo, A.S.; Killian, J.K.; LaQuaglia, M.P.; Weldon, C.B.; George, S.; Trent, J.C.; von Mehren, M.; Wright, J.A.; Schiffman, J.D.; et al. Molecular Subtypes of KIT/PDGFRA Wild-Type Gastrointestinal Stromal Tumors. JAMA Oncol. 2016, 2, 922. [Google Scholar] [CrossRef]
- Pantaleo, M.A.; Astolfi, A.; Urbini, M.; Nannini, M.; Paterini, P.; Indio, V.; Saponara, M.; Formica, S.; Ceccarelli, C.; Casadio, R.; et al. Analysis of all subunits, SDHA, SDHB, SDHC, SDHD, of the succinate dehydrogenase complex in KIT/PDGFRA wild-type GIST. Eur. J. Hum. Genet. 2013, 22, 32–39. [Google Scholar] [CrossRef]
- Wang, Y.-M.; Gu, M.-L.; Ji, F. Succinate dehydrogenase-deficient gastrointestinal stromal tumors. World J. Gastroenterol. 2015, 21, 2303. [Google Scholar] [CrossRef]
- Killian, J.K.; Kim, S.Y.; Miettinen, M.; Smith, C.; Merino, M.; Tsokos, M.; Quezado, M.; Smith, W.I., Jr.; Jahromi, M.S.; Xekouki, P.; et al. Succinate Dehydrogenase Mutation Underlies Global Epigenomic Divergence in Gastrointestinal Stromal Tumor. Cancer Discov. 2013, 3, 648–657. [Google Scholar] [CrossRef]
- Boikos, S.A.; Helman, L.J.; Stratakis, C.A.; Pediatric and Wildtype GIST Clinic at the National Institutes of Health. Thyroid Hormone Inactivation in Gastrointestinal Stromal Tumors. N. Engl. J. Med. 2014, 371, 84–87. [Google Scholar]
- McWhinney, S.R.; Pasini, B.; Stratakis, C.A. Familial Gastrointestinal Stromal Tumors and Germ-Line Mutations. N. Engl. J. Med. 2007, 357, 1054–1056. [Google Scholar] [CrossRef]
- Benn, D.E.; Gimenez-Roqueplo, A.-P.; Reilly, J.R.; Bertherat, J.; Burgess, J.; Byth, K.; Croxson, M.; Dahia, P.L.M.; Elston, M.; Gimm, O.; et al. Clinical Presentation and Penetrance of Pheochromocytoma/Paraganglioma Syndromes. J. Clin. Endocrinol. Metab. 2006, 91, 827–836. [Google Scholar] [CrossRef]
- Recht, H.S.; Fishman, E.K. Carney-Stratakis syndrome: A dyad of familial paraganglioma and gastrointestinal stromal tumor. Radiol. Case Rep. 2020, 15, 2071–2075. [Google Scholar] [CrossRef]
- Carney, J.A.; Stratakis, C.A. Familial paraganglioma and gastric stromal sarcoma: A new syndrome distinct from the Carney triad. Am. J. Med. Genet. 2002, 108, 132–139. [Google Scholar] [CrossRef]
- Italiano, A.; Chen, C.-L.; Sung, Y.-S.; Singer, S.; DeMatteo, R.P.; LaQuaglia, M.P.; Besmer, P.; Socci, N.; Antonescu, C.R. SDHA loss of function mutations in a subset of young adult wild-type gastrointestinal stromal tumors. BMC Cancer 2012, 12, 408. [Google Scholar] [CrossRef]
- von Mehren, M.; Joensuu, H. Gastrointestinal Stromal Tumors. J. Clin. Oncol. 2018, 36, 136–143. [Google Scholar] [CrossRef]
- Wada, R.; Arai, H.; Kure, S.; Peng, W.; Naito, Z. “Wild type” GIST: Clinicopathological features and clinical practice. Pathol. Int. 2016, 66, 431–437. [Google Scholar] [CrossRef]
- Zöller, M.E.; Rembeck, B.; Odén, A.; Samuelsson, M.; Angervall, L. Malignant and benign tumors in patients with neurofibromatosis type 1 in a defined Swedish population. Cancer 1997, 79, 2125–2131. [Google Scholar] [CrossRef]
- Scherübl, H. Management of early asymptomatic gastrointestinal stromal tumors of the stomach. World J. Gastrointest Endosc. 2014, 6, 266. [Google Scholar] [CrossRef]
- Park, C.H.; Kim, E.H.; Jung, D.H.; Chung, H.; Park, J.C.; Shin, S.K.; Lee, Y.C.; Kim, H.; Kil Lee, S. Impact of Periodic Endoscopy on Incidentally Diagnosed Gastric Gastrointestinal Stromal Tumors: Findings in Surgically Resected and Confirmed Lesions. Ann. Surg. Oncol. 2015, 22, 2933–2939. [Google Scholar] [CrossRef]
- Bratu, O.G.; Cherciu, A.I.; Bumbu, A.; Lupu, S.; Marcu, D.R.; Radu, F.I.; Manea, M.; Furau, C.; Diaconu, C.C.; Mischianu, D.L.D. Retroperitoneal Tumours–Treatment and Prognosis of Tumour Recurrence. Rev. Chim. 2019, 70, 191–194. [Google Scholar] [CrossRef]
- Ghanem, N.; Altehoefer, C.; Furtwängler, A.; Winterer, J.; Schäfer, O.; Springer, O.; Kotter, E.; Langer, M. Computed tomography in gastrointestinal stromal tumors. Eur. Radiol. 2003, 13, 1669–1678. [Google Scholar] [CrossRef]
- Kim, H.; Kim, S.; Park, S.; Lee, J.; Lee, M.; Han, J.; Choi, B. Small gastrointestinal stromal tumours with focal areas of low attenuation on CT: Pathological correlation. Clin. Radiol. 2005, 60, 384–388. [Google Scholar] [CrossRef]
- Sandrasegaran, K.; Rajesh, A.; Rushing, D.A.; Rydberg, J.; Akisik, F.M.; Henley, J.D. Gastrointestinal stromal tumors: CT and MRI findings. Eur. Radiol. 2005, 15, 1407–1414. [Google Scholar] [CrossRef]
- Catalano, O.; Castelguidone, E.D.L.D.; Nunziata, A.; De Rosa, V.; Siani, A. Gastrointestinal stromal tumours: Pictorial review. Radiol. Med. 2006, 110, 484–491. [Google Scholar]
- Chourmouzi, D.; Sinakos, E.; Papalavrentios, L.; Akriviadis, E.; Drevelegas, A. Gastrointestinal stromal tumors: A pictorial review. J. Gastrointestin. Liver Dis. 2009, 18, 379–383. [Google Scholar]
- Bartolotta, T.V.; Taibbi, A.; Galia, M.; Cannella, I.; Re, G.L.; Sparacia, G.; Midiri, M.; Lagalla, R. Gastrointestinal stromal tumour: 40–row multislice computed tomography findings. Radiol. Med. 2006, 111, 651–660. [Google Scholar] [CrossRef]
- Kochhar, R.; Manoharan, P.; Leahy, M.; Taylor, M. Imaging in gastrointestinal stromal tumours: Current status and future directions. Clin. Radiol. 2010, 65, 584–592. [Google Scholar] [CrossRef]
- Levy, A.D.; Remotti, H.E.; Thompson, W.M.; Sobin, L.H.; Miettinen, M. From the Archives of the AFIP. Radiographics 2003, 23, 283–304. [Google Scholar] [CrossRef]
- Amano, M.; Okuda, T.; Amano, Y.; Tajiri, T.; Kumazaki, T. Magnetic resonance imaging of gastrointestinal stromal tumor in the abdomen and pelvis. Clin. Imaging 2006, 30, 127–131. [Google Scholar] [CrossRef]
- Yu, M.H.; Lee, J.M.; Baek, J.H.; Han, J.K.; Choi, B.-I. MRI Features of Gastrointestinal Stromal Tumors. Am. J. Roentgenol. 2014, 203, 980–991. [Google Scholar] [CrossRef]
- Scarpa, M.; Bertin, M.; Ruffolo, C.; Polese, L.; D’Amico, D.F.; Angriman, I. A systematic review on the clinical diagnosis of gastrointestinal stromal tumors. J. Surg. Oncol. 2008, 98, 384–392. [Google Scholar] [CrossRef]
- Lau, S.; Tam, K.; Kam, C.; Lui, C.; Siu, C.; Lam, H.; Mak, K. Imaging of gastrointestinal stromal tumour (GIST). Clin. Radiol. 2004, 59, 487–498. [Google Scholar] [CrossRef]
- Schaefer, I.M.; Mariño-Enríquez, A.; Fletcher, J.A. What is New in Gastrointestinal Stromal Tumor? Adv. Anat. Pathol. 2017, 24, 259–267. [Google Scholar] [CrossRef]
- Hamed, H.; Wahab, M.A.; Elmahdy, Y.; El-Wahab, R.M.A.; El-Magd, E.-S.A. Gastrointestinal stromal tumors of the small intestine: The challenge of diagnosis and the outcome of management. World J. Surg. Oncol. 2023, 21, 85. [Google Scholar] [CrossRef]
- Nishida, T.; Goto, O.; Raut, C.P.; Yahagi, N. Diagnostic and treatment strategy for small gastrointestinal stromal tumors. Cancer 2016, 122, 3110–3118. [Google Scholar] [CrossRef]
- Agaimy, A.; Wünsch, P.H.; Dirnhofer, S.; Bihl, M.P.; Terracciano, L.M.; Tornillo, L. Microscopic Gastrointestinal Stromal Tumors in Esophageal and Intestinal Surgical Resection Specimens. Am. J. Surg. Pathol. 2008, 32, 867–873. [Google Scholar] [CrossRef]
- Abraham, S.C.; Krasinskas, A.M.; Hofstetter, W.L.; Swisher, S.G.; Wu, T.-T. “Seedling” Mesenchymal Tumors (Gastrointestinal Stromal Tumors and Leiomyomas) are Common Incidental Tumors of the Esophagogastric Junction. Am. J. Surg. Pathol. 2007, 31, 1629–1635. [Google Scholar] [CrossRef]
- Kawanowa, K.; Sakuma, Y.; Sakurai, S.; Hishima, T.; Iwasaki, Y.; Saito, K.; Hosoya, Y.; Nakajima, T.; Funata, N. High incidence of microscopic gastrointestinal stromal tumors in the stomach. Hum. Pathol. 2006, 37, 1527–1535. [Google Scholar] [CrossRef]
- Hedenbro, J.L.; Ekelund, M.; Wetterberg, P. Endoscopic diagnosis of submucosal gastric lesions. Surg. Endosc. 1991, 5, 20–23. [Google Scholar] [CrossRef]
- Rossi, S.; Gasparotto, D.; Toffolatti, L.; Pastrello, C.; Gallina, G.; Marzotto, A.; Sartor, C.; Barbareschi, M.; Cantaloni, C.; Messerini, L.; et al. Molecular and Clinicopathologic Characterization of Gastrointestinal Stromal Tumors (GISTs) of Small Size. Am. J. Surg. Pathol. 2010, 34, 1480–1491. [Google Scholar] [CrossRef]
- Nishida, T.; Kawai, N.; Yamaguchi, S.; Nishida, Y. Submucosal tumors: Comprehensive guide for the diagnosis and therapy of gastrointestinal submucosal tumors. Dig. Endosc. 2013, 25, 479–489. [Google Scholar] [CrossRef]
- Lupescu, I.G.; Grasu, M.; Boros, M.; Gheorghe, C.; Ionescu, M.; Popescu, I.; Herlea, V.; Georgescu, S.A. Gastrointestinal stromal tumors: Retrospective analysis of the computer–tomographic aspects. J. Gastrointestin. Liver Dis. 2007, 16, 147–151. [Google Scholar]
- Levy, A.D.; Remotti, H.E.; Thompson, W.M.; Sobin, L.H.; Miettinen, M. Gastrointestinal stromal tumors: Radiologic features with pathologic correlation. Radiographics 2003, 23, 283–304. [Google Scholar] [CrossRef]
- Menge, F.; Jakob, J.; Kasper, B.; Smakic, A.; Gaiser, T.; Hohenberger, P. Clinical Presentation of Gastrointestinal Stromal Tumors. Visc. Med. 2018, 34, 335–340. [Google Scholar] [CrossRef]
- Aghdassi, A.; Christoph, A.; Dombrowski, F.; Döring, P.; Barth, C.; Christoph, J.; Lerch, M.M.; Simon, P. Gastrointestinal Stromal Tumors: Clinical Symptoms, Location, Metastasis Formation, and Associated Malignancies in a Single Center Retrospective Study. Dig. Dis. 2018, 36, 337–345. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Q. Prognostic Indicators for Gastrointestinal Stromal Tumors: A Review. Transl. Oncol. 2020, 13, 100812. [Google Scholar] [CrossRef]
- Martin-Broto, J.; Gutierrez, A.; Garcia-Del-Muro, X.; Lopez-Guerrero, J.A.; Martinez-Trufero, J.; de Sande, L.M.; Lainez, N.; Maurel, J.; De Juan, A.; Losa, F.; et al. Prognostic time dependence of deletions affecting codons 557 and/or 558 of KIT gene for relapse–free survival (RFS) in localized GIST: A Spanish Group for Sarcoma Research (GEIS) Study. Ann. Oncol. 2010, 21, 1552–1557. [Google Scholar] [CrossRef]
- Joensuu, H.; Wardelmann, E.; Sihto, H.; Eriksson, M.; Hall, K.S.; Reichardt, A.; Hartmann, J.T.; Pink, D.; Cameron, S.; Hohenberger, P.; et al. Effect of KIT and PDGFRA Mutations on Survival in Patients With Gastrointestinal Stromal Tumors Treated With Adjuvant Imatinib: An Exploratory Analysis of a Randomized Clinical Trial. JAMA Oncol. 2017, 3, 602. [Google Scholar] [CrossRef]
- Wozniak, A.; Rutkowski, P.; Schöffski, P.; Ray-Coquard, I.; Hostein, I.; Schildhaus, H.U.; Le Cesne, A.; Bylina, E.; Limon, J.; Blay, J.-Y.; et al. Tumor Genotype Is an Independent Prognostic Factor in Primary Gastrointestinal Stromal Tumors of Gastric Origin: A European Multicenter Analysis Based on ConticaGIST. Clin. Cancer Res. 2014, 20, 6105–6116. [Google Scholar] [CrossRef]
- Wada, N.; Kurokawa, Y.; Takahashi, T.; Hamakawa, T.; Hirota, S.; Naka, T.; Miyazaki, Y.; Makino, T.; Yamasaki, M.; Nakajima, K.; et al. Detecting Secondary C–KIT Mutations in the Peripheral Blood of Patients with Imatinib–Resistant Gastrointestinal Stromal Tumor. Oncology 2016, 90, 112–117. [Google Scholar] [CrossRef]
- Miettinen, M.; Majidi, M.; Lasota, J. Pathology and diagnostic criteria of gastrointestinal stromal tumors (GISTs): A review. Eur. J. Cancer 2002, 38, S39–S51. [Google Scholar] [CrossRef]
- Lasota, J.; Corless, C.L.; Heinrich, M.C.; Debiec-Rychter, M.; Sciot, R.; Wardelmann, E.; Merkelbach-Bruse, S.; Schildhaus, H.-U.; E Steigen, S.; Stachura, J.; et al. Clinicopathologic profile of gastrointestinal stromal tumors (GISTs) with primary KIT exon 13 or exon 17 mutations: A multicenter study on 54 cases. Mod. Pathol. 2008, 21, 476–484. [Google Scholar] [CrossRef]
- Bednarski, B.K.; Araujo, D.M.; Yi, M.; Torres, K.E.; Lazar, A.; Trent, J.C.; Cormier, J.N.; Pisters, P.W.T.; Lev, D.C.; Pollock, R.E.; et al. Analysis of Prognostic Factors Impacting Oncologic Outcomes After Neoadjuvant Tyrosine Kinase Inhibitor Therapy for Gastrointestinal Stromal Tumors. Ann. Surg. Oncol. 2014, 21, 2499–2505. [Google Scholar] [CrossRef]
- Kang, G.; Kang, Y.; Ha, S.Y.; Kim, J.Y.; Shim, Y.M.; Heinrich, M.C.; Kim, K.; Corless, C.L. Gastrointestinal stromal tumours of the oesophagus: A clinicopathological and molecular analysis of 27 cases. Histopathology 2017, 71, 805–812. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Griffith, D.J.; Druker, B.J.; Wait, C.L.; Ott, K.A.; Zigler, A.J. Inhibition of c–kit receptor tyrosine kinase activity by STI 571, a selective tyrosine kinase inhibitor. Blood 2000, 96, 925–932. [Google Scholar] [CrossRef]
- Nishida, T.; Yoshinaga, S.; Takahashi, T.; Naito, Y. Recent Progress and Challenges in the Diagnosis and Treatment of Gastrointestinal Stromal Tumors. Cancers 2021, 13, 3158. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Owzar, K.; Corless, C.L.; Hollis, D.; Borden, E.C.; Fletcher, C.D.; Ryan, C.W.; von Mehren, M.; Blanke, C.D.; Rankin, C.; et al. Correlation of Kinase Genotype and Clinical Outcome in the North American Intergroup Phase III Trial of Imatinib Mesylate for Treatment of Advanced Gastrointestinal Stromal Tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J. Clin. Oncol. 2008, 26, 5360–5367. [Google Scholar] [CrossRef]
- Demetri, G.D.; Von Mehren, M.; Blanke, C.D.; Van den Abbeele, A.D.; Eisenberg, B.; Roberts, P.J.; Heinrich, M.C.; Tuveson, D.A.; Singer, S.; Janicek, M.; et al. Efficacy and Safety of Imatinib Mesylate in Advanced Gastrointestinal Stromal Tumors. N. Engl. J. Med. 2002, 347, 472–480. [Google Scholar] [CrossRef]
- Joensuu, H.; Roberts, P.J.; Sarlomo–Rikala, M.; Andersson, L.C.; Tervahartiala, P.; Tuveson, D.; Silberman, S.L.; Capdeville, R.; Dimitrijevic, S.; Druker, B.; et al. Effect of the Tyrosine Kinase Inhibitor STI571 in a Patient with a Metastatic Gastrointestinal Stromal Tumor. N. Engl. J. Med. 2001, 344, 1052–1056. [Google Scholar] [CrossRef]
- Balachandran, V.P.; DeMatteo, R.P. Gastrointestinal Stromal Tumors. Adv. Surg. 2014, 48, 165–183. [Google Scholar] [CrossRef]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: A randomised controlled trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Maki, R.G.; Corless, C.L.; Antonescu, C.R.; Harlow, A.; Griffith, D.; Town, A.; Mckinley, A.; Ou, W.-B.; Fletcher, J.A.; et al. Primary and Secondary Kinase Genotypes Correlate With the Biological and Clinical Activity of Sunitinib in Imatinib–Resistant Gastrointestinal Stromal Tumor. J. Clin. Oncol. 2008, 26, 5352–5359. [Google Scholar] [CrossRef]
- Reichardt, P.; Demetri, G.D.; Gelderblom, H.; Rutkowski, P.; Im, S.A.; Gupta, S.; Kang, Y.K.; Schoffski, P.; Schuette, J.; Soulieres, D.; et al. Correlation of KIT and PDGFRA mutational status with clinical benefit in patients with gastrointestinal stromal tumor treated with sunitinib in a worldwide treatment–use trial. BMC Cancer 2016, 16, 22. [Google Scholar] [CrossRef]
- Yoon, D.H.; Ryu, M.-H.; Ryoo, B.-Y.; Beck, M.; Choi, D.R.; Cho, Y.; Lee, J.-L.; Chang, H.-M.; Kim, T.W.; Kang, Y.-K. Sunitinib as a second–line therapy for advanced GISTs after failure of imatinib: Relationship between efficacy and tumor genotype in Korean patients. Investig. New Drugs 2010, 30, 819–827. [Google Scholar] [CrossRef]
- Demetri, G.D.; Reichardt, P.; Kang, Y.-K.; Blay, J.-Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; Von Mehren, M.; Joensuu, H.; et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo–controlled, phase 3 trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef]
- Wilhelm, S.M.; Dumas, J.; Adnane, L.; Lynch, M.; Carter, C.A.; Schütz, G.; Thierauch, K.; Zopf, D. Regorafenib (BAY 73–4506): A new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int. J. Cancer 2010, 129, 245–255. [Google Scholar] [CrossRef]
- Grellety, T.; Kind, M.; Coindre, J.-M.; Italiano, A. Clinical activity of regorafenib in PDGFRA–mutated gastrointestinal stromal tumor. Future Sci. OA 2015, 1, FSO33. [Google Scholar] [CrossRef]
- Evans, E.K.; Gardino, A.K.; Kim, J.L.; Hodous, B.L.; Shutes, A.; Davis, A.; Zhu, X.J.; Schmidt–Kittler, O.; Wilson, D.; Wilson, K.; et al. A precision therapy against cancers driven by KIT/PDGFRA mutations. Sci. Transl. Med. 2017, 9, eaao1690. [Google Scholar] [CrossRef]
- Zalcberg, J.R.; Heinrich, M.C.; George, S.; Bauer, S.; Schöffski, P.; Serrano, C.; Gelderblom, H.; Jones, R.L.; Attia, S.; D’Amato, G.; et al. Clinical Benefit of Ripretinib Dose Escalation After Disease Progression in Advanced Gastrointestinal Stromal Tumor: An Analysis of the INVICTUS Study. Oncologist 2021, 26, e2053–e2060. [Google Scholar] [CrossRef]
- Smith, B.D.; Kaufman, M.D.; Lu, W.-P.; Gupta, A.; Leary, C.B.; Wise, S.C.; Rutkoski, T.J.; Ahn, Y.M.; Al-Ani, G.; Bulfer, S.L.; et al. Ripretinib (DCC–2618) Is a Switch Control Kinase Inhibitor of a Broad Spectrum of Oncogenic and Drug–Resistant KIT and PDGFRA Variants. Cancer Cell 2019, 35, 738–751.e9. [Google Scholar] [CrossRef]
- Dhillon, S. Ripretinib: First Approval. Drugs 2020, 80, 1133–1138. [Google Scholar] [CrossRef]
- Blay, J.-Y.; Serrano, C.; Heinrich, M.C.; Zalcberg, J.; Bauer, S.; Gelderblom, H.; Schöffski, P.; Jones, R.L.; Attia, S.; D’Amato, G.; et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): A double–blind, randomised, placebo–controlled, phase 3 trial. Lancet Oncol. 2020, 21, 923–934. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Jones, R.L.; von Mehren, M.; Schöffski, P.; Serrano, C.; Kang, Y.-K.; Cassier, P.A.; Mir, O.; Eskens, F.; Tap, W.D.; et al. Avapritinib in advanced PDGFRA D842V–mutant gastrointestinal stromal tumour (NAVIGATOR): A multicentre, open-label, phase 1 trial. Lancet Oncol. 2020, 21, 935–946. [Google Scholar] [CrossRef]
- Gebreyohannes, Y.K.; Wozniak, A.; Zhai, M.E.; Wellens, J.; Cornillie, J.; Vanleeuw, U.; Evans, E.; Gardino, A.K.; Lengauer, C.; Debiec-Rychter, M.; et al. Robust Activity of Avapritinib, Potent and Highly Selective Inhibitor of Mutated KIT, in Patient–derived Xenograft Models of Gastrointestinal Stromal Tumors. Clin. Cancer Res. 2019, 25, 609–618. [Google Scholar] [CrossRef]
- Jones, R.L.; Serrano, C.; von Mehren, M.; George, S.; Heinrich, M.C.; Kang, Y.-K.; Schöffski, P.; Cassier, P.A.; Mir, O.; Chawla, S.P.; et al. Avapritinib in unresectable or metastatic PDGFRA D842V–mutant gastrointestinal stromal tumours: Long-term efficacy and safety data from the NAVIGATOR phase I trial. Eur. J. Cancer 2021, 145, 132–142. [Google Scholar] [CrossRef]
- Hashimoto, T.; Nakamura, Y.; Komatsu, Y.; Yuki, S.; Takahashi, N.; Okano, N.; Hirano, H.; Ohtsubo, K.; Ohta, T.; Oki, E.; et al. Different efficacy of tyrosine kinase inhibitors by KIT and PGFRA mutations identified in circulating tumor DNA for the treatment of refractory gastrointestinal stromal tumors. BJC Rep. 2024, 2, 54. [Google Scholar] [CrossRef]
- Grunewald, S.; Klug, L.R.; Mühlenberg, T.; Lategahn, J.; Falkenhorst, J.; Town, A.; Ehrt, C.; Wardelmann, E.; Hartmann, W.; Schildhaus, H.-U.; et al. Resistance to Avapritinib in PDGFRA–Driven GIST Is Caused by Secondary Mutations in the PDGFRA Kinase Domain. Cancer Discov. 2021, 11, 108–125. [Google Scholar] [CrossRef]
- Chen, K.; Zhang, B.; Liang, Y.-L.; Ji, L.; Xia, S.-J.; Pan, Y.; Zheng, X.-Y.; Wang, X.-F.; Cai, X.-J. Laparoscopic versus Open Resection of Small Bowel Gastrointestinal Stromal Tumors. Chin. Med. J. 2017, 130, 1595–1603. [Google Scholar] [CrossRef]
- Koo, D.-H.; Ryu, M.-H.; Kim, K.-M.; Yang, H.-K.; Sawaki, A.; Hirota, S.; Zheng, J.; Zhang, B.; Tzen, C.-Y.; Yeh, C.-N.; et al. Asian Consensus Guidelines for the Diagnosis and Management of Gastrointestinal Stromal Tumor. Cancer Res. Treat. 2016, 48, 1155–1166. [Google Scholar] [CrossRef]
- Liao, C.-H.; Liu, Y.-Y.; Chen, C.-C.; Wang, S.-Y.; Ooyang, C.-H.; Kuo, I.-M.; Yeh, T.-S. Single-Incision Laparoscopic-Assisted Surgery for Small Bowel Obstruction. J. Laparoendosc. Adv. Surg. Tech. 2012, 22, 957–961. [Google Scholar] [CrossRef]
- Stiekema, J.; Luttikhold, J.; Heineman, D.; Neerincx, M.; Daams, F. Minimally invasive technique for gastric GIST at challenging locations: Single incision surgical gastroscopy. Updat. Surg. 2023, 75, 953–958. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).