
Mesenchymal Tumors of the Gastrointestinal Tract—Beyond GIST—A Review



Abstract
1. Introduction
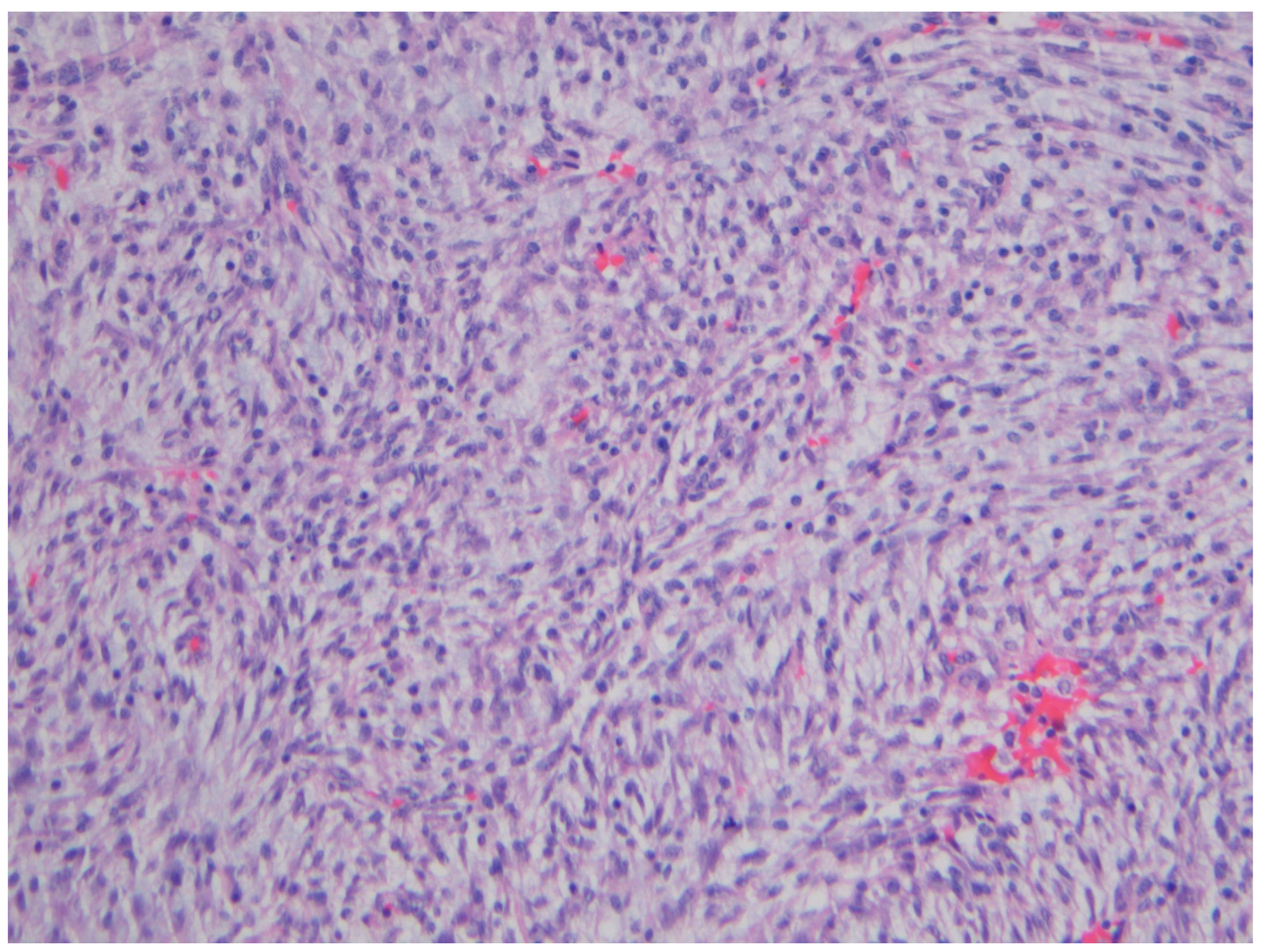
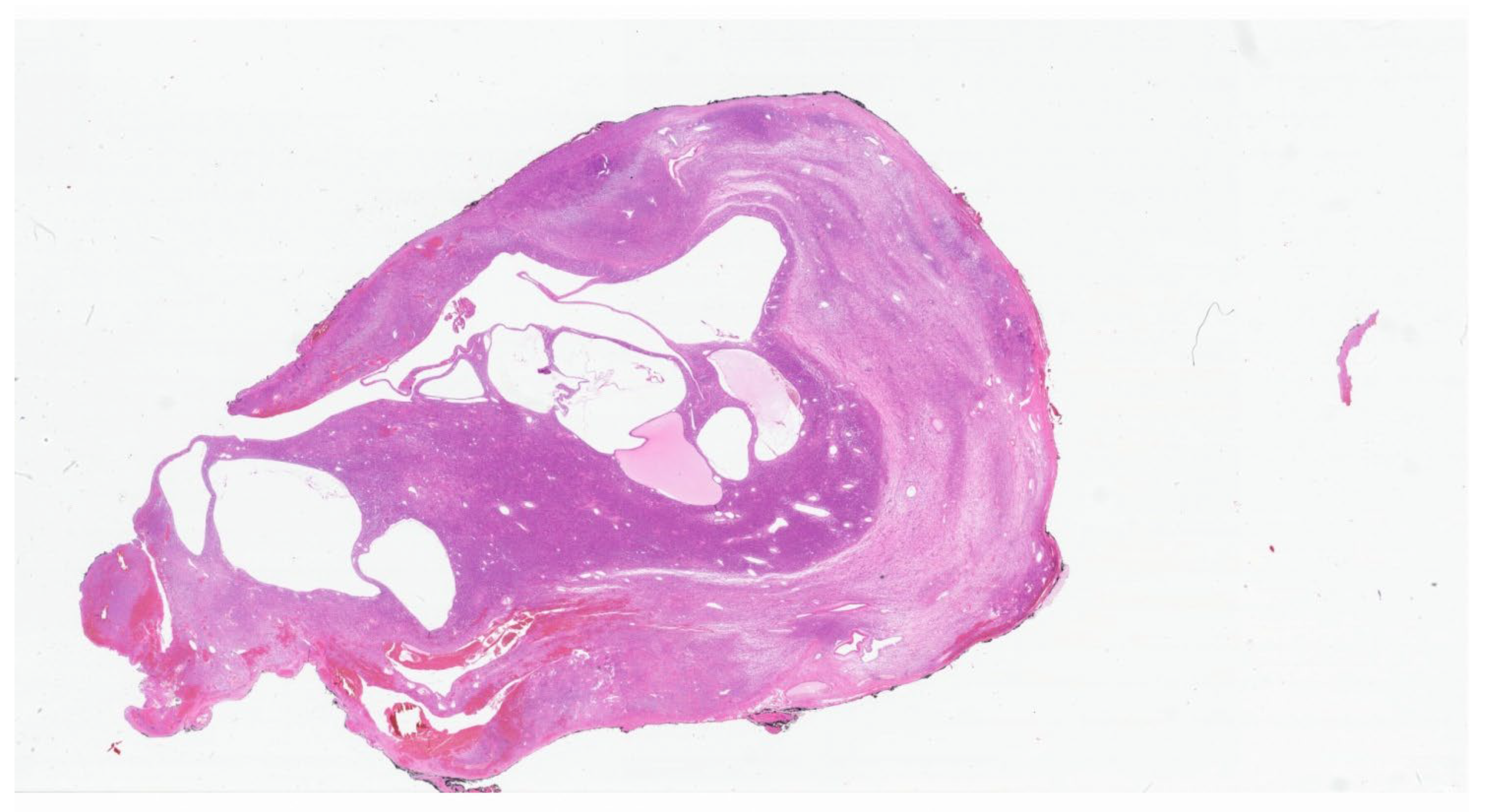
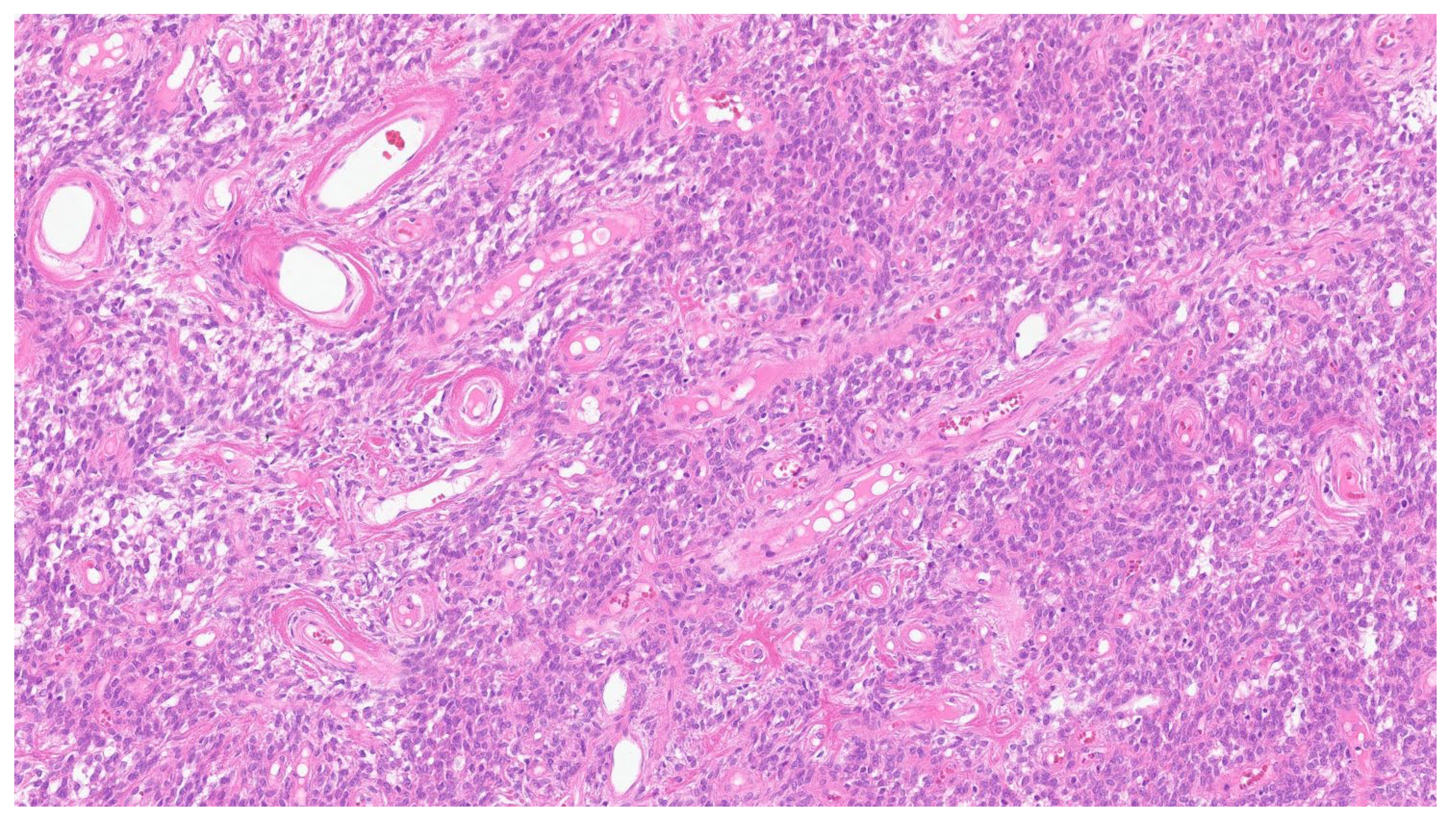
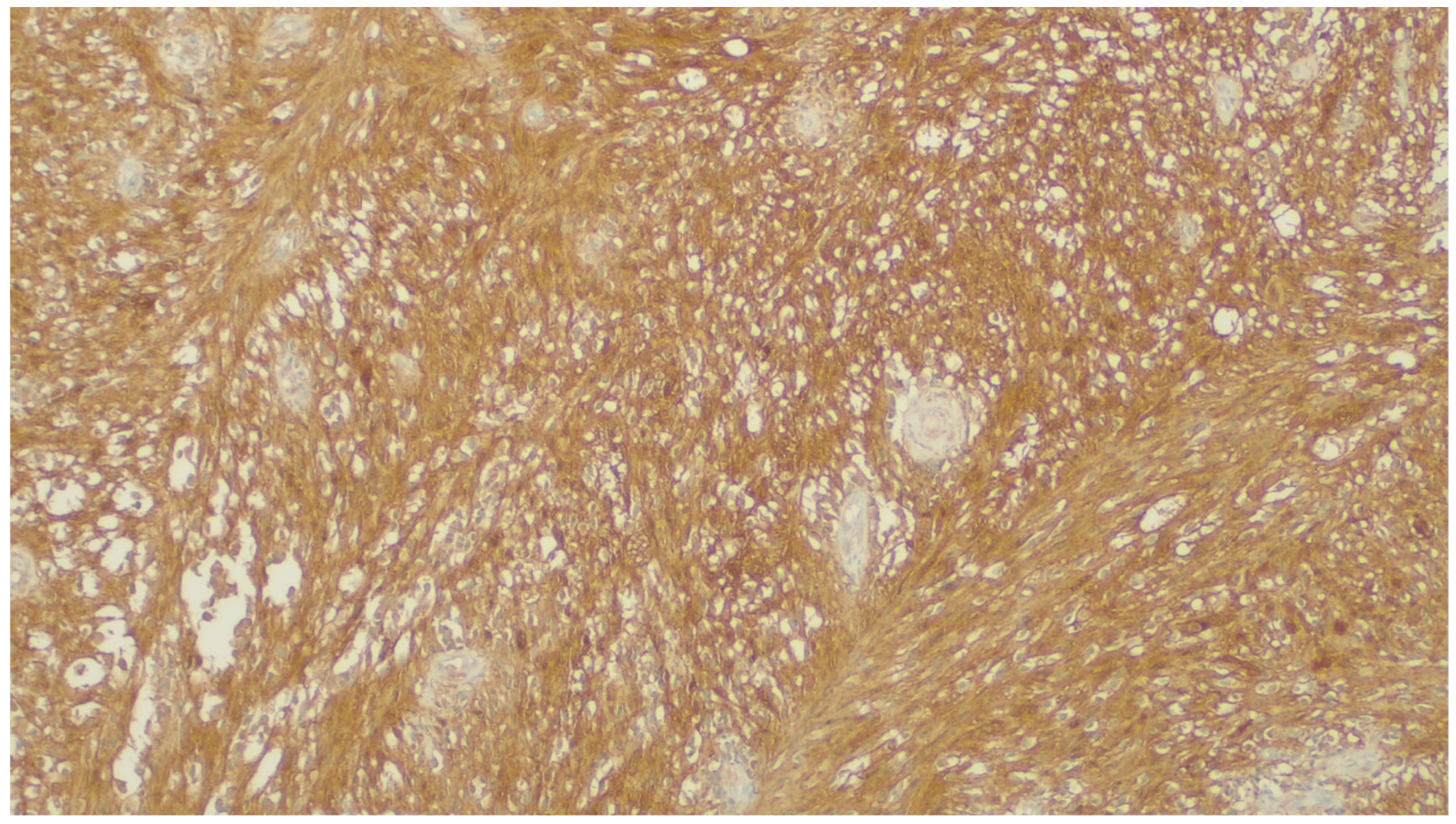
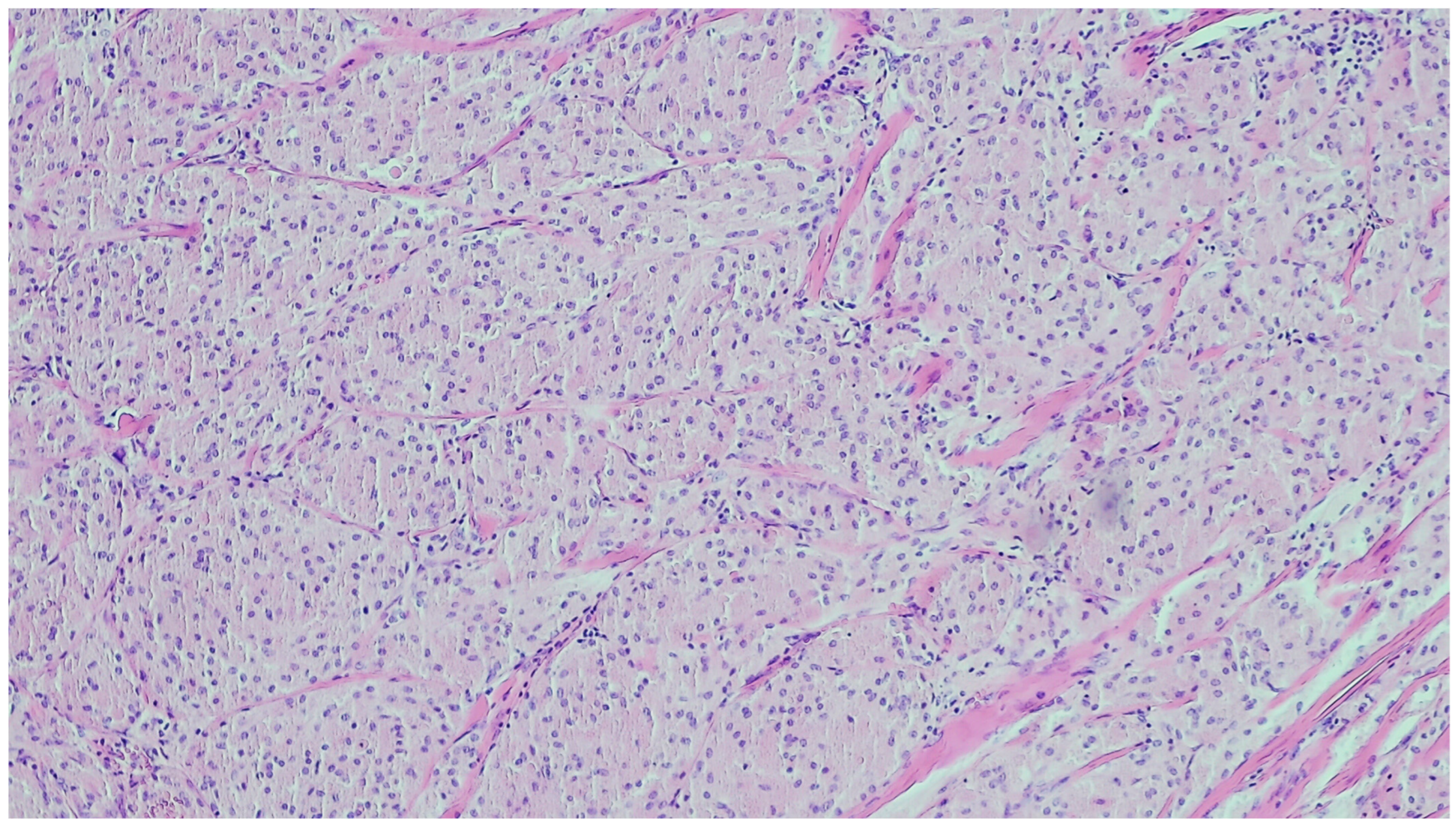
2. Inflammatory Myofibroblastic Tumor
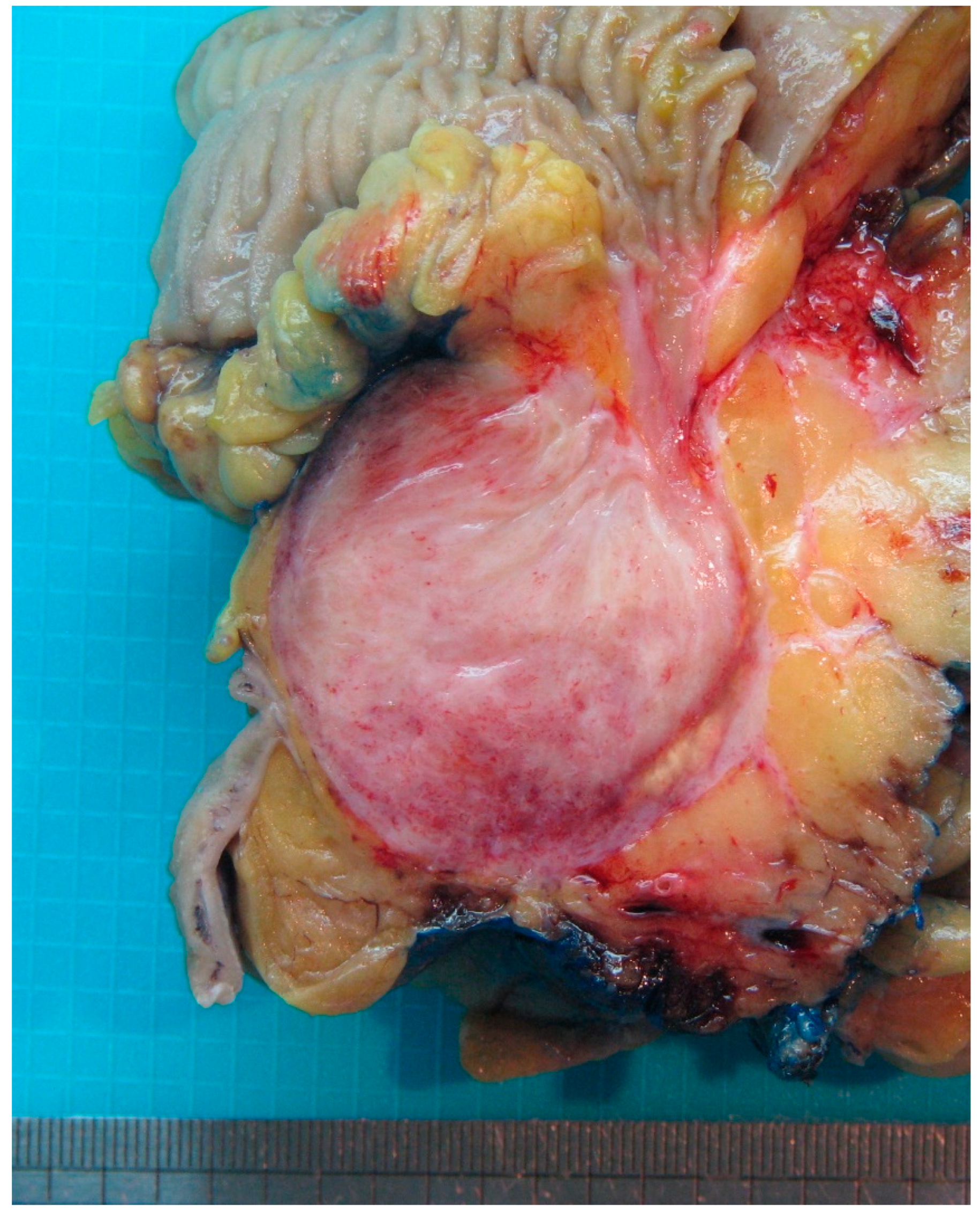
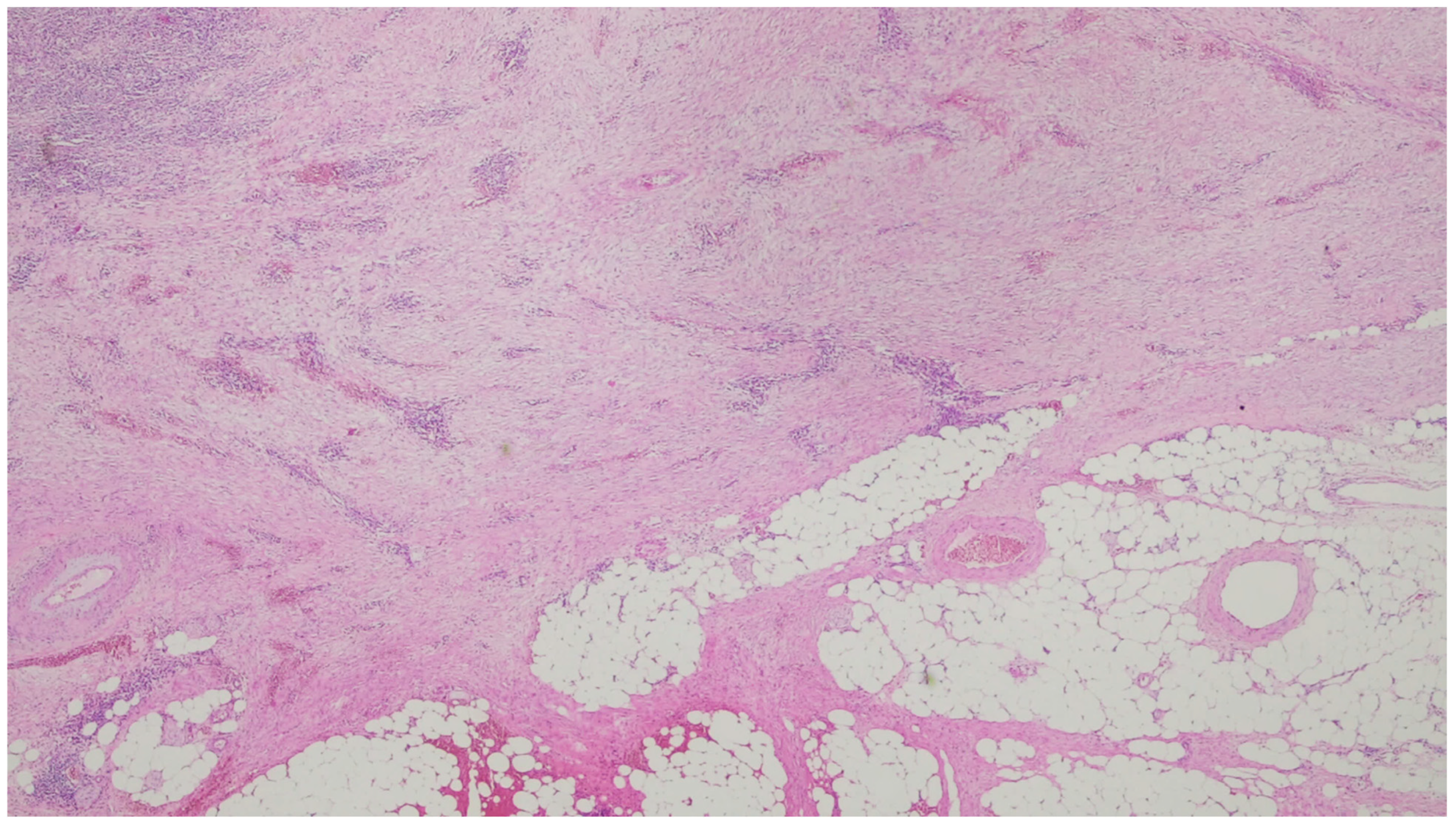
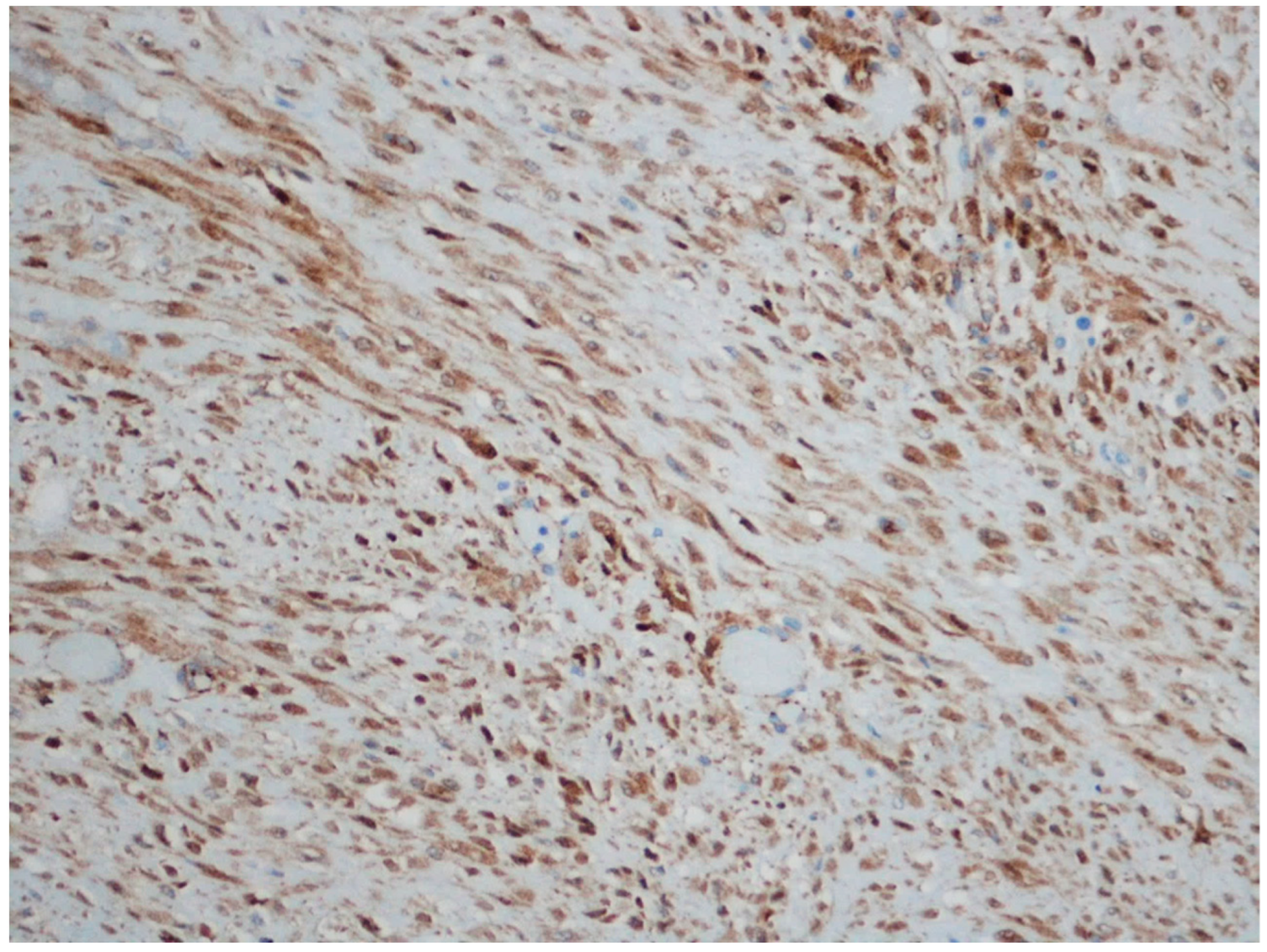
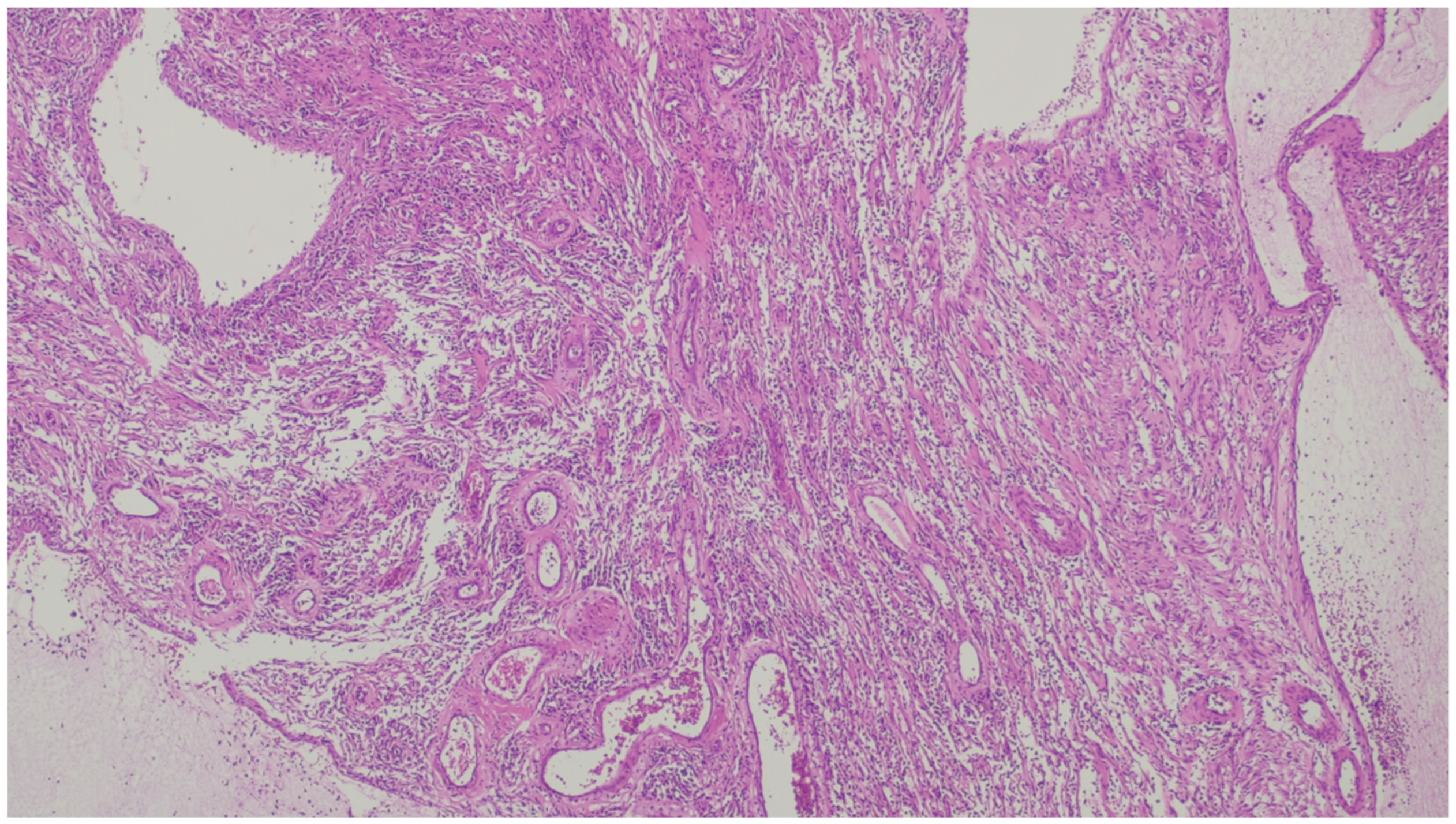
3. Desmoid Fibromatosis
4. Solitary Fibrous Tumor
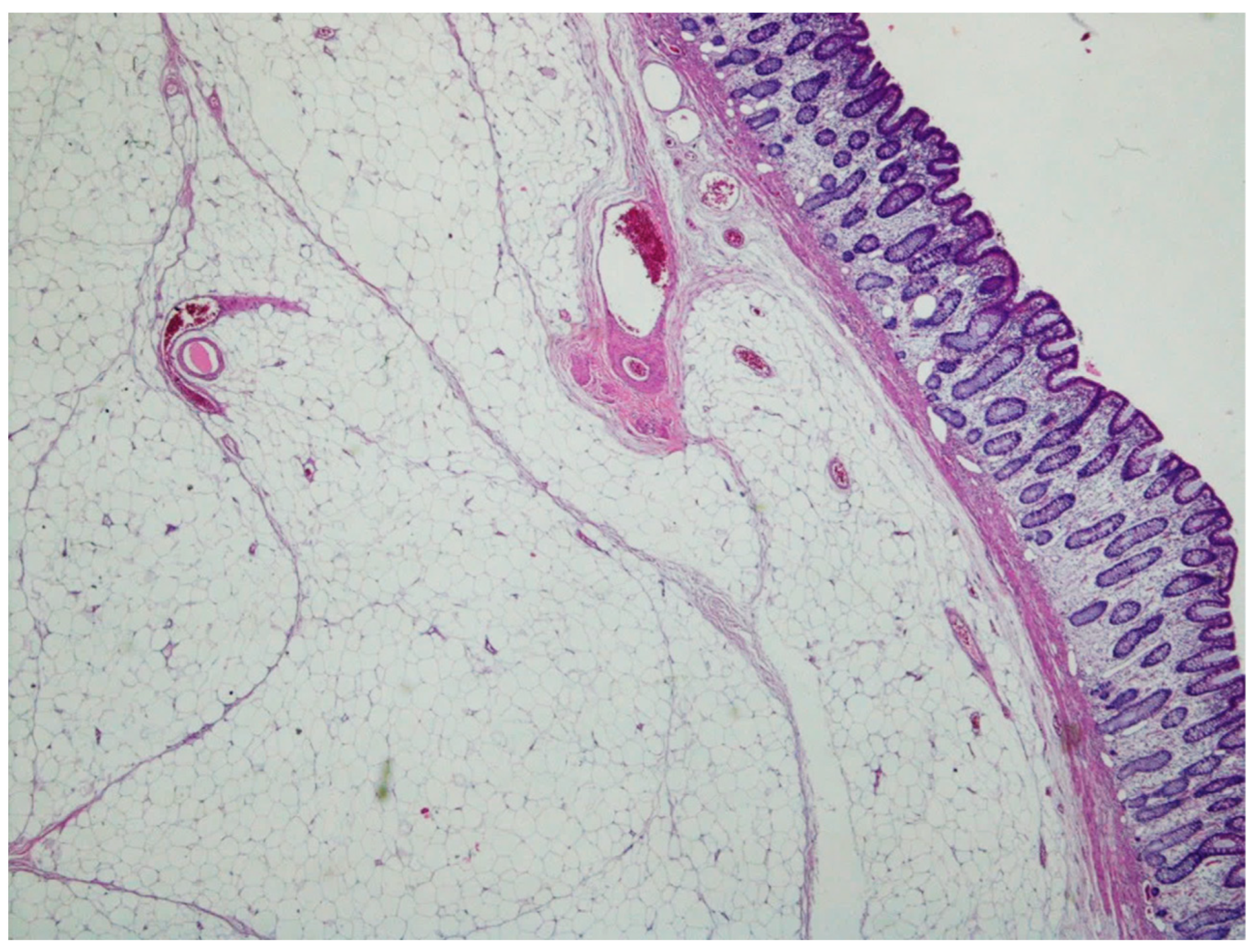
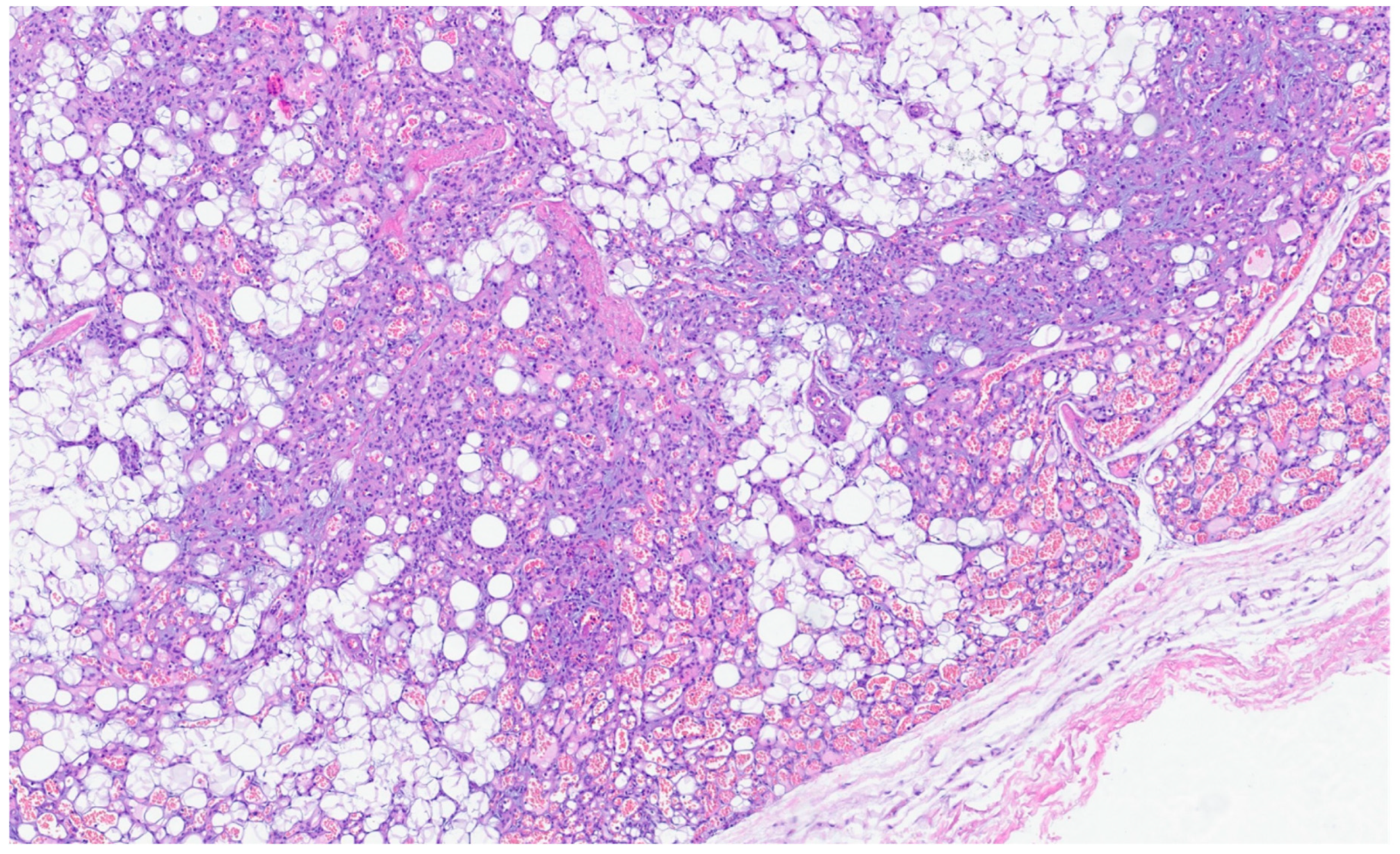
5. Lipoma
6. Inflammatory Fibroid Polyp
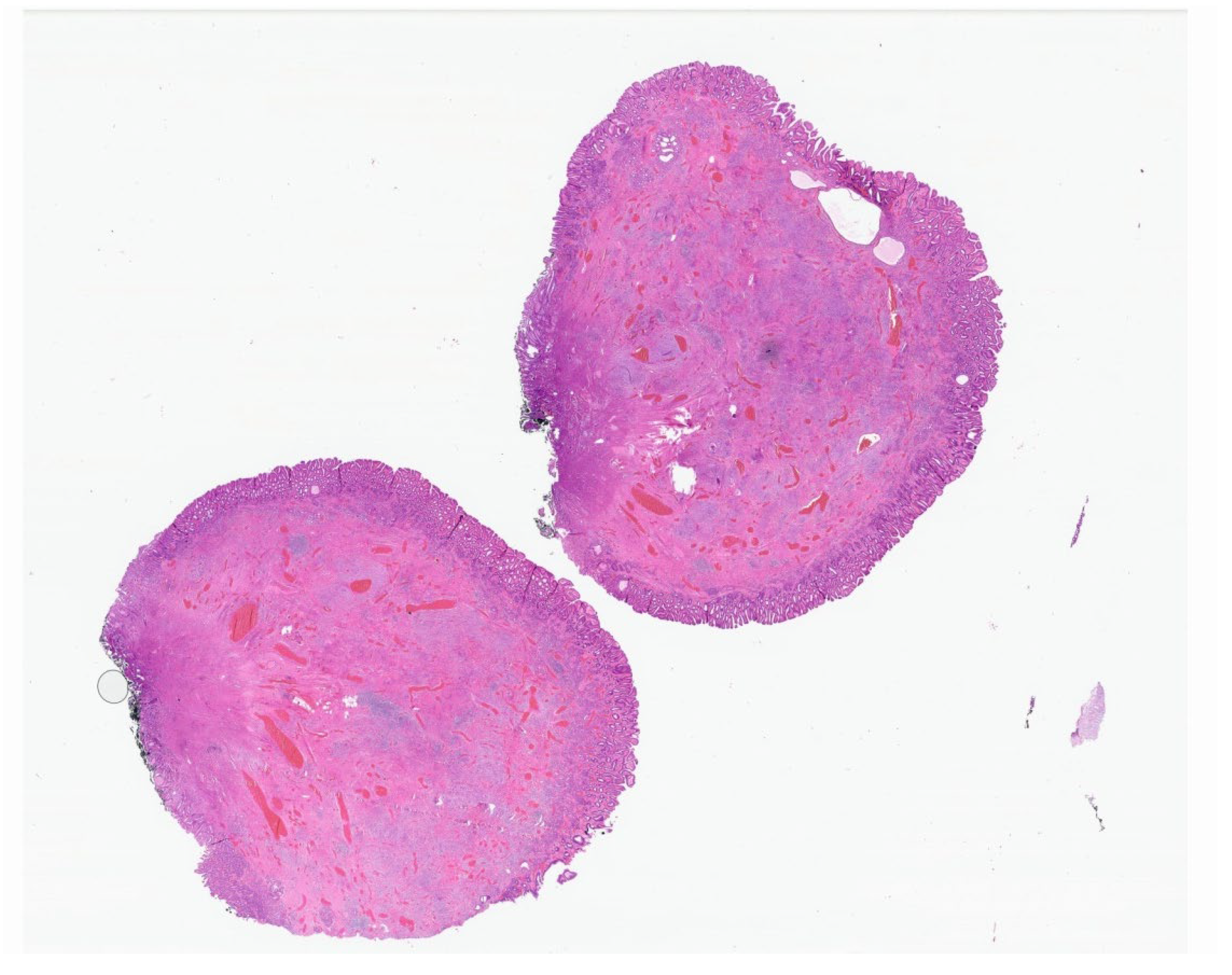
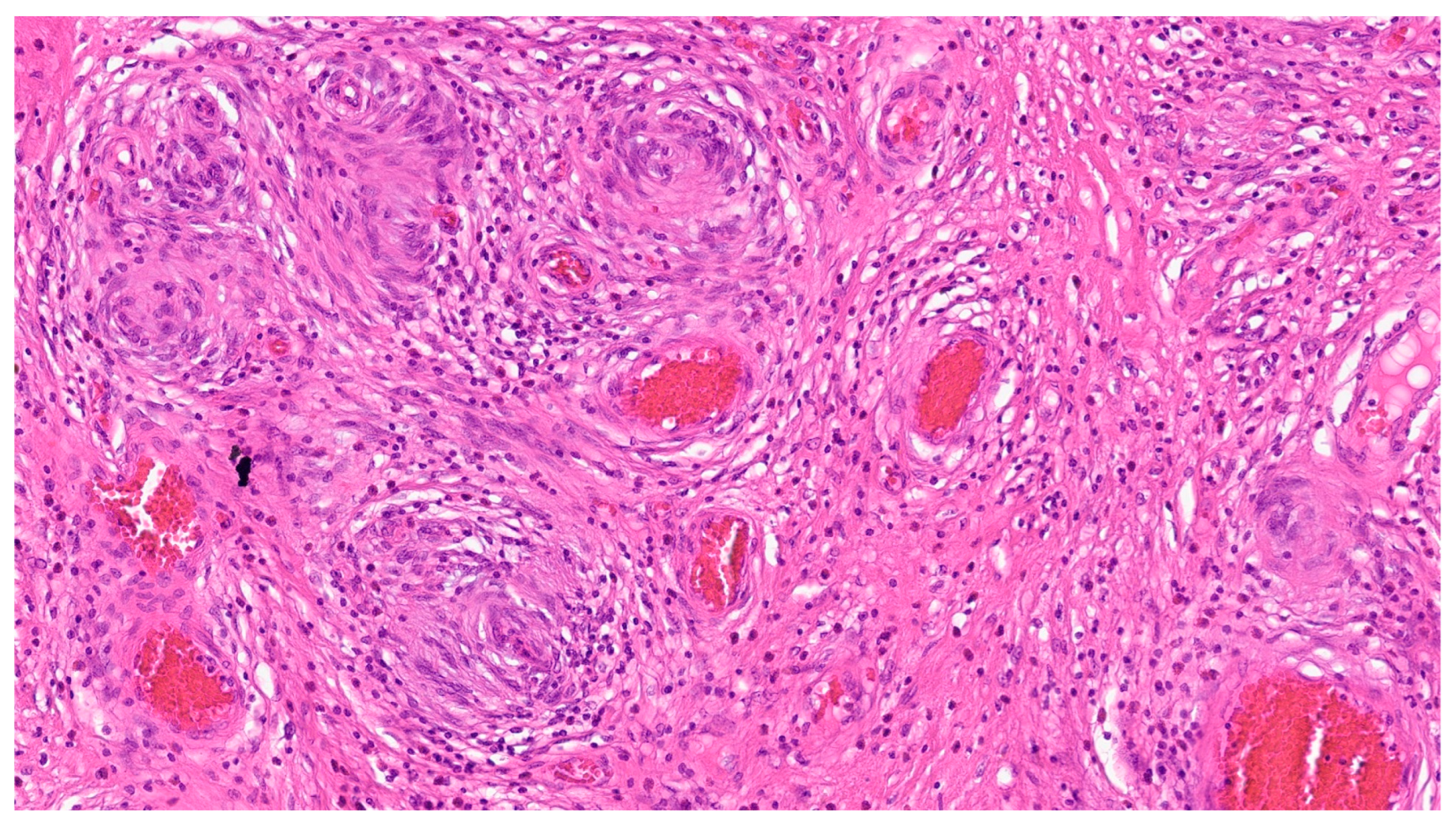
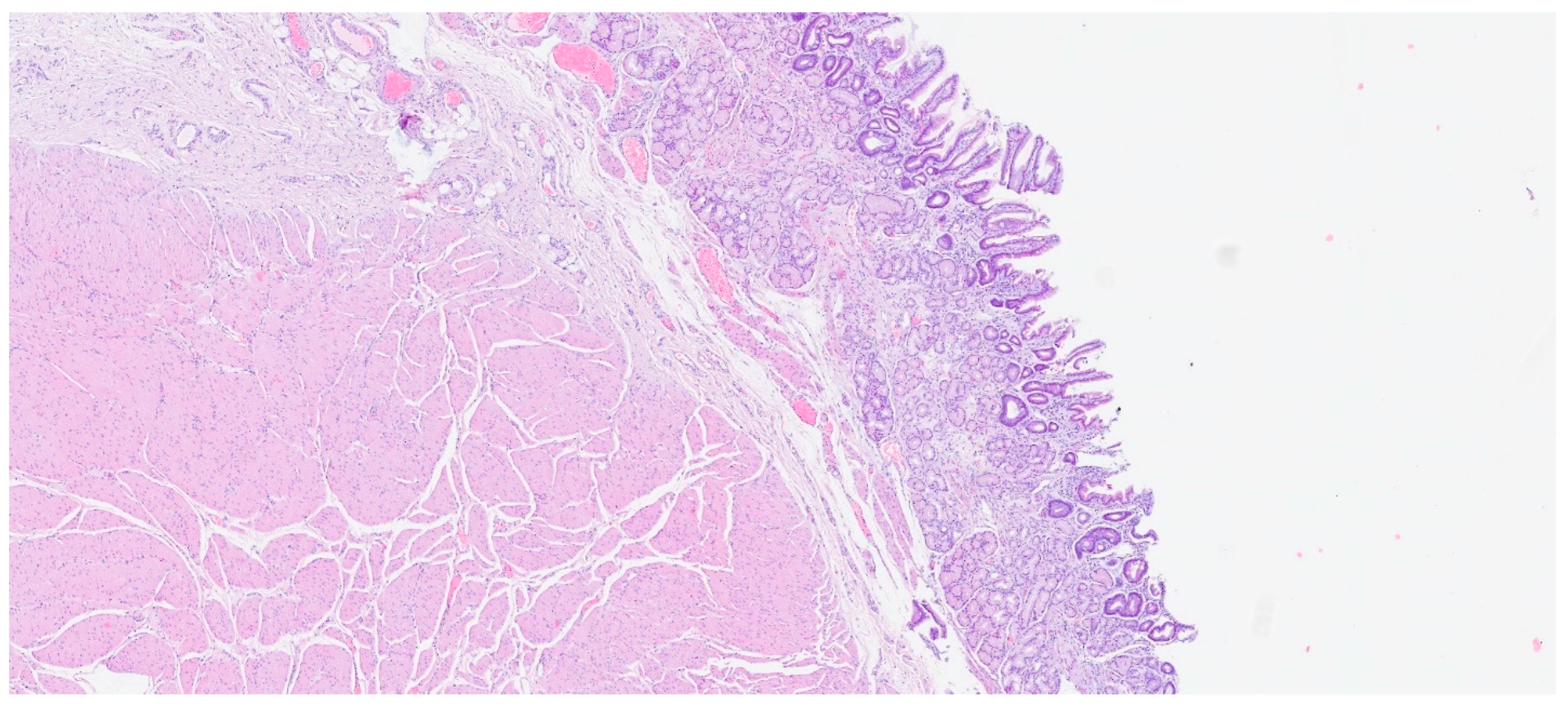
7. Plexiform Fibromyxoma
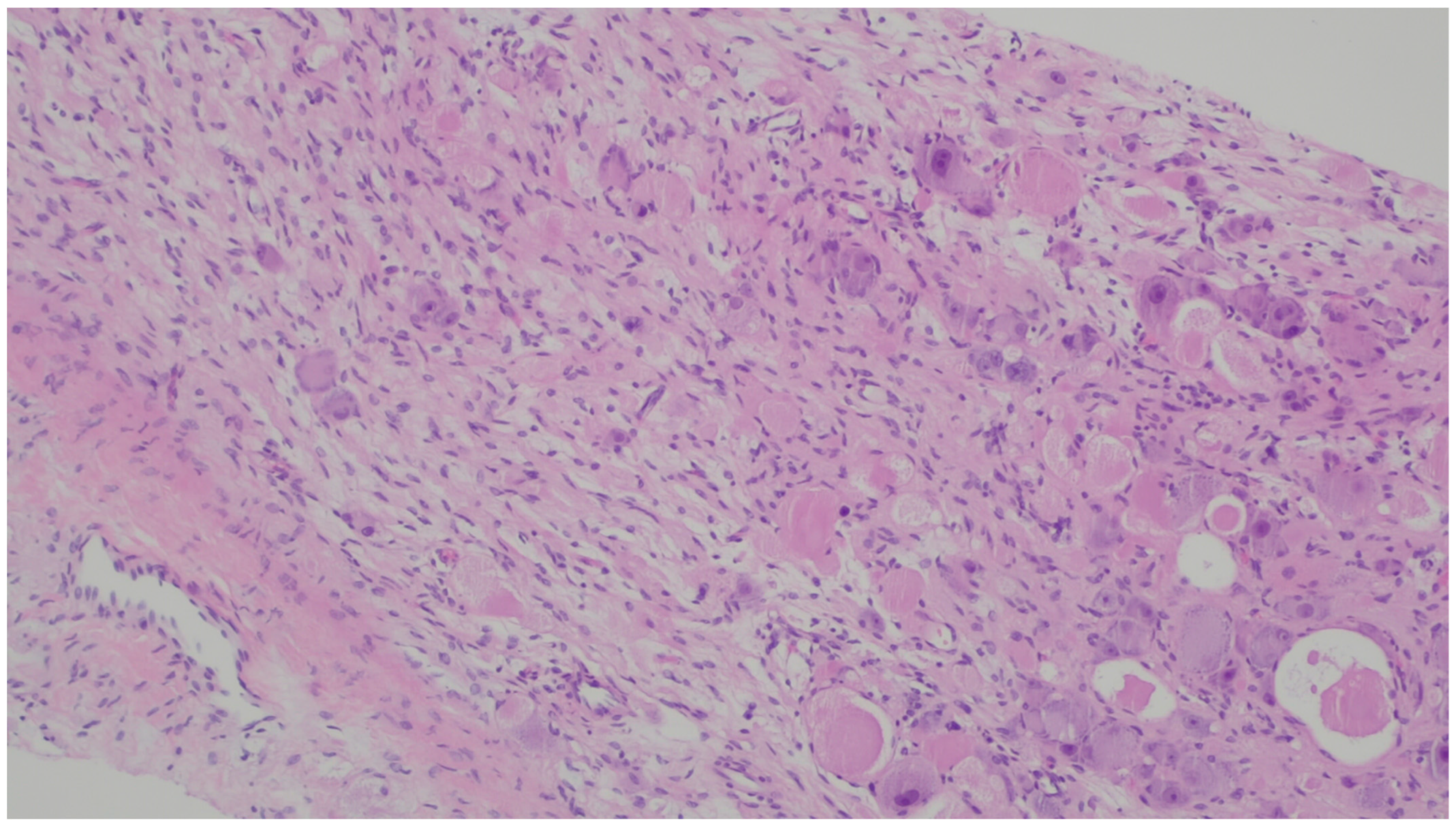
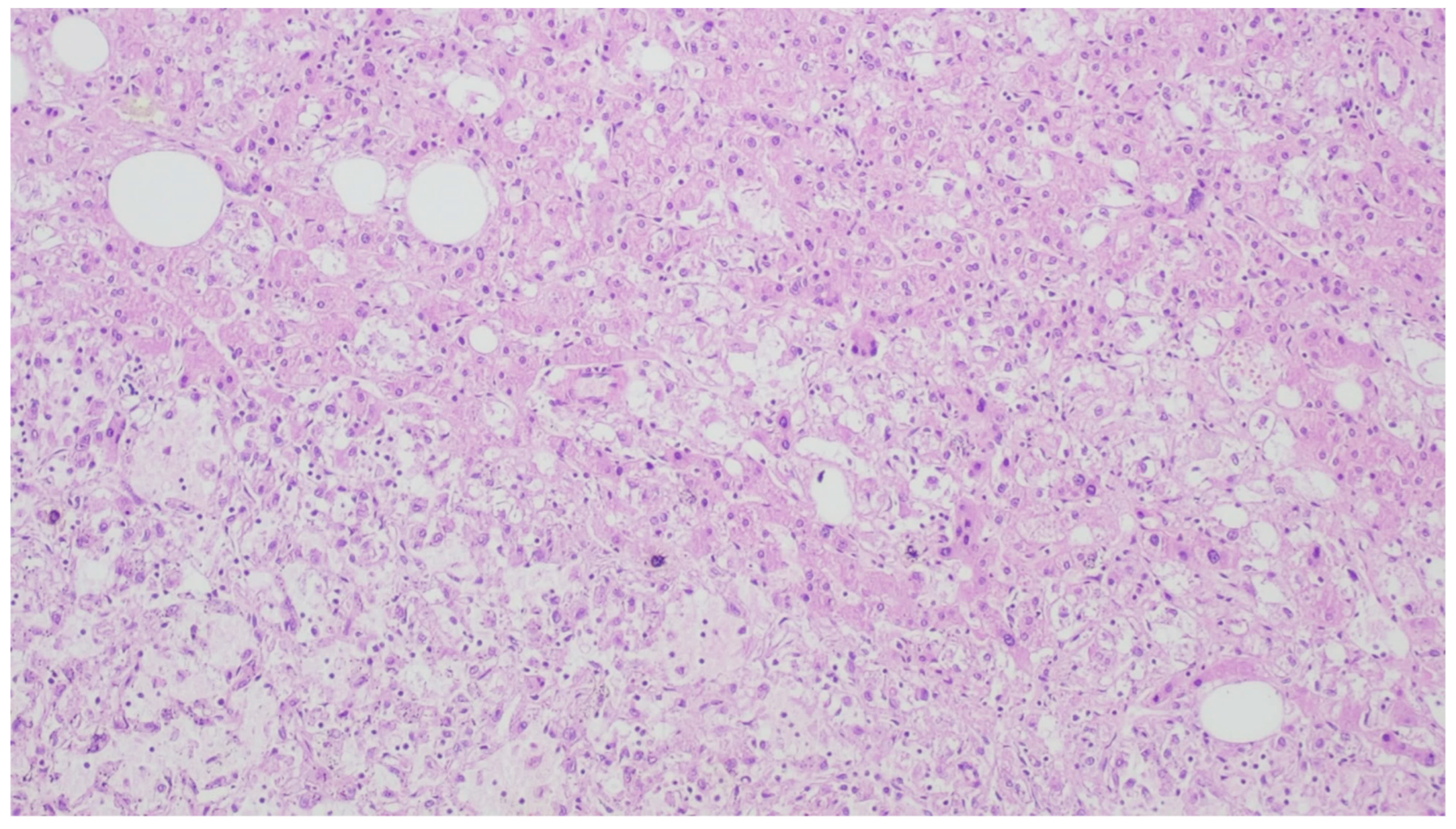
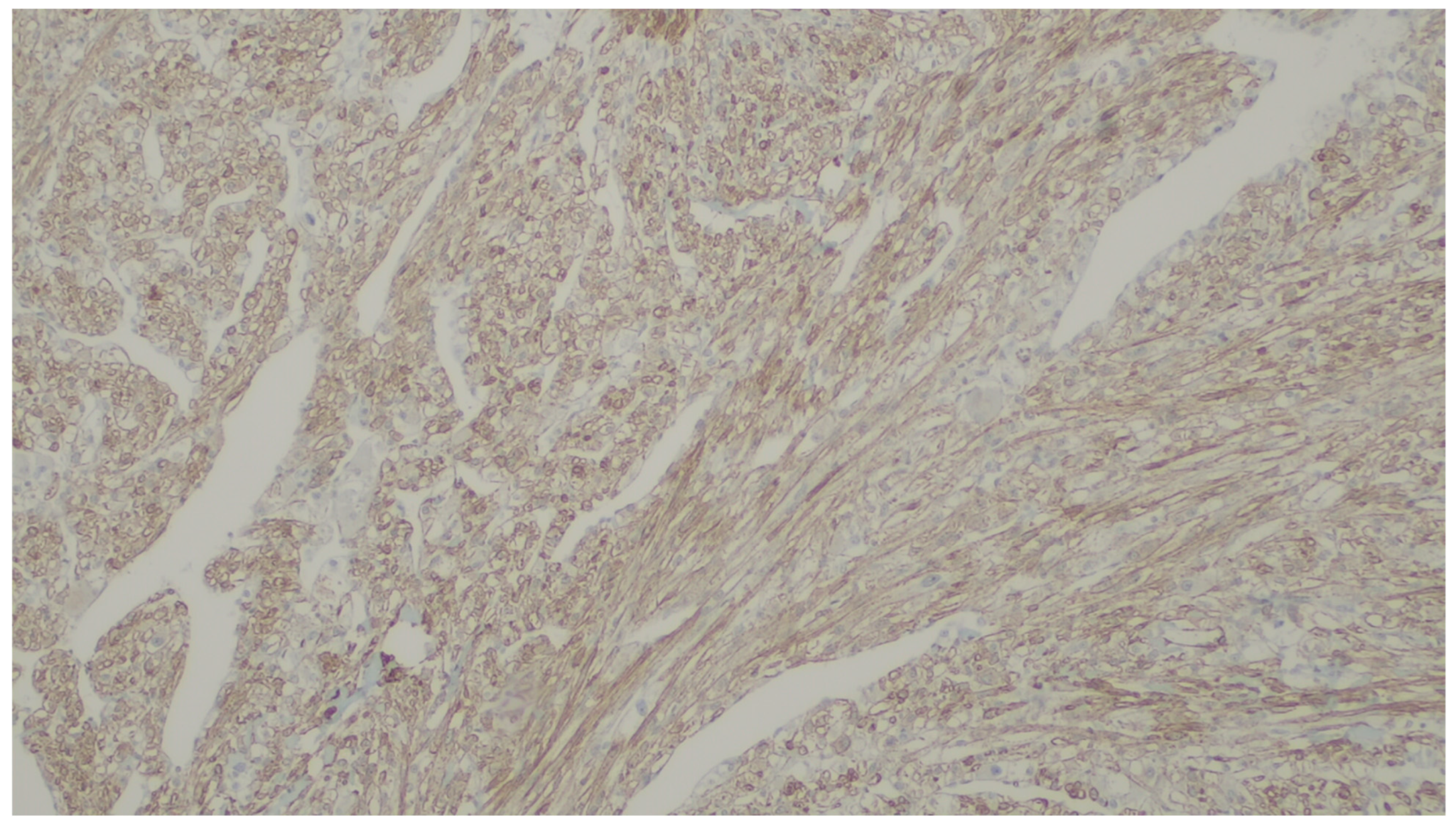
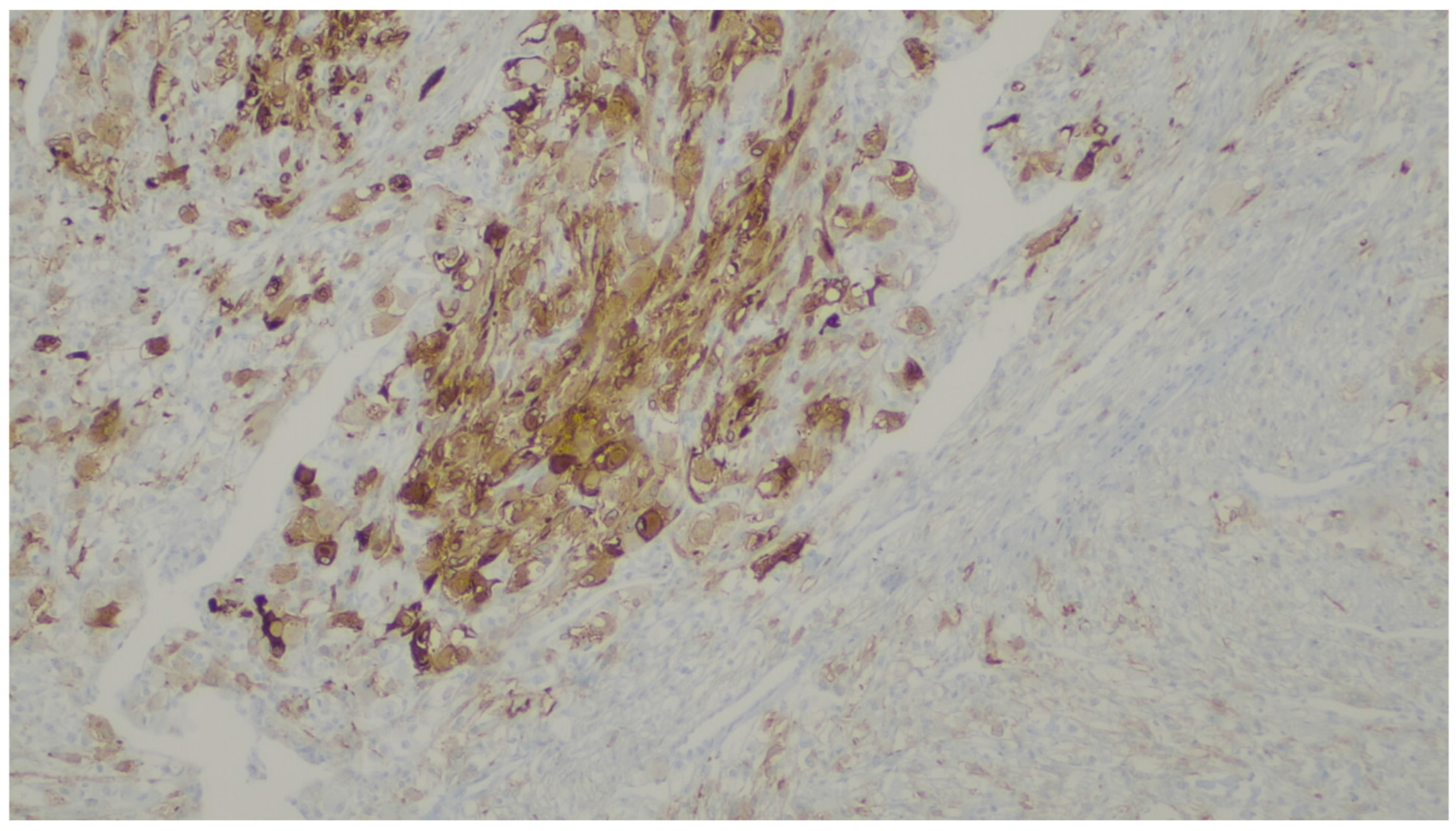
8. Leiomyoma
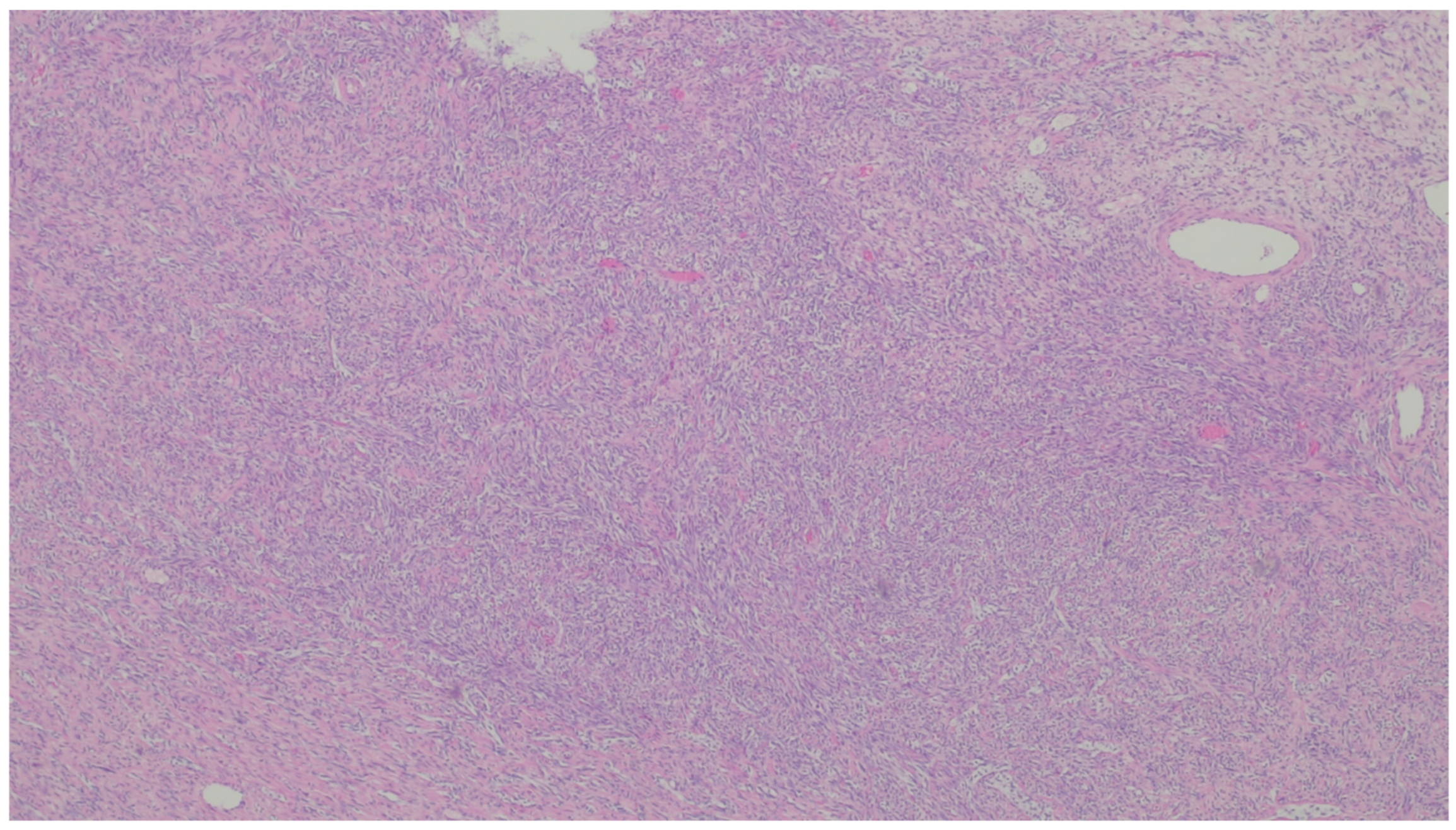
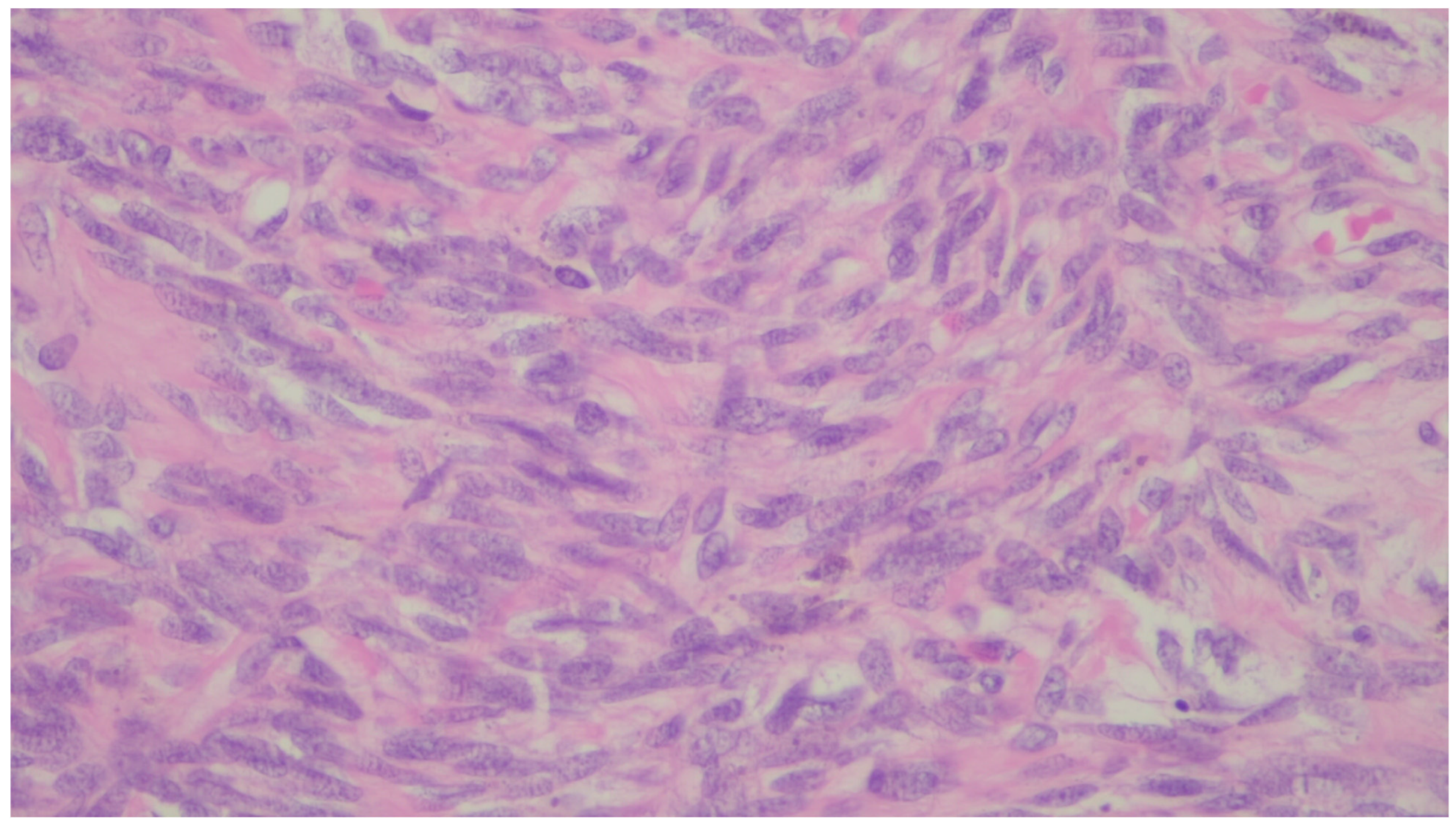
9. Leiomyosarcoma
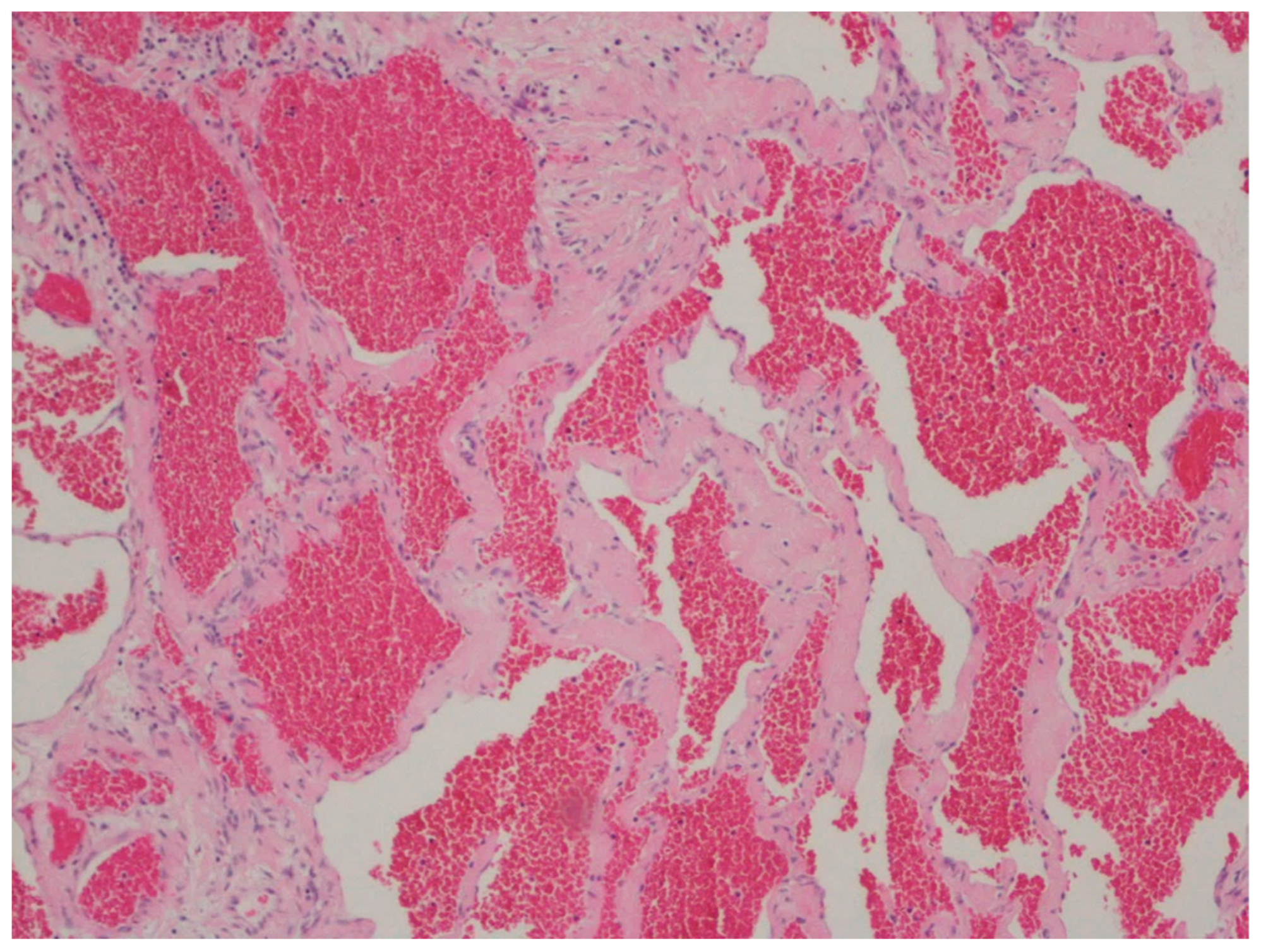
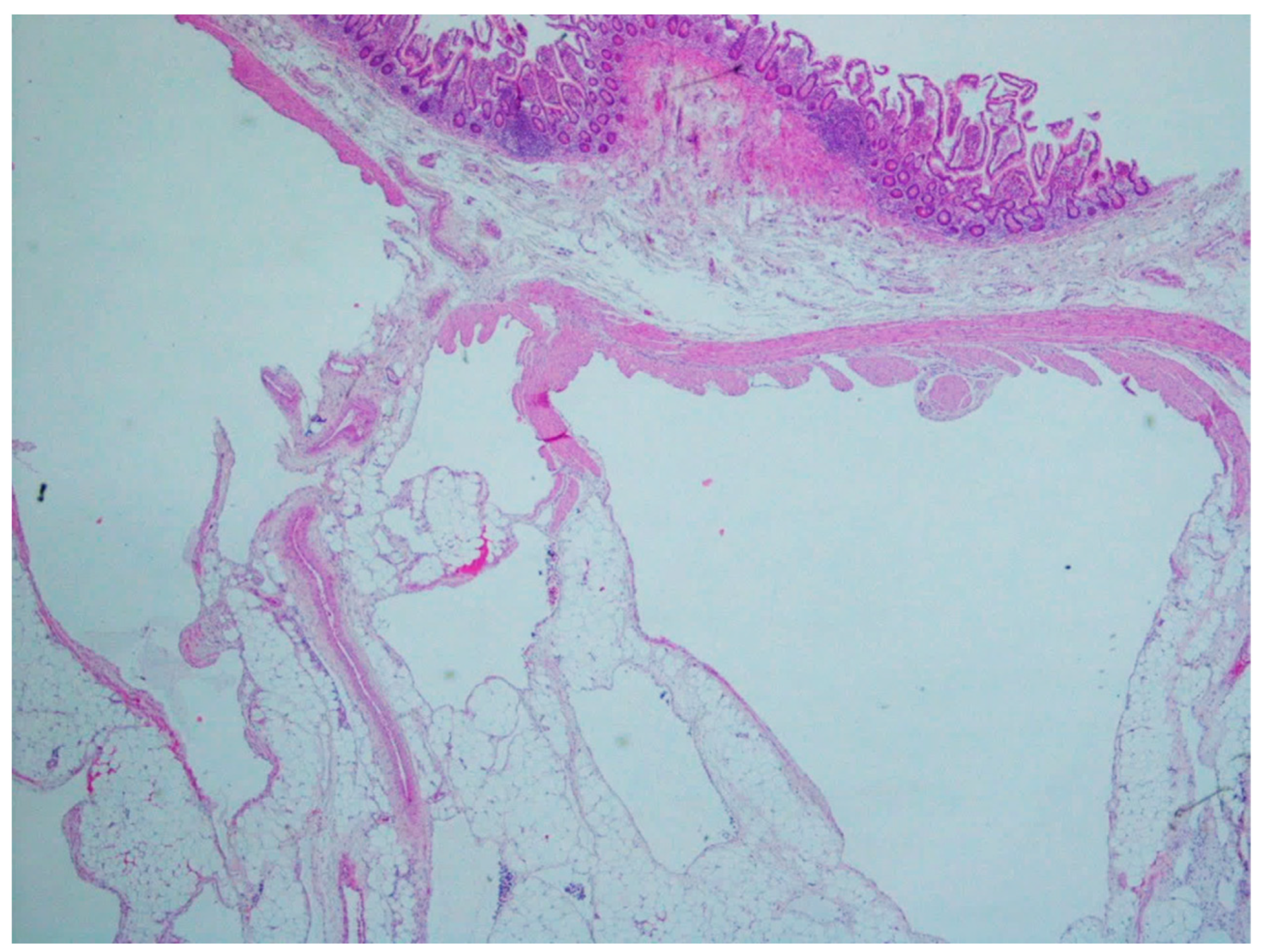
10. Hemangioma
11. Lymphangiomas
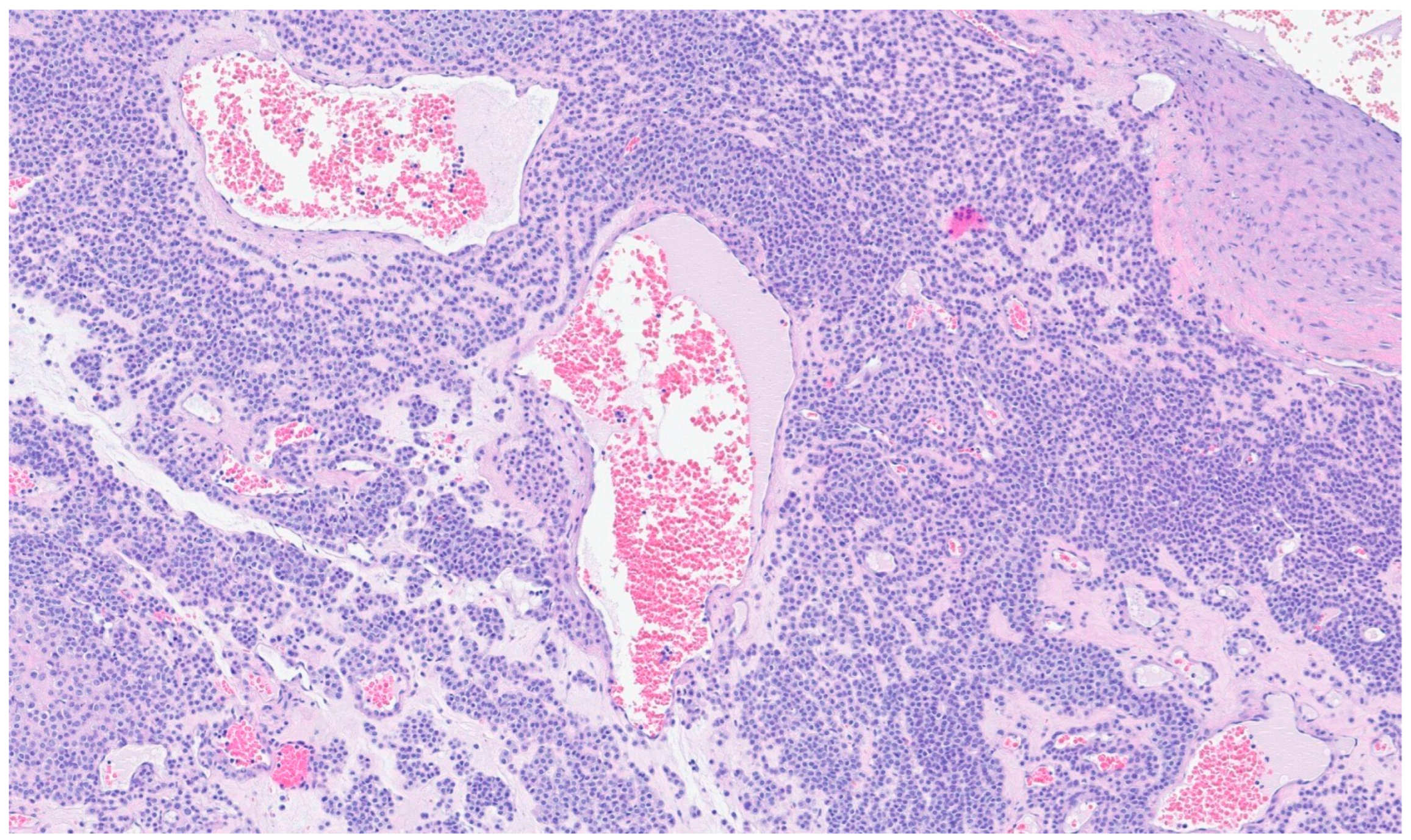
12. Kaposi Sarcoma
13. Angiosarcoma
14. Glomus Tumor
15. Schwannoma
16. Granular Cell Tumor
17. Perineurioma
18. Ganglioneuroma
19. PEComa
20. NUTM1-Rearranged Colorectal Sarcoma
21. Mesenchymal Tumors Associated with NTRK Rearrangements
22. Synovial Sarcoma
23. Gastrointestinal Clear Cell Sarcoma/Malignant Gastrointestinal Neuroectodermal Tumor
24. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| a-SMA | Alpha-smooth muscle actin |
| APC | Adenomatous polyposis coli |
| FISH | Fluorescence In Situ Hybridization |
| GI | Gastro intestinal |
| GIST | Gastrointestinal stromal tumor |
| IMT | inflammatory myofibroblastic tumor |
| LMS | Leiomyosarcoma |
| MEN | Multiple endocrine neoplasia |
| PEComas | Perivascular epithelioid cell tumors |
| VM | Vascular Malformations |
References
- Siegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2023. CA A Cancer J. Clin. 2023, 73, 233–254. [Google Scholar] [CrossRef]
- Waidhauser, J.; Bornemann, A.; Trepel, M.; Märkl, B. Frequency, localization, and types of gastrointestinal stromal tumor-associated neoplasia. World J. Gastroenterol. 2019, 25, 4261–4277. [Google Scholar] [CrossRef] [PubMed]
- Güller, U.; Tarantino, I.; Cerny, T.; Schmied, B.M.; Warschkow, R. Population-based SEER trend analysis of overall and cancer-specific survival in 5138 patients with gastrointestinal stromal tumor. BMC Cancer 2015, 15, 557. [Google Scholar] [CrossRef] [PubMed]
- Sbaraglia, M.; Businello, G.; Bellan, E.; Fassan, M.; Tos, A.P.D. Mesenchymal tumours of the gastrointestinal tract. Pathologica 2021, 113, 230–251. [Google Scholar] [CrossRef]
- Shi, H.; Wei, L.; Sun, L.; Guo, A. Primary gastric inflammatory myofibroblastic tumor: A clinicopathologic and immunohistochemical study of 5 cases. Pathol. Res. Pract. 2010, 206, 287–291. [Google Scholar] [CrossRef]
- Cheng, B.; Yang, C.; Liu, Z.; Liu, L.; Zhou, L. Primary gastric inflammatory myofibroblastic tumor. Medicine 2018, 97, e13423. [Google Scholar] [CrossRef]
- Antonescu, C.R.; Suurmeijer, A.J.; Zhang, L.; Sung, Y.-S.; Jungbluth, A.A.; Travis, W.D.; Al-Ahmadie, H.; Fletcher, C.D.; Alaggio, R. Molecular Characterization of Inflammatory Myofibroblastic Tumors With Frequent ALK and ROS1 Gene Fusions and Rare Novel RET Rearrangement. Am. J. Surg. Pathol. 2015, 39, 957–967. [Google Scholar] [CrossRef]
- Lee, C.-J.; Schöffski, P.; Modave, E.; van Wezel, T.; Boeckx, B.; Sufliarsky, J.; Gelderblom, H.; Blay, J.-Y.; Debiec-Rychter, M.; Sciot, R.; et al. Comprehensive Molecular Analysis of Inflammatory Myofibroblastic Tumors Reveals Diverse Genomic Landscape and Potential Predictive Markers for Response to Crizotinib. Clin. Cancer Res. 2021, 27, 6737–6748. [Google Scholar] [CrossRef]
- Harada, R.; Ohtaki, M.; Hashizume, N.; Takeshi, A.; Murai, E.; Shirahata, Y.; Suzuki, S.; Uchigasaki, S.; Yoshida, H. Inflammatory myofibroblastic tumor of the small intestine with intussusception in a child: Case report and literature review. J. Pediatr. Surg. Case Rep. 2020, 56, 101423. [Google Scholar] [CrossRef]
- Ribeiro, M.C.B.; Lopes, L.R.; Neto, J.C.d.S.; Meirelles, L.R.; de Carvalho, R.B.; Andreollo, N.A. Rare Gastric Inflammatory Myofibroblastic Tumor in an Adult Woman: A Case Report with Review of the Literature. Case Rep. Med. 2012, 2012, 374070. [Google Scholar] [CrossRef]
- Abrantes, C.F.C.; Silva, M.R.; Oliveira, R.C.; Eloy, C.; Cipriano, M.A.; Castro, L.P. Inflammatory myofibroblastic tumour arising incidentally as a polypoid lesion in the gallbladder Tumor miofibroblástico inflamatório surgindo incidentalmente como lesão polipoide na vesícula biliar. J. Bras. Patol. Med. Lab. 2015, 51, 422–426. [Google Scholar] [CrossRef]
- Siemion, K.; Reszec-Gielazyn, J.; Kisluk, J.; Roszkowiak, L.; Zak, J.; Korzynska, A. What do we know about inflammatory myofibroblastic tumors?—A systematic review. Adv. Med. Sci. 2022, 67, 129–138. [Google Scholar] [CrossRef]
- Nagtegaal, I.D.; Odze, R.D.; Klimstra, D.; Paradis, V.; Rugge, M.; Schirmacher, P.; Washington, K.M.; Carneiro, F.; Cree, I.A.; the WHO Classification of Tumours Editorial Board. The 2019 WHO classification of tumours of the digestive system. Histopathology 2020, 76, 182–188. [Google Scholar] [CrossRef]
- Gros, L.; Tos, A.P.D.; Jones, R.L.; Digklia, A. Inflammatory Myofibroblastic Tumour: State of the Art. Cancers 2022, 14, 3662. [Google Scholar] [CrossRef]
- Xu, P.; Shen, P.; Jin, Y.; Wang, L.; Wu, W. Epithelioid inflammatory myofibroblastic sarcoma of stomach: Diagnostic pitfalls and clinical characteristics. Int. J. Clin. Exp. Pathol. 2019, 12, 1738–1744. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6947137/ (accessed on 6 December 2023).
- Ganeshan, D.; Amini, B.; Nikolaidis, P.; Assing, M.; Vikram, R. Current Update on Desmoid Fibromatosis. J. Comput. Assist. Tomogr. 2019, 43, 29–38. [Google Scholar] [CrossRef]
- Xiao, J.; Mao, J.; Li, B. Clinical Characteristics and Treatment of Intra-abdominal Aggressive Fibromatosis: A Retrospective Study of 16 Patients. Front. Med. 2020, 7, 2. [Google Scholar] [CrossRef]
- Lips, D.; Barker, N.; Clevers, H.; Hennipman, A. The role of APC and beta-catenin in the aetiology of aggressive fibromatosis (Desmoid tumors). Eur. J. Surg. Oncol. (EJSO) 2009, 35, 3–10. [Google Scholar] [CrossRef]
- Sioda, N.A.; Wakim, A.A.; Wong, T.; Patel, S.; Coan, K.; Row, D. A Large Sporadic Intra-abdominal Desmoid-Type Fibromatosis in a Young Male: A Case Report. Front. Surg. 2020, 7, 60. [Google Scholar] [CrossRef]
- Enzo, M.; Rastrelli, M.; Rossi, C.; Hladnik, U.; Segat, D. The Wnt/β-catenin pathway in human fibrotic-like diseases and its eligibility as a therapeutic target. Mol. Cell. Ther. 2015, 3, 1. [Google Scholar] [CrossRef]
- McLean, T.D.; Duchi, S.; Di Bella, C. Molecular Pathogenesis of Sporadic Desmoid Tumours and Its Implications for Novel Therapies: A Systematised Narrative Review. Target. Oncol. 2022, 17, 223–252. [Google Scholar] [CrossRef]
- Zreik, R.T.; Fritchie, K.J. Morphologic Spectrum of Desmoid-Type Fibromatosis. Am. J. Clin. Pathol. 2016, 145, 332–340. [Google Scholar] [CrossRef]
- Bhattacharya, B.; Dilworth, H.P.; Iacobuzio-Donahue, C.; Ricci, F.; Weber, K.; A Furlong, M.; Fisher, C.M.; Montgomery, E. Nuclear β-Catenin Expression Distinguishes Deep Fibromatosis From Other Benign and Malignant Fibroblastic and Myofibroblastic Lesions. Am. J. Surg. Pathol. 2005, 29, 653–659. [Google Scholar] [CrossRef]
- Gao, C.; Wang, Y.; Broaddus, R.; Sun, L.; Xue, F.; Zhang, W. Exon 3 mutations of CTNNB1 drive tumorigenesis: A review. Oncotarget 2017, 9, 5492–5508. [Google Scholar] [CrossRef]
- Sturt, N.J.H.; Gallagher, M.C.; Bassett, P.; Philp, C.R.; Neale, K.F.; Tomlinson, I.P.M.; Silver, A.R.J.; Phillips, R.K.S. Evidence for genetic predisposition to desmoid tumours in familial adenomatous polyposis independent of the germline APC mutation. Gut 2004, 53, 1832–1836. [Google Scholar] [CrossRef]
- Howard, J.H.; Pollock, R.E. Intra-Abdominal and Abdominal Wall Desmoid Fibromatosis. Oncol. Ther. 2016, 4, 57–72. [Google Scholar] [CrossRef]
- Ghert, M.; Yao, X.; Corbett, T.; Gupta, A.; Kandel, R.; Verma, S.; Werier, J. Treatment and Follow-Up Strategies in Desmoid Tumours: A Practice Guideline. Curr. Oncol. 2014, 21, 642–649. [Google Scholar] [CrossRef]
- Zhang, Z.; Shi, J.; Yang, T.; Liu, T.; Zhang, K. Management of aggressive fibromatosis (Review). Oncol. Lett. 2020, 21, 1. [Google Scholar] [CrossRef]
- Testa, S.; Bui, N.Q.; Charville, G.W.; Avedian, R.S.; Steffner, R.; Ghanouni, P.; Mohler, D.G.; Ganjoo, K.N. Management of Patients with Newly Diagnosed Desmoid Tumors in a First-Line Setting. Cancers 2022, 14, 3907. [Google Scholar] [CrossRef]
- Van Broekhoven, D.L.M.; Verhoef, C.; Grünhagen, D.J.; van Gorp, J.M.H.H.; Bakker, M.A.D.; Hinrichs, J.W.J.; de Voijs, C.M.A.; van Dalen, T. Prognostic Value of CTNNB1 Gene Mutation in Primary Sporadic Aggressive Fibromatosis. Ann. Surg. Oncol. 2014, 22, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Martin-Broto, J.; Mondaza-Hernandez, J.L.; Moura, D.S.; Hindi, N. A Comprehensive Review on Solitary Fibrous Tumor: New Insights for New Horizons. Cancers 2021, 13, 2913. [Google Scholar] [CrossRef] [PubMed]
- Inayat, F.; Hussain, Q.; Shafique, K.; Hurairah, A.; Grossman, E.B. Solitary Fibrous Tumor of the Stomach. ACG Case Rep. J. 2017, 4, e35. [Google Scholar] [CrossRef]
- Kimmel, J.; Dikman, A.; Hajdu, C. Gastric Solitary Fibrous Tumor Causing Upper Gastrointestinal Bleeding. ACG Case Rep. J. 2019, 6, e00005. [Google Scholar] [CrossRef]
- Vogels, R.J.; Vlenterie, M.; Versleijen-Jonkers, Y.M.; Ruijter, E.; Bekers, E.M.; Verdijk, M.A.; Link, M.M.; Bonenkamp, J.J.; van der Graaf, W.T.; Slootweg, P.J.; et al. Solitary fibrous tumor—Clinicopathologic, immunohistochemical and molecular analysis of 28 cases. Diagn. Pathol. 2014, 9, 224. [Google Scholar] [CrossRef]
- Park, H.K.; Yu, D.B.; Sung, M.; Oh, E.; Kim, M.; Song, J.-Y.; Lee, M.-S.; Jung, K.; Noh, K.-W.; An, S.; et al. Molecular changes in solitary fibrous tumor progression. J. Mol. Med. 2019, 97, 1413–1425. [Google Scholar] [CrossRef] [PubMed]
- Georgiesh, T.; Namløs, H.M.; Sharma, N.; Lorenz, S.; Myklebost, O.; Bjerkehagen, B.; Meza-Zepeda, L.A.; Boye, K. Clinical and molecular implications of NAB2-STAT6 fusion variants in solitary fibrous tumour. Pathology 2021, 53, 713–719. [Google Scholar] [CrossRef]
- Lee, J.-C.; Fletcher, C.D. Malignant Fat-Forming Solitary Fibrous Tumor (so-called “Lipomatous Hemangiopericytoma”). Am. J. Surg. Pathol. 2011, 35, 1177–1185. [Google Scholar] [CrossRef]
- A Doyle, L.; Vivero, M.; Fletcher, C.D.; Mertens, F.; Hornick, J.L. Nuclear expression of STAT6 distinguishes solitary fibrous tumor from histologic mimics. Mod. Pathol. 2014, 27, 390–395. [Google Scholar] [CrossRef] [PubMed]
- Demicco, E.G.; Wagner, M.J.; Maki, R.G.; Gupta, V.; Iofin, I.; Lazar, A.J.; Wang, W.-L. Risk assessment in solitary fibrous tumors: Validation and refinement of a risk stratification model. Mod. Pathol. 2017, 30, 1433–1442. [Google Scholar] [CrossRef]
- Salas, S.; Resseguier, N.; Blay, J.Y.; Le Cesne, A.; Italiano, A.; Chevreau, C.; Rosset, P.; Isambert, N.; Soulie, P.; Cupissol, D.; et al. Prediction of local and metastatic recurrence in solitary fibrous tumor: Construction of a risk calculator in a multicenter cohort from the French Sarcoma Group (FSG) database. Ann. Oncol. 2017, 28, 1779–1787. [Google Scholar] [CrossRef]
- Voth, E.; Serio, S.; Gross, J.; Singh, A.; Dietz, N.; Nandipati, K. Solitary fibrous tumor of the stomach with high-grade sarcomatous dedifferentiation. J. Surg. Case Rep. 2018, 2018, rjy307. [Google Scholar] [CrossRef] [PubMed]
- Davanzo, B.; Emerson, R.E.; Lisy, M.; Koniaris, L.G.; Kays, J.K. Solitary fibrous tumor. Transl. Gastroenterol. Hepatol. 2018, 3, 94. [Google Scholar] [CrossRef]
- Kumar, S.; Harisankar, A.; Singh, R.; Kumar, A.; Kumar, B.; Mandal, M. Lipoma of the gastrointestinal tract: A tertiary care centre experience. Ind. Mark. Manag. 2023. [Google Scholar] [CrossRef]
- Caliskan, A.; Kohlmann, W.K.; E Affolter, K.; Downs-Kelly, E.; Kanth, P.; Bronner, M.P. Intramucosal lipomas of the colon implicate Cowden syndrome. Mod. Pathol. 2018, 31, 643–651. [Google Scholar] [CrossRef]
- Kastanakis, M.; Anyfantakis, D.; Symvoulakis, E.K.; Katsougris, N.; Papadomichelakis, A.; Kokkinos, I.; Petrakis, G.; Bobolakis, E. Cecal Lipoma Presenting as Acute Intestinal Obstruction in an Elderly Woman: A Case Report. Case Rep. Surg. 2013, 2013, 1–3. [Google Scholar] [CrossRef]
- Rezaii, S.; Enshaii, A.; Zahedi, A.; Amestejani, M.; Herik Dizaji, M. Gastrointestinal bleeding due to gastrointestinal lipoma: A case report. Casp. J. Intern. Med. 2021, 12, 622. [Google Scholar] [CrossRef]
- Garmpis, N.; Damaskos, C.; Garmpi, A.; Georgakopoulou, V.E.; Sakellariou, S.; Liakea, A.; Schizas, D.; Diamantis, E.; Farmaki, P.; Voutyritsa, E.; et al. Inflammatory Fibroid Polyp of the Gastrointestinal Tract: A Systematic Review for a Benign Tumor. In Vivo 2021, 35, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Klingbeil, K.D.; Balaban, A.; Fertig, R.M.; Gamret, A.C.; Gong, Y.; Torres, C.; Satahoo, S.S. Inflammatory fibroid polyp of the gastric antrum presenting as hypovolemic shock: Case report and literature review. Intractable Rare Dis. Res. 2017, 6, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Gadoura, A.; Mohammed, F.; Abdulkarim, M.; Ibn Yasir, A.; Shani, D.; Salih, N. Inflammatory fibroid polyp (Vanek’s tumor) causing double compound ileo-ileal intussusception in an adult patient, a case report. Int. J. Surg. Case Rep. 2022, 93, 106947. [Google Scholar] [CrossRef]
- Maccagno, A.; Sander, B.; Dintner, S.; Harloff, M.; Füzesi, L.; Märkl, B. Inflammatory fibroid polyp: A series of 29 cases and a systematic review of the literature. Hum. Pathol. Rep. 2023, 32. [Google Scholar] [CrossRef]
- Huss, S.; Wardelmann, E.; Goltz, D.; Binot, E.; Hartmann, W.; Merkelbach-Bruse, S.; Büttner, R.; Schildhaus, H. Activating PDGFRA mutations in inflammatory fibroid polyps occur in exons 12, 14 and 18 and are associated with tumour localization. Histopathology 2012, 61, 59–68. [Google Scholar] [CrossRef]
- Almaghrabi, H.; Almaghrabi, M.; Al-Maghrabi, H. Giant Ileocecal Inflammatory Fibroid Polyp: Unique Clinical Presentation. Case Rep. Surg. 2020, 2020, 8811404. [Google Scholar] [CrossRef]
- Lasota, J.; Wang, Z.-F.; Sobin, L.H.; Miettinen, M. Gain-of-function PDGFRA mutations, earlier reported in gastrointestinal stromal tumors, are common in small intestinal inflammatory fibroid polyps. A study of 60 cases. Mod. Pathol. 2009, 22, 1049–1056. [Google Scholar] [CrossRef]
- Tang, Y.-S.; Liu, L.; Gao, Y.; He, Q.-C.; Guo, H.-M.; Zhao, Z.-F. Minimally invasive colonoscopy treatment of inflammatory fibroid polyps in the terminal ileum. Sci. Rep. 2023, 13, 4929. [Google Scholar] [CrossRef]
- Lin, M.M.; Song, L.M.; Qin, S.M.; Li, D.M.; Hou, G.B.; Li, X. Plexiform fibromyxoma. Medicine 2021, 100, e27164. [Google Scholar] [CrossRef]
- Higashi, M.; Hamada, T.; Sasaki, K.; Tsuruda, Y.; Shimonosono, M.; Kitazono, I.; Kirishima, M.; Tasaki, T.; Noguchi, H.; Tabata, K.; et al. Esophageal plexiform fibromyxoma: A case report with molecular analysis for MALAT1-GLI1 fusion. Pathol. - Res. Pract. 2022, 233, 153878. [Google Scholar] [CrossRef]
- Arslan, M.E.; Li, H.; Fu, Z.; A Jennings, T.; Lee, H. Plexiform fibromyxoma: Review of rare mesenchymal gastric neoplasm and its differential diagnosis. World J. Gastrointest. Oncol. 2021, 13, 409–423. [Google Scholar] [CrossRef]
- Sugimura, N.; Kubota, E.; Sasaki, M.; Fukusada, S.; Mizuno, Y.; Iwasaki, H.; Tanaka, M.; Ozeki, K.; Shimura, T.; Kataoka, H. A case of asymptomatic gastric plexiform fibromyxoma followed up for 3 years. DEN Open 2023, 4, e291. [Google Scholar] [CrossRef] [PubMed]
- Altameemi, H.; A Asghar, I. Gastric Plexiform Fibromyxoma: A Rare Clinical Entity. Am. J. Clin. Pathol. 2022, 158, S55–S56. [Google Scholar] [CrossRef]
- Miettinen, M.; Makhlouf, H.R.; Sobin, L.H.; Lasota, J. Plexiform fibromyxoma: A distinctive benign gastric antral neoplasm not to be confused with a myxoid GIST. Am. J. Surg. Pathol. 2009, 33, 1624–1632. [Google Scholar] [CrossRef] [PubMed]
- Su, H.-A.; Yen, H.-H.; Chen, C.-J. An Update on Clinicopathological and Molecular Features of Plexiform Fibromyxoma. Can. J. Gastroenterol. Hepatol. 2019, 2019, 3960920. [Google Scholar] [CrossRef]
- Senapathi, H.; Morada, A.; Perry, M.; Bertram, C.; Yeung, E.; Sultany, M.; Bertsch, D.; Cagir, B. Prognostic Factors in Gastrointestinal Leiomyosarcomas: An Analysis Using the Surveillance, Epidemiology, and End Results (SEER) Database. Cureus 2021, 13. [Google Scholar] [CrossRef]
- Yamamoto, H.; Handa, M.; Tobo, T.; Setsu, N.; Fujita, K.; Oshiro, Y.; Mihara, Y.; Yoshikawa, Y.; Oda, Y. Clinicopathological features of primary leiomyosarcoma of the gastrointestinal tract following recognition of gastrointestinal stromal tumours. Histopathology 2013, 63, 194–207. [Google Scholar] [CrossRef]
- Hilal, L.; Barada, K.; Mukherji, D.; Temraz, S.; Shamseddine, A. Gastrointestinal (GI) leiomyosarcoma (LMS) case series and review on diagnosis, management, and prognosis. Med. Oncol. 2016, 33, 20. [Google Scholar] [CrossRef]
- Alpert, L.; Al-Sabti, R.; Graham, R.P.; Pai, R.K.; Gonzalez, R.S.; Zhang, X.; Smith, V.; Wang, H.L.; Westbrook, L.; Goldblum, J.R.; et al. Smooth muscle tumors of the gastrointestinal tract: An analysis of prognostic features in 407 cases. Mod. Pathol. 2020, 33, 1410–1419. [Google Scholar] [CrossRef]
- Demicco, E.G.; Boland, G.M.; Savannah, K.J.B.; Lusby, K.; Young, E.D.; Ingram, D.; Watson, K.L.; Bailey, M.; Guo, X.; Hornick, J.L.; et al. Progressive loss of myogenic differentiation in leiomyosarcoma has prognostic value. Histopathology 2014, 66, 627–638. [Google Scholar] [CrossRef]
- Aggarwal, G.; Sharma, S.; Zheng, M.; Reid, M.D.; Crosby, J.H.; Chamberlain, S.M.; Nayak-Kapoor, A.; Lee, J.R. Primary leiomyosarcomas of the gastrointestinal tract in the post–gastrointestinal stromal tumor era. Ann. Diagn. Pathol. 2012, 16, 532–540. [Google Scholar] [CrossRef]
- Agaimy, A.; Wünsch, P.H. True smooth muscle neoplasms of the gastrointestinal tract: Morphological spectrum and classification in a series of 85 cases from a single institute. Langenbeck’s Arch. Surg. 2006, 392, 75–81. [Google Scholar] [CrossRef]
- Mellish, R. Multiple hemangiomas of the gastrointestinal tract in children. Am. J. Surg. 1971, 121, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S. GI-Associated Hemangiomas and Vascular Malformations. Clin. Colon Rectal Surg. 2011, 24, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Handra-Luca, A.; Montgomery, E. Vascular malformations and hemangiolymphangiomas of the gastrointestinal tract: Morphological features and clinical impact. Int. J. Clin. Exp. Pathol. 2011, 4, 430–443. [Google Scholar]
- Roisman, I.; Manny, J.; Fields, S.; Shiloni, E. Intra-abdominal lymphangioma. Br. J. Surg. 1989, 76, 485–489. [Google Scholar] [CrossRef]
- Wiegand, S.; Eivazi, B.; Barth, P.J.; von Rautenfeld, D.B.; Folz, B.J.; Mandic, R.; Werner, J.A. Pathogenesis of lymphangiomas. Virchows Arch. 2008, 453, 1–8. [Google Scholar] [CrossRef]
- Hornick, J.L.; Fletcher, C.D. Intraabdominal cystic lymphangiomas obscured by marked superimposed reactive changes: Clinicopathological analysis of a series. Hum. Pathol. 2005, 36, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Ho-Yen, C.; Chang, F.; van der Walt, J.; Lucas, S. Gastrointestinal Malignancies in HIV-infected or Immunosuppressed Patients. Adv. Anat. Pathol. 2007, 14, 431–443. [Google Scholar] [CrossRef]
- Hengge, U.R.; Ruzicka, T.; Tyring, S.K.; Stuschke, M.; Roggendorf, M.; A Schwartz, R.; Seeber, S. Update on Kaposi’s sarcoma and other HHV8 associated diseases. Part 1: Epidemiology, environmental predispositions, clinical manifestations, and therapy. Lancet Infect. Dis. 2002, 2, 281–292. [Google Scholar] [CrossRef]
- Barrison, I.G.; Foster, S.; Harris, J.W.; Pinching, A.J.; Walker, J.G. Upper gastrointestinal Kaposi’s sarcoma in patients positive for HIV antibody without cutaneous disease. BMJ 1988, 296, 92–93. [Google Scholar] [CrossRef]
- Nagata, N.; Shimbo, T.; Yazaki, H.; Asayama, N.; Akiyama, J.; Teruya, K.; Igari, T.; Ohmagari, N.; Oka, S.; Uemura, N. Predictive Clinical Factors in the Diagnosis of Gastrointestinal Kaposi’s Sarcoma and Its Endoscopic Severity. PLoS ONE 2012, 7, e46967. [Google Scholar] [CrossRef]
- Zheng, W.; Obeng, R.C.; Graham, R.P.M.; Lui, S.; Cheng, J.; Alexiev, B.A.; Quigley, B.; Krasinskas, A.; Yang, G.-Y.; Escobar, D.; et al. Histologic Variants of Kaposi Sarcoma in the Gastrointestinal Tract. Am. J. Surg. Pathol. 2022, 46, 1500–1506. [Google Scholar] [CrossRef] [PubMed]
- Robin, Y.-M.; Guillou, L.; Michels, J.-J.; Coindre, J.-M. Human Herpesvirus 8 Immunostaining: A Sensitive and Specific Method for Diagnosing Kaposi Sarcoma in Paraffin-Embedded Sections. Am. J. Clin. Pathol. 2004, 121, 330–334. [Google Scholar] [CrossRef]
- IARC. WHO Classification of Soft Tissue and Bone Tumours; IARC: Geneva, Switzerland, 2020. [Google Scholar]
- Allison, K.H.; Yoder, B.J.; Bronner, M.P.; Goldblum, J.R.; Rubin, B.P. Angiosarcoma Involving the Gastrointestinal Tract. Am. J. Surg. Pathol. 2004, 28, 298–307. [Google Scholar] [CrossRef]
- Schizas, D.; Mastoraki, A.; Giannakodimos, I.; Giannakodimos, A.; Ziogou, A.; Katsaros, I.; Frountzas, M.; Koutelidakis, I.; Vassiliu, P.; Pikoulis, E. Primary Angiosarcoma of the Gastrointestinal Tract: A Systematic Review of the Literature. J. Investig. Surg. 2020, 35, 400–408. [Google Scholar] [CrossRef]
- Guillou, L.; Aurias, A. Soft tissue sarcomas with complex genomic profiles. Virchows Arch. 2009, 456, 201–217. [Google Scholar] [CrossRef]
- Italiano, A.; Chen, C.; Thomas, R.; Breen, M.; Bonnet, F.; Sevenet, N.; Longy, M.; Maki, R.G.; Coindre, J.; Antonescu, C.R. Alterations of the p53 and PIK3CA/AKT/mTOR pathways in angiosarcomas: A pattern distinct from other sarcomas with complex genomics. Cancer 2012, 118, 5878–5887. [Google Scholar] [CrossRef]
- Itakura, E.; Yamamoto, H.; Oda, Y.; Tsuneyoshi, M. Detection and characterization of vascular endothelial growth factors and their receptors in a series of angiosarcomas. J. Surg. Oncol. 2007, 97, 74–81. [Google Scholar] [CrossRef]
- Young, R.J.; Brown, N.J.; Reed, M.W.; Hughes, D.; Woll, P.J. Angiosarcoma. Lancet Oncol. 2010, 11, 983–991. [Google Scholar] [CrossRef]
- Huang, S.-C.; Zhang, L.; Sung, Y.-S.M.; Chen, C.-L.M.; Kao, Y.-C.; Agaram, N.P.M.; Singer, S.; Tap, W.D.; D’angelo, S.; Antonescu, C.R. Recurrent CIC Gene Abnormalities in Angiosarcomas. Am. J. Surg. Pathol. 2016, 40, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Tarpey, P.S.; Sheldon, H.; Martincorena, I.; Van Loo, P.; Gundem, G.; Wedge, D.C.; Ramakrishna, M.; Cooke, S.L.; Pillay, N.; et al. Recurrent PTPRB and PLCG1 mutations in angiosarcoma. Nat. Genet. 2014, 46, 376–379. [Google Scholar] [CrossRef]
- Hogeboom-Gimeno, A.; van Ravensteijn, S.; Desar, I.; Hillebrandt-Roeffen, M.; van Cleef, P.; Bonenkamp, J.; Flucke, U.; Versleijen-Jonkers, Y. MYC amplification in angiosarcoma depends on etiological/clinical subgroups–Diagnostic and prognostic value. Ann. Diagn. Pathol. 2023, 63, 152096. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gong, H.; Wang, Y.; Zhang, G.; Hou, P. Angiosarcoma of the visceral organs: A morphological, immunohistochemical, and C-MYC status analysis. Pathol.-Res. Pract. 2022, 238, 154118. [Google Scholar] [CrossRef]
- Meis-Kindblom, J.M.; Kindblom, L.-G. Angiosarcoma of Soft Tissue. Am. J. Surg. Pathol. 1998, 22, 683–697. [Google Scholar] [CrossRef]
- Pusztaszeri, M.P.; Seelentag, W.; Bosman, F.T. Immunohistochemical Expression of Endothelial Markers CD31, CD34, von Willebrand Factor, and Fli-1 in Normal Human Tissues. J. Histochem. Cytochem. 2006, 54, 385–395. [Google Scholar] [CrossRef]
- Miettinen, M.; Wang, Z.-F.; Paetau, A.; Tan, S.-H.; Dobi, A.; Srivastava, S.; Sesterhenn, I. ERG Transcription Factor as an Immunohistochemical Marker for Vascular Endothelial Tumors and Prostatic Carcinoma. Am. J. Surg. Pathol. 2011, 35, 432–441. [Google Scholar] [CrossRef]
- Folpe, A.L.; Chand, E.M.; Goldblum, J.R.; Weiss, S.W. Expression of Fli-1, a Nuclear Transcription Factor, Distinguishes Vascular Neoplasms from Potential Mimics. Am. J. Surg. Pathol. 2001, 25, 1061–1066. [Google Scholar] [CrossRef]
- Alles, J.U.; Bosslet, K. Immunocytochemistry of Angiosarcomas: A Study of 19 Cases with Special Emphasis on the Applicability of Endothelial Cell Specific Markers to Routinely Prepared Tissues. Am. J. Clin. Pathol. 1988, 89, 463–471. [Google Scholar] [CrossRef]
- Fayette, J.; Martin, E.; Piperno-Neumann, S.; Le Cesne, A.; Robert, C.; Bonvalot, S.; Ranchère, D.; Pouillart, P.; Coindre, J.M.; Blay, J.Y. Angiosarcomas, a heterogeneous group of sarcomas with specific behavior depending on primary site: A retrospective study of 161 cases. Ann. Oncol. 2007, 18, 2030–2036. [Google Scholar] [CrossRef]
- Girard, N.; Marin, C.; Hélias-Rodzewicz, Z.; Villa, C.; Julié, C.; de Lajarte-Thirouard, A.; de Beauce, S.M.; Lagorce-Pages, C.; Renaud, F.; Cazals-Hatem, D.; et al. CARMN-NOTCH2 fusion transcript drives high NOTCH2 expression in glomus tumors of the upper digestive tract. Genes Chromosom. Cancer 2021, 60, 723–732. [Google Scholar] [CrossRef]
- Miettinen, M.M.; Paal, E.M.; Lasota, J.M.; Sobin, L.H.M. Gastrointestinal glomus tumors: A clinicopathologic, immunohistochemical, and molecular genetic study of 32 cases. Am. J. Surg. Pathol. 2002, 26, 301–311. [Google Scholar] [CrossRef]
- Folpe, A.L.M.; Fanburg–Smith, J.C.M.; Miettinen, M.M.; Weiss, S.W.M. Atypical and malignant glomus tumors: Analysis of 52 cases, with a proposal for the reclassification of glomus tumors. Am. J. Surg. Pathol. 2001, 25, 1–12. [Google Scholar] [CrossRef]
- Bozdogan, N.; Dilek, G.B.; Benzer, E.; Karadeniz, M.; Bozdogan, O. Transducing-Like Enhancer of Split 1: A Potential Immunohistochemical Marker for Glomus Tumor. Am. J. Dermatopathol. 2017, 39, 524–527. [Google Scholar] [CrossRef]
- Dervan, P.A.; Tobbia, I.N.; Casey, M.; O’Loughlin, J.; O’Brien, M. Glomus tumours: An immunohistochemical profile of 11 cases. Histopathology 1989, 14, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Mosquera, J.; Sboner, A.; Zhang, L.; Chen, C.; Sung, Y.; Chen, H.; Agaram, N.P.; Briskin, D.; Basha, B.M.; Singer, S.; et al. Novel MIR143-NOTCH fusions in benign and malignant glomus tumors. Genes, Chromosom. Cancer 2013, 52, 1075–1087. [Google Scholar] [CrossRef]
- Agaram, N.P.M.; Zhang, L.; Jungbluth, A.A.; Dickson, B.C.; Antonescu, C.R. A Molecular Reappraisal of Glomus Tumors and Related Pericytic Neoplasms with Emphasis on NOTCH-gene Fusions. Am. J. Surg. Pathol. 2020, 44, 1556–1562. [Google Scholar] [CrossRef]
- Chakrapani, A.; Warrick, A.; Nelson, D.; Beadling, C.; Corless, C.L. BRAF and KRAS Mutations in Sporadic Glomus Tumors. Am. J. Dermatopathol. 2012, 34, 533–535. [Google Scholar] [CrossRef]
- Dashti, N.K.; Bahrami, A.; Lee, S.J.; Jenkins, S.M.; Rodriguez, F.J.; Folpe, A.L.; Boland, J.M. BRAF V600E Mutations Occur in a Subset of Glomus Tumors, and Are Associated With Malignant Histologic Characteristics. Am. J. Surg. Pathol. 2017, 41, 1532–1541. [Google Scholar] [CrossRef]
- Papke, D.J.J.; Sholl, L.M.; Doyle, L.A.; Fletcher, C.D.M.; Hornick, J.L. Gastroesophageal Glomus Tumors: Clinicopathologic and Molecular Genetic Analysis of 26 Cases with a Proposal for Malignancy Criteria. Am. J. Surg. Pathol. 2022, 46, 1436–1446. [Google Scholar] [CrossRef]
- Folpe, A.L.; Mentzel, T.; Lehr, H.-A.; Fisher, C.; Balzer, B.L.; Weiss, S.W. Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: A clinicopathologic study of 26 cases and review of the literature. Am. J. Surg. Pathol. 2005, 29, 1558–1575. [Google Scholar] [CrossRef]
- Lomdo, M.; Setti, K.; Oukabli, M.; Moujahid, M.; Bounaim, A. Gastric schwannoma: A diagnosis that should be known in 2019. J. Surg. Case Rep. 2020, 2020, rjz382. [Google Scholar] [CrossRef]
- Mekras, A.; Krenn, V.; Perrakis, A.; Croner, R.S.; Kalles, V.; Atamer, C.; Grützmann, R.; Vassos, N. Gastrointestinal schwannomas: A rare but important differential diagnosis of mesenchymal tumors of gastrointestinal tract. BMC Surg. 2018, 18, 47. [Google Scholar] [CrossRef]
- Crinò, S.; Bernardoni, L.; Manfrin, E.; Parisi, A.; Gabbrielli, A. Endoscopic ultrasound features of pancreatic schwannoma. Endosc. Ultrasound 2016, 5, 396–398. [Google Scholar] [CrossRef]
- Kudose, S.; Kyriakos, M.; Awad, M.M. Gastric plexiform schwannoma in association with neurofibromatosis type 2. Clin. J. Gastroenterol. 2016, 9, 352–357. [Google Scholar] [CrossRef]
- Yoon, H.Y.; Kim, C.B.; Lee, Y.H.; Kim, H.G. Gastric Schwannoma. Yonsei Med. J. 2008, 49, 1052–1054. [Google Scholar] [CrossRef]
- Radaelli, F.; Minoli, G. Granular Cell Tumors of the Gastrointestinal Tract: Questions and Answers. Gastroenterol. Hepatol. 2009, 5, 798. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2886372/ (accessed on 9 December 2023).
- Chen, Y.; Chen, Y.; Chen, X.; Chen, L.; Liang, W. Colonic granular cell tumor: Report of 11 cases and management with review of the literature. Oncol. Lett. 2018, 16, 1419–1424. [Google Scholar] [CrossRef]
- Marcoval, J.; Bauer-Alonso, A.; Llobera-Ris, C.; Moreno-Vílchez, C.; Penín, R.; Bermejo, J. Granular Cell Tumor: A Clinical Study of 81 Patients. Actas Dermo-Sifiliográficas 2021, 112, 441–446. [Google Scholar] [CrossRef]
- Castagna, J.; Clerc, J.; Dupond, A.-S.; Laresche, C. Multiple granular cell tumours in a patient with Noonan’s syndrome and juvenile myelomonocytic leukemia. Ann. Dermatol. Venereol. 2017, 144, 705–711. [Google Scholar] [CrossRef]
- Schrader, K.; Nelson, T.; De Luca, A.; Huntsman, D.; McGillivray, B. Multiple granular cell tumors are an associated feature of LEOPARD syndrome caused by mutation in PTPN11. Clin. Genet. 2009, 75, 185–189. [Google Scholar] [CrossRef]
- Pareja, F.; Brandes, A.H.; Basili, T.; Selenica, P.; Geyer, F.C.; Fan, D.; Da Cruz Paula, A.; Kumar, R.; Brown, D.N.; Gularte-Mérida, R.; et al. Loss-of-function mutations in ATP6AP1 and ATP6AP2 in granular cell tumors. Nat. Commun. 2018, 9, 3533. [Google Scholar] [CrossRef]
- Katiyar, V.; Vohra, I.; Uprety, A.; Yin, W.; Gupta, S. Recurrent Unresectable Malignant Granular Cell Tumor with Response to Pazopanib. Cureus 2020, 12, e8287. [Google Scholar] [CrossRef]
- Morita, S.; Hiramatsu, M.; Sugishita, M.; Gyawali, B.; Shibata, T.; Shimokata, T.; Urakawa, H.; Mitsuma, A.; Moritani, S.; Kubota, T.; et al. Pazopanib monotherapy in a patient with a malignant granular cell tumor originating from the right orbit: A case report. Oncol. Lett. 2015, 10, 972–974. [Google Scholar] [CrossRef]
- van Wyk, A.C.; van Zyl, H.; Rigby, J. Colonic perineurioma (benign fibroblastic polyp): Case report and review of the literature. Diagn. Pathol. 2018, 13, 16. [Google Scholar] [CrossRef]
- Tsuchiya, T.; Iwaya, Y.; Iwaya, M.; Okamura, T.; Nagaya, T.; Umemura, T. A Case of Multiple Perineuriomas in the Colon with Underlying Neurofibromatosis Type I. ACG Case Rep. J. 2021, 8, e00665. [Google Scholar] [CrossRef]
- Agaimy, A.; Stoehr, R.; Vieth, M.; Hartmann, A. Benign Serrated Colorectal Fibroblastic Polyps/Intramucosal Perineuriomas Are True Mixed Epithelial-stromal Polyps (Hybrid Hyperplastic Polyp/Mucosal Perineurioma) With Frequent BRAF Mutations. Am. J. Surg. Pathol. 2010, 34, 1663–1671. [Google Scholar] [CrossRef]
- Baiomi, A.; Abbas, H.; Niazi, M.; Remotti, H.; Daniel, M.; Balar, B. Colonic Ganglioneuroma: A Rare Lesion with Extremely Different Presentations and Outcomes in Two Patients. Gastroenterol. Res. 2021, 14, 194–198. [Google Scholar] [CrossRef]
- Mauro, A.; Zenzeri, L.; Esposito, F.; Gaglione, G.; Strisciuglio, C.; Pilozzi, E.; Corleto, V.D.; Ziparo, C.; Di Nardo, G. Isolated intestinal Ganglioneuromatosis: Case report and literature review. Ital. J. Pediatr. 2021, 47, 80. [Google Scholar] [CrossRef]
- Lu, C.; Qiu, Y.; Lu, X.; Li, G.; Bu, H. Synchronous diffuse ganglioneuromatosis and multiple schwannomas of the colon: A case report and literature review. Exp. Ther. Med. 2015, 9, 733–736. [Google Scholar] [CrossRef][Green Version]
- Pistorius, S.; Klink, B.; Pablik, J.; Rump, A.; Aust, D.; Garzarolli, M.; Schröck, E.; Schackert, H.K. An unusual case of Cowden syndrome associated with ganglioneuromatous polyposis. Hered. Cancer Clin. Pract. 2016, 14, 11. [Google Scholar] [CrossRef][Green Version]
- Bonetti, F.M.; Pea, M.M.; Martignoni, G.M.; Zamboni, G.M. PEC and Sugar. Am. J. Surg. Pathol. 1992, 16, 307–308. [Google Scholar] [CrossRef]
- Thway, K.; Fisher, C. PEComa: Morphology and genetics of a complex tumor family. Ann. Diagn. Pathol. 2015, 19, 359–368. [Google Scholar] [CrossRef]
- Pan, C.; Chung, M.; Ng, K.; Liu, C.; Wang, J.; Chai, C.; Huang, S.; Chen, P.; Ho, D. Constant allelic alteration on chromosome 16p (TSC2 gene) in perivascular epithelioid cell tumour (PEComa): Genetic evidence for the relationship of PEComa with angiomyolipoma. J. Pathol. 2008, 214, 387–393. [Google Scholar] [CrossRef]
- Agaram, N.P.; Sung, Y.-S.; Zhang, L.; Chen, C.-L.; Chen, H.-W.; Singer, S.; Dickson, M.A.; Berger, M.F.; Antonescu, C.R. Dichotomy of Genetic Abnormalities in PEComas With Therapeutic Implications. Am. J. Surg. Pathol. 2015, 39, 813–825. [Google Scholar] [CrossRef]
- Doyle, L.A.; Hornick, J.L.; Fletcher, C.D. PEComa of the Gastrointestinal tract: Clinicopathologic study of 35 cases with evaluation of prognostic parameters. Am. J. Surg. Pathol. 2013, 37, 1769–1782. [Google Scholar] [CrossRef]
- Park, S.J.; Han, D.K.; Baek, H.J.; Chung, S.Y.; Nam, J.H.; Kook, H.; Hwang, T.J. Perivascular epithelioid cell tumor (PEComa) of the ascending colon: The implication of IFN-α2b treatment. Korean J. Pediatr. 2010, 53, 975–978. [Google Scholar] [CrossRef]
- Baek, J.-H.; Chung, M.G.; Jung, D.H.; Oh, J.H. Perivascular Epithelioid Cell Tumor (Pecoma) in the Transverse Colon of an Adolescent: A Case Report. Tumori J. 2007, 93, 106–108. [Google Scholar] [CrossRef]
- Pereira, K.; Inamdar, A.A.; Zaveri, A.; Teitelbaum, J.E.; Shertz, W.; Belitsis, K. A Rare Case of a Translocation-Associated Perivascular Epithelioid Cell Neoplasm (PEComa). Case Rep. Pediatr. 2022, 2022, 7519456. [Google Scholar] [CrossRef]
- Bleeker, J.S.; Quevedo, J.F.; Folpe, A.L. “Malignant” Perivascular Epithelioid Cell Neoplasm: Risk Stratification and Treatment Strategies. Sarcoma 2012, 2012, 541626. [Google Scholar] [CrossRef]
- Argani, P.; Aulmann, S.; Illei, P.B.; Netto, G.J.; Ro, J.; Cho, H.-Y.; Dogan, S.; Ladanyi, M.; Martignoni, G.; Goldblum, J.R.; et al. A Distinctive Subset of PEComas Harbors TFE3 Gene Fusions. Am. J. Surg. Pathol. 2010, 34, 1395–1406. [Google Scholar] [CrossRef]
- Italiano, A.; Delcambre, C.; Hostein, I.; Cazeau, A.L.; Marty, M.; Avril, A.; Coindre, J.-M.; Bui, B. Treatment with the mTOR inhibitor temsirolimus in patients with malignant PEComa. Ann. Oncol. 2010, 21, 1135–1137. [Google Scholar] [CrossRef]
- Wagner, A.J.; Malinowska-Kolodziej, I.; Morgan, J.A.; Qin, W.; Fletcher, C.D.; Vena, N.; Ligon, A.H.; Antonescu, C.R.; Ramaiya, N.H.; Demetri, G.D.; et al. Clinical Activity of mTOR Inhibition with Sirolimus in Malignant Perivascular Epithelioid Cell Tumors: Targeting the Pathogenic Activation of mTORC1 in Tumors. J. Clin. Oncol. 2010, 28, 835–840. [Google Scholar] [CrossRef]
- French, C.A. NUT Carcinoma: Clinicopathologic features, pathogenesis, and treatment. Pathol. Int. 2018, 68, 583–595. [Google Scholar] [CrossRef]
- Dickson, B.C.; Sung, Y.-S.M.; Rosenblum, M.K.; Reuter, V.E.; Harb, M.; Wunder, J.S.; Swanson, D.B.; Antonescu, C.R. NUTM1 Gene Fusions Characterize a Subset of Undifferentiated Soft Tissue and Visceral Tumors. Am. J. Surg. Pathol. 2018, 42, 636–645. [Google Scholar] [CrossRef]
- Sekine, S.; Kiyono, T.; Ryo, E.; Ogawa, R.; Wakai, S.; Ichikawa, H.; Suzuki, K.; Arai, S.; Tsuta, K.; Ishida, M.; et al. Recurrent YAP1-MAML2 and YAP1-NUTM1 fusions in poroma and porocarcinoma. J. Clin. Investig. 2019, 129, 3827–3832. [Google Scholar] [CrossRef]
- Le Loarer, F.; Pissaloux, D.; Watson, S.; Godfraind, C.; Galmiche-Rolland, L.; Silva, K.; Mayeur, L.; Italiano, A.; Michot, A.; Pierron, G.; et al. Clinicopathologic Features of CIC-NUTM1 Sarcomas, a New Molecular Variant of the Family of CIC-Fused Sarcomas. Am. J. Surg. Pathol. 2019, 43, 268–276. [Google Scholar] [CrossRef]
- Van Treeck, B.J.; Thangaiah, J.J.; Torres-Mora, J.; Stevens, T.M.; Rothermundt, C.; Fassan, M.; Loupakis, F.; Diebold, J.; Hornick, J.L.; Halling, K.C.; et al. NUTM1-rearranged colorectal sarcoma: A clinicopathologically and genetically distinctive malignant neoplasm with a poor prognosis. Mod. Pathol. 2021, 34, 1547–1557. [Google Scholar] [CrossRef]
- Folpe, A.L. ‘I Can’t Keep Up!’: An update on advances in soft tissue pathology occurring after the publication of the 2020 World Health Organization classification of soft tissue and bone tumours. Histopathology 2021, 80, 54–75. [Google Scholar] [CrossRef]
- Yoshida, A. NUT carcinoma and thoracic SMARCA4-deficient undifferentiated tumour: Facts and controversies. Histopathology 2023, 84, 86–101. [Google Scholar] [CrossRef]
- Luo, W.; Stevens, T.M.; Stafford, P.; Miettinen, M.; Gatalica, Z.; Vranic, S. NUTM1-Rearranged Neoplasms—A Heterogeneous Group of Primitive Tumors with Expanding Spectrum of Histology and Molecular Alterations—An Updated Review. Curr. Oncol. 2021, 28, 4485–4503. [Google Scholar] [CrossRef]
- Atiq, M.A.; Davis, J.L.; Hornick, J.L.; Dickson, B.C.; Fletcher, C.D.M.; Fletcher, J.A.; Folpe, A.L.; Mariño-Enríquez, A. Mesenchymal tumors of the gastrointestinal tract with NTRK rearrangements: A clinicopathological, immunophenotypic, and molecular study of eight cases, emphasizing their distinction from gastrointestinal stromal tumor (GIST). Mod. Pathol. 2020, 34, 95–103. [Google Scholar] [CrossRef]
- Brenca, M.; Rossi, S.; Polano, M.; Gasparotto, D.; Zanatta, L.; Racanelli, D.; Valori, L.; Lamon, S.; Tos, A.P.D.; Maestro, R. Transcriptome sequencing identifies ETV6–NTRK3 as a gene fusion involved in GIST. J. Pathol. 2015, 238, 543–549. [Google Scholar] [CrossRef]
- Shi, E.; Chmielecki, J.; Tang, C.-M.; Wang, K.; Heinrich, M.C.; Kang, G.; Corless, C.L.; Hong, D.; Fero, K.E.; Murphy, J.D.; et al. FGFR1 and NTRK3 actionable alterations in “Wild-Type” gastrointestinal stromal tumors. J. Transl. Med. 2016, 14, 339. [Google Scholar] [CrossRef]
- Sinniah, R.P.; Roche, E.; Cameron, D. GI Synovial Sarcomas. Clin. Transl. Gastroenterol. 2012, 3, e11. [Google Scholar] [CrossRef]
- Romeo, S.; Rossi, S.; Marín, M.A.; Canal, F.; Sbaraglia, M.; Laurino, L.; Mazzoleni, G.; Montesco, M.C.; Valori, L.; Dell’orto, M.C.; et al. Primary Synovial Sarcoma (SS) of the digestive system: A molecular and clinicopathological study of fifteen cases. Clin. Sarcoma Res. 2015, 5, 7. [Google Scholar] [CrossRef]
- Requena, D.O.; Longacre, T.A.; Rosenberg, A.E.; Torres, J.M.V.; Yanchenko, N.; Garcia-Buitrago, M.T.; Voltaggio, L.; Montgomery, E.A. Synovial Sarcoma of the Gastrointestinal Tract. Mod. Pathol. 2024, 37, 100383. [Google Scholar] [CrossRef]
































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
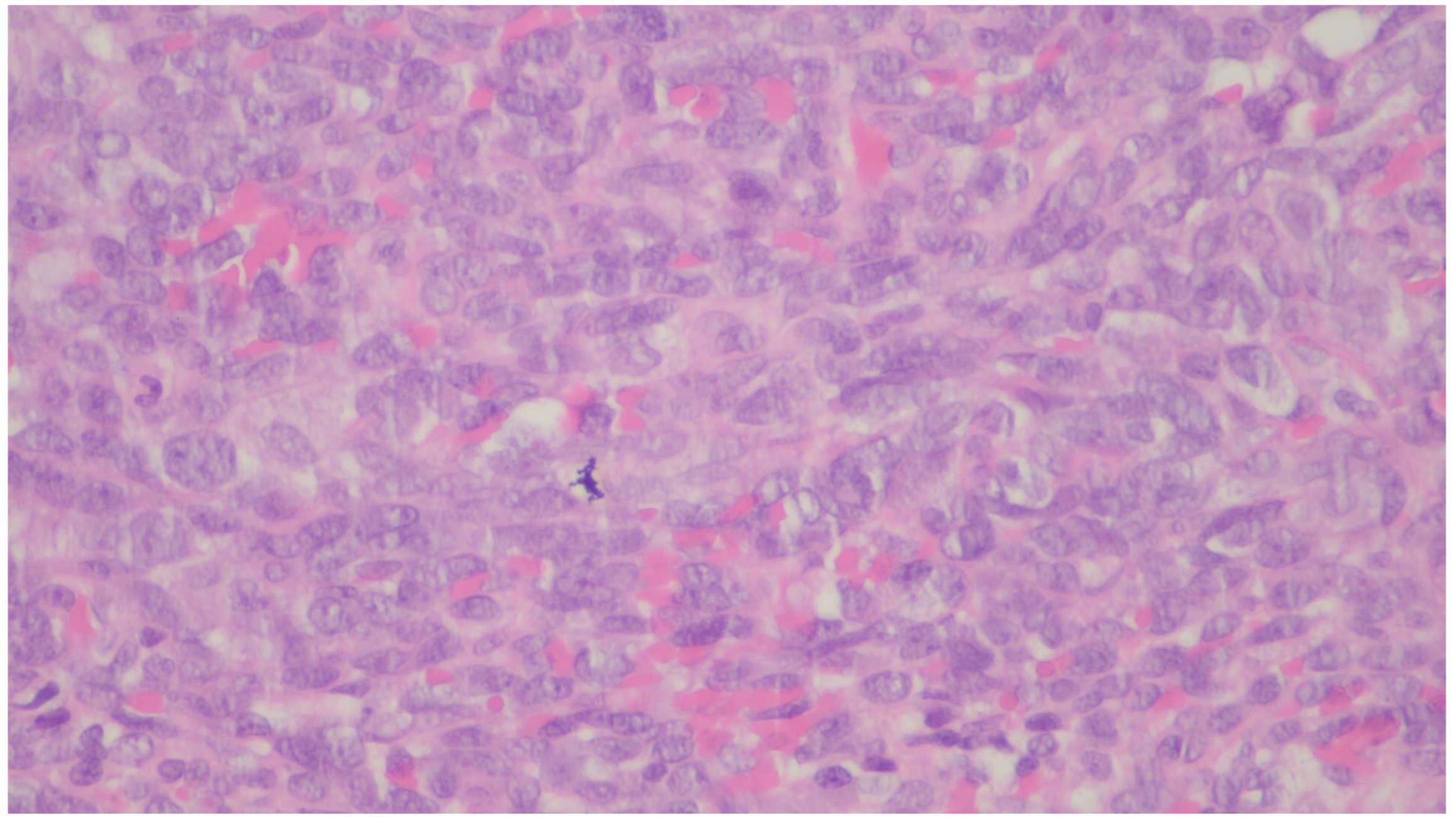{kind=link}
{kind=link}
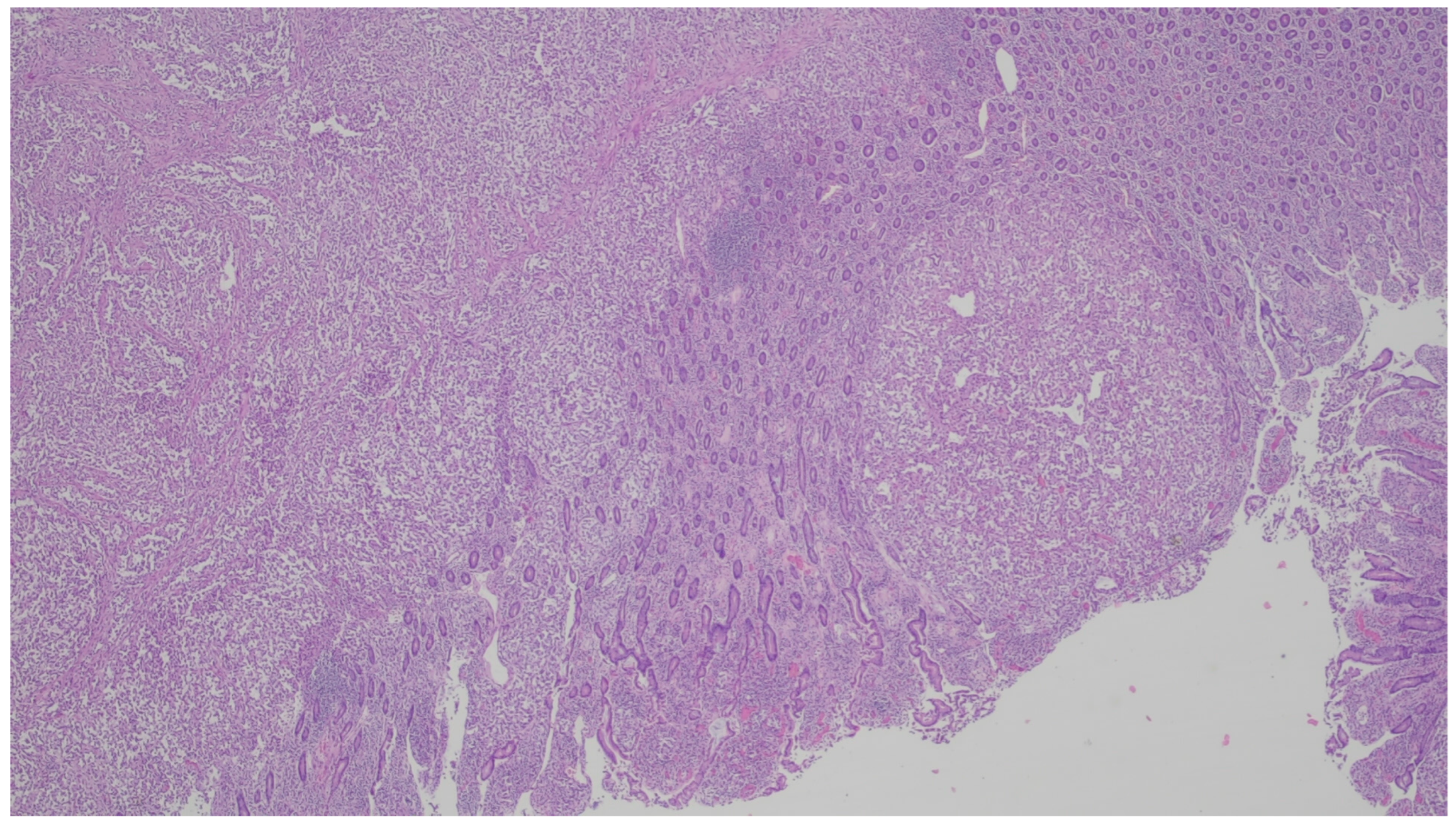{kind=link}
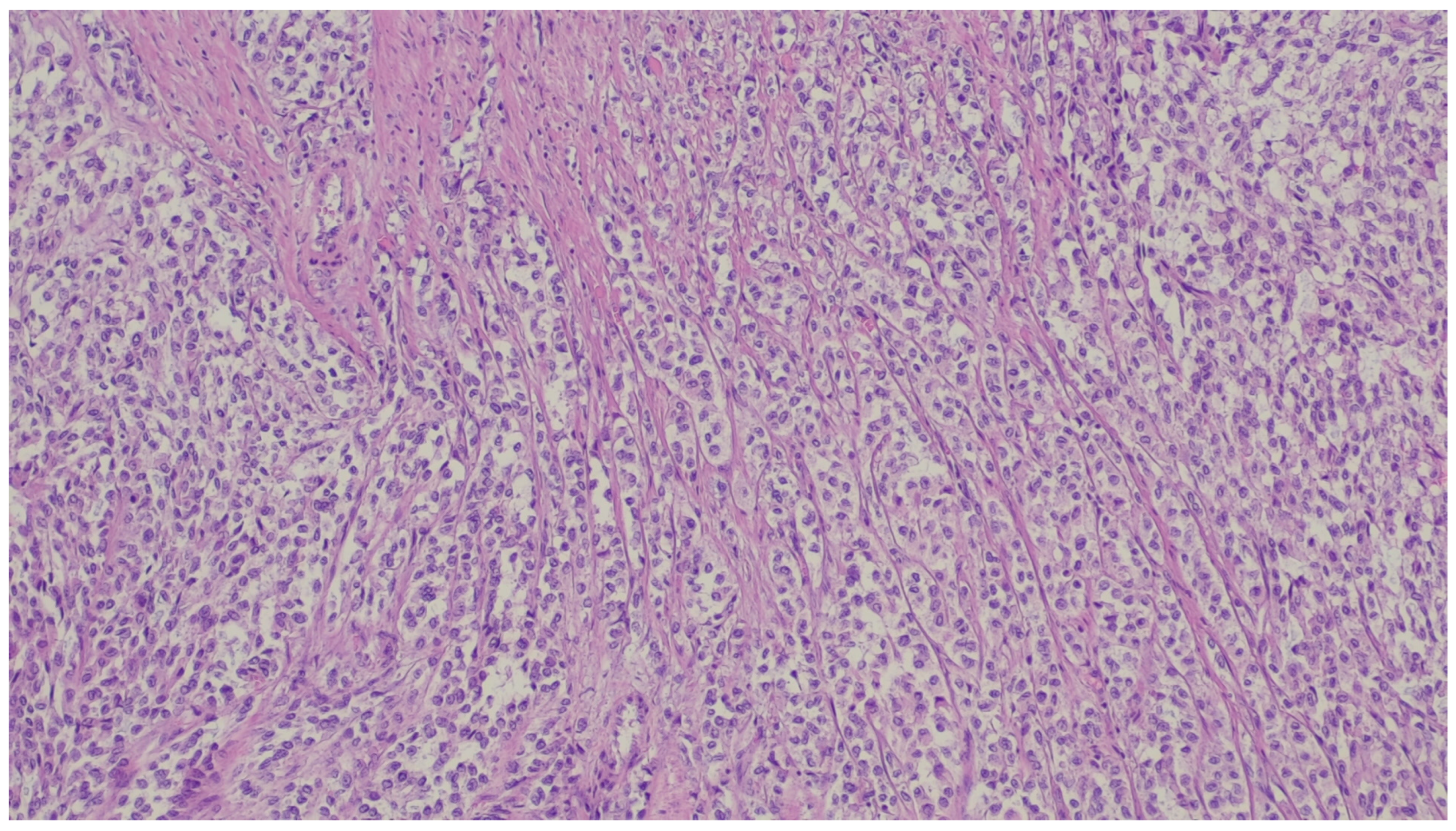{kind=link}
| Entity | Demographics | Site | Clinical Presentation | Histology | Immunohistochemistry |
|---|---|---|---|---|---|
| Inflammatory myofibroblastic tumor | Children and young adults | Colon and small intestine | Variable, from abdominal pain to obstruction | Uniform, plump spindle cells with pale cytoplasm organized into loose fascicles; colagenous or myxoid stroma with inflammation, mainly lymhocytes and plasma cells | α-SMA, ALK positive |
| Fibromatosis | 30 and 40 years | Mesentery of the small bowel | Abdominal pain | Long fascicles of spindle cells with ovoid nuclei without atypia or necrosis | Β-catenin |
| Solitary fibrous tumor | Adults, with a wide range of ages | Anywhere in the GI tract | Symptoms are unspecific, usually associated with compression and, sometimes, hemorrhage | Ovoid/spindle cells arranged in poorly defined fascicles, supported by thin-walled vessels, dilated in a staghorn pattern | STAT6 |
| Lipoma | Adults | Anywhere in the GI tract | Asymptomatic; some cases in the ileocecal valve with obstruction | Mature adipose tissue | S100, Rb1 is retaned, CD34 is negative |
| Inflammatory fibroid polyp | Adults | Stomach | Incidental findings; some cases with abdominal pain, hemorrhage, or obstruction | Proliferation of spindle and stellate cells in haphazard distribution in the submucosa | CD34; PDGFRA |
| Plexiform fibromyxoma | Wide range of age | Antrum and pyloric region | Asymptomatic; some cases with ulceration | Bland spindle cells, supported by a myxoid/fibromyxoid/collagenous stroma and with a rich vascular network of thin-walled vessels in a multinodular pattern | SMA and, occasionally desmin positive. ALK, CD34, keratins, KIT, and DOG1 are negative |
| Leiomyoma | Wide range of age | Esophagus, colon, and rectum | Asympromatic | Well-differentiated fascicles of bland, fusiform cells with elongated nuclei with tapered ends with eosinophilic cytoplasm | α-SMA, desmin, caldesmon |
| Leiomyosarcoma | Very rare in the GI tract | Small intestine | Abdominal pain, obstruction | Of fascicles of spindle cells with atypia, centrally placed cigar-shaped nuclei, and eosinophilic fibrillary cytoplasm with high mitotic counts | α-sma, desmin, caldesmon |
| Hemangioma | Uncommon | Anywhere in the GI tract | Occult or acute GI bleeding | Large and dilated vessels with a thin wall | CD31, CD34 and ERG |
| Lymphangiomas | Children and young adults | Small intestine | Anemia or acute abdomen | Thin-walled, cystically dilated lymphatic spaces lined by endothelial cells with lymph fluid | D2-40, CD31, CD34 |
| Kaposi sarcoma | 25 to 72 years (median 34 years), with a male predominance | Stomach, duodenum, and esophagus | Frequently asymptomatic, when symptomatic, the most common manifestations are diarrhea, abdominal pain, and gi bleeding | Proliferation of blood vessels and spindle cells with minimal atypia organized in vague fascicles, slit-like spaces with extravasated red blood cells with hemosiderin deposition, associated with lymphocytic and plasma cell infiltrate | CD31, CD34, ERG, D2-40, HHV-8 |
| Angiosarcoma | Male predominance and more commonly affect adults with a peak in the seventh decade | Small and large intestine | GI bleeding, abdominal pain, mass or ascites | A wide variation in morphology, from a well-formed network of vessels to sheets of epithelioid and spindle atypical cells without a clear formation of vessels, with necrosis | CD31, CD34, ERG, D2-40 |
| Glomus tumor | Equal predominance, with a paper showing female predominance | Stomach | Upper GI bleeding | Cells with a round and uniform shape and sharply defined borders organized in sheets and nests surrounding vascular spaces | SMA, calponin, TLE1, caldesmon. Variable desmin. S100 and c-kit are absent. |
| Schwannoma | Marked female predominance | Stomach | Mass-like symptoms | Spindle cells distributed in hypo and hypercellular areas; older lesions may show cystic changes, hemorrhage, and hyaline thick- walled blood vessels | S100, SOX-10 |
| Granular cell tumor | Sixth decade of life | Esophagus | Asymptomatic | Monotonous and epithelioid cells with abundant eosinophilic and granular cytoplasm | S100 and calretinin |
| Perineurioma | Female predominance and appear in middle-aged adults | Rectosigmoid colon | Asymptomatic, GI bleeding | Show expansion of the lamina propria by uniform and bland spindle cells, entrapping colonic cripts | EMA, claudin-1, and GLUT1 |
| Ganglioneuroma | Wide age range | Large intestine, with a predilection for the left side and rectum | They present in colorectal cancer screening as small polypoid lesions (inferior to 2 cm) and can be solitary, multiple, or diffuse; however symptms are not specific | Expansion of the lamina propria by ganglion and schwan cells, accompanied by eosinophils, sometimes with cystic colonic glands | S100, SOX10 |
| PEComa | Rare | Colon and small intestine | Symptoms are not specific | Nested, trabecular, and alveolar architecture, surrounded by a delicate vasculature; the cells are usually epithelioid with round to oval nuclei with a granular eosinophilic cytoplasm, but a minority of spindle cells can be seen | α-SMA, desmin, HMB-45 Melan-A, MiTF |
| NUTM1-rearranged colorectal sarcoma | Teens and young adults; more common in females | Colorectal | Symptoms are not specific | Three distinctive patterns: intersecting fascicles with uniform cells pattern, a hyalinized/nested pattern, and an epithelioid and rhabdoid pattern | nuclear expression of NUT, variable keratin expression and expression of CD117 and DOG1, retained expression of SMARCB1 and SMARCA4 |
| Mesenchymal tumors associated with NTRK rearrangements | - | - | Symptoms are not specific | Spindle cells tumors | CD34, S100, NTRK |
| Synovial sarcoma | Middle-aged to older adults | Stomach | Polypoid or an ulcerated lesion | Monophasic with uniform spindle cells with scarce matrix or biphasic with glandular elements | SS18/SSX positivity. Focal positivity for keratins and EMA in monophasic lesions and strong staining in the biphasic version |
| Gastrointestinal clear cell sarcoma/malignant gastrointestinal neuroectodermal tumor | Young adults | More common in the small intestine, stomach, and colon | Polypoid or mural in appearance, with ulceration resembling carcinomas | Nested or pseudopapillary pattern and sometimes a spindle phenotype | S100, SOX-10, Melan-A, HMB45, MiTF |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gama, J.M.; Oliveira, R.C. Mesenchymal Tumors of the Gastrointestinal Tract—Beyond GIST—A Review. Gastrointest. Disord. 2024, 6, 257-291. https://doi.org/10.3390/gidisord6010019
Gama JM, Oliveira RC. Mesenchymal Tumors of the Gastrointestinal Tract—Beyond GIST—A Review. Gastrointestinal Disorders. 2024; 6(1):257-291. https://doi.org/10.3390/gidisord6010019
Chicago/Turabian StyleGama, João Martins, and Rui Caetano Oliveira. 2024. "Mesenchymal Tumors of the Gastrointestinal Tract—Beyond GIST—A Review" Gastrointestinal Disorders 6, no. 1: 257-291. https://doi.org/10.3390/gidisord6010019
APA StyleGama, J. M., & Oliveira, R. C. (2024). Mesenchymal Tumors of the Gastrointestinal Tract—Beyond GIST—A Review. Gastrointestinal Disorders, 6(1), 257-291. https://doi.org/10.3390/gidisord6010019