Mismatch Repair System Genomic Scars in Gastroesophageal Cancers: Biology and Clinical Testing





Abstract
1. Introduction
2. Clinicopathologic and Molecular Features of MMR-Deficient Gastroesophageal Cancers
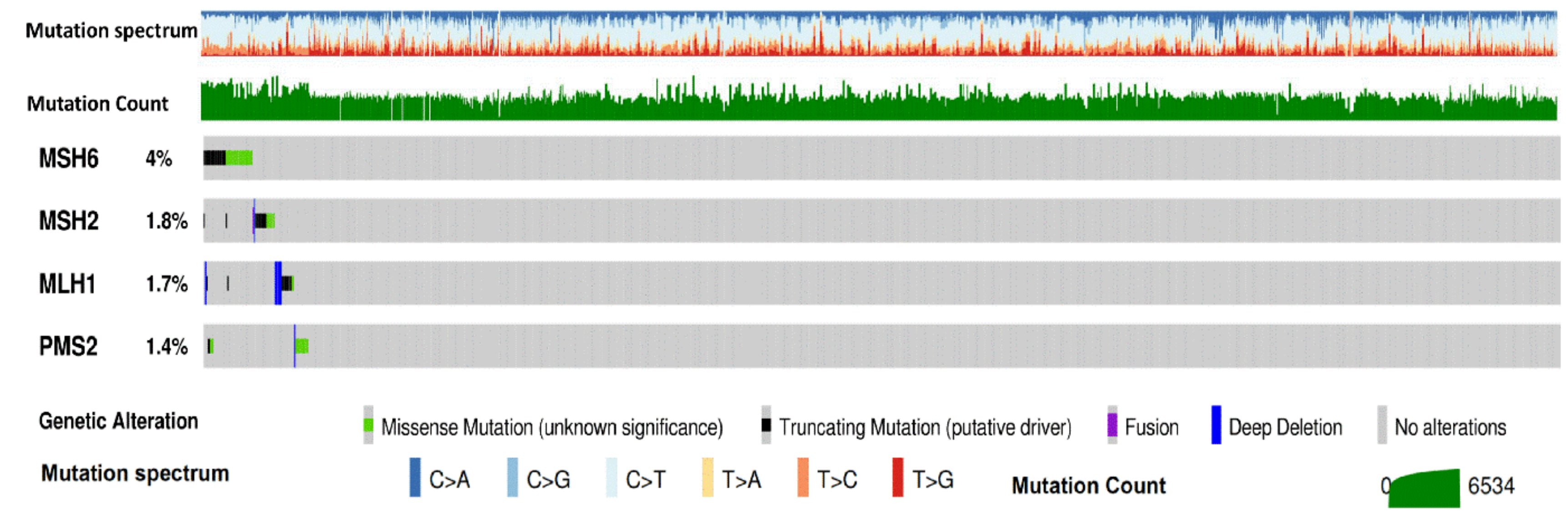
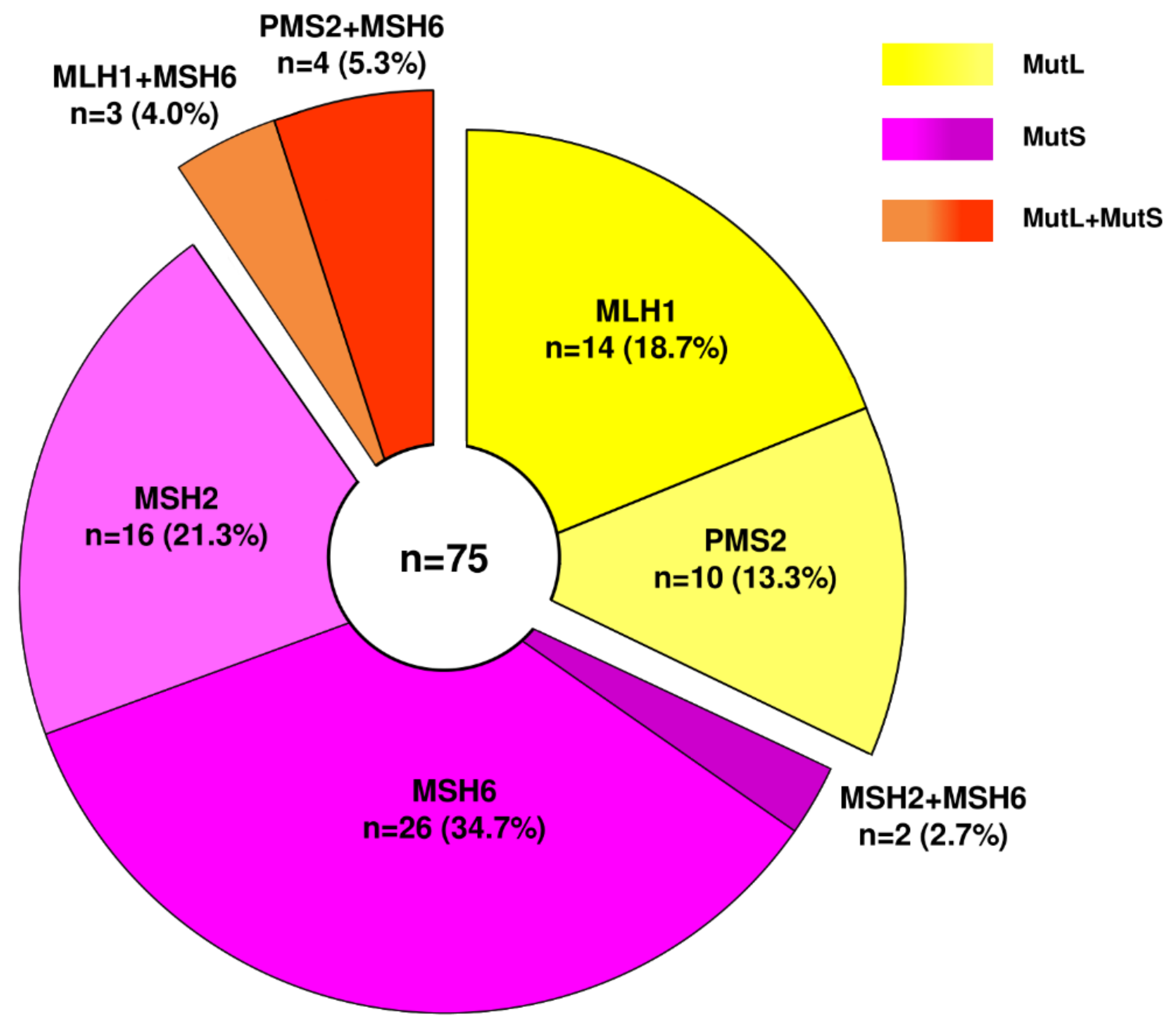
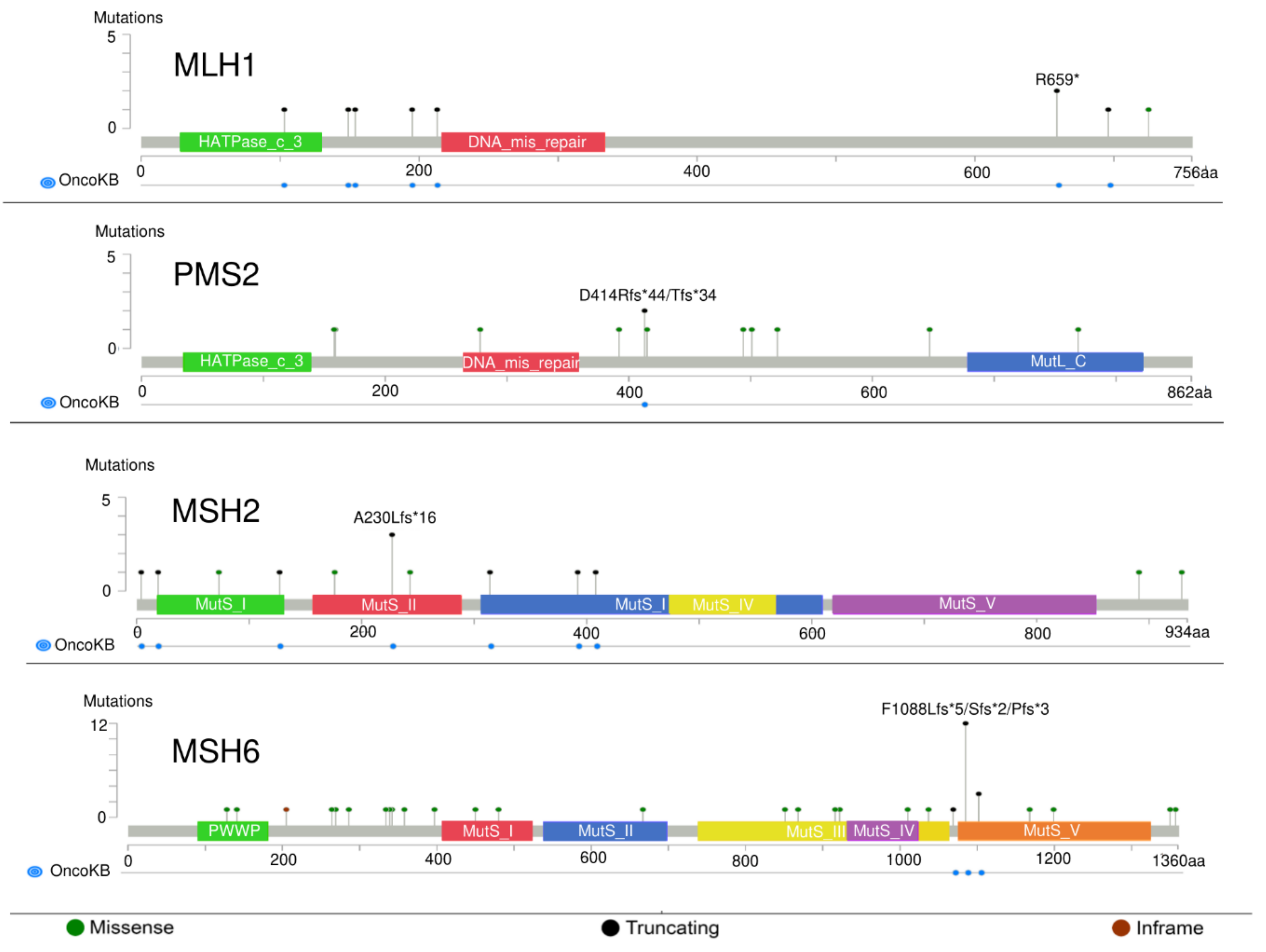
3. Types and Frequency of MMR Alterations in Gastroesophageal Cancers: Analysis of Publicly Available Genomic Datasets
4. MMR Clinical Testing in GEC: Rationale, Currently Available Strategies, Unaddressed Issues
5. MMR Status Assessment to Select GEC Patients for Immunotherapy
6. Future Prospects for MMR Testing and Precision Cancer Medicine
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| MMR | Mismatch repair |
| dMMR | MMR deficiency |
| MSI | Microsatellite instability |
| GEC | Gastroesophageal cancers |
References
- Corti, C.; Sajjadi, E.; Fusco, N. Determination of Mismatch Repair Status in Human Cancer and Its Clinical Significance: Does One Size Fit All? Adv. Anat. Pathol. 2019, 26, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Bradford, K.C.; Wilkins, H.; Hao, P.; Li, Z.M.; Wang, B.; Burke, D.; Wu, D.; Smith, A.E.; Spaller, L.; Du, C.; et al. Dynamic human MutSα–MutLα complexes compact mismatched DNA. Proc. Natl. Acad. Sci. USA 2020, 117, 16302–16312. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Luo, H.; Huang, L.; Zhu, X. Microsatellite instability: a review of what the oncologist should know. Cancer Cell Int. 2020, 20, 16. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, G.M. DNA mismatch repair in the context of chromatin. Cell Biosci. 2020, 10, 10. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Yuza, K.; Nagahashi, M.; Watanabe, S.; Takabe, K.; Wakai, T. Hypermutation and microsatellite instability in gastrointestinal cancers. Oncotarget 2017, 8, 112103–112115. [Google Scholar] [CrossRef]
- Kim, S.M.; An, J.Y.; Byeon, S.J.; Lee, J.; Kim, K.M.; Choi, M.G.; Lee, J.H.; Sohn, T.S.; Bae, J.M.; Kim, S. Prognostic value of mismatch repair deficiency in patients with advanced gastric cancer, treated by surgery and adjuvant 5-fluorouracil and leucovorin chemoradiotherapy. Eur. J. Surg. Oncol. 2020, 46, 189–194. [Google Scholar] [CrossRef]
- Smyth, E.C.; Wotherspoon, A.; Peckitt, C.; Gonzalez, D.; Hulkki-Wilson, S.; Eltahir, Z.; Fassan, M.; Rugge, M.; Valeri, N.; Okines, A.; et al. Mismatch Repair Deficiency, Microsatellite Instability, and Survival: An Exploratory Analysis of the Medical Research Council Adjuvant Gastric Infusional Chemotherapy (MAGIC) Trial. JAMA Oncol. 2017, 3, 1197–1203. [Google Scholar] [CrossRef]
- Sundar, R.; Smyth, E.C.; Peng, S.; Yeong, J.P.S.; Tan, P. Predictive Biomarkers of Immune Checkpoint Inhibition in Gastroesophageal Cancers. Front. Oncol. 2020, 10, 763. [Google Scholar] [CrossRef]
- Brar, G.; Shah, M.A. The role of pembrolizumab in the treatment of PD-L1 expressing gastric and gastroesophageal junction adenocarcinoma. Therap. Adv. Gastroenterol. 2019, 12, 1756284819869767. [Google Scholar] [CrossRef]
- An, J.Y.; Choi, Y.Y.; Lee, J.; Hyung, W.J.; Kim, K.M.; Noh, S.H.; Choi, M.G.; Cheong, J.H. A Multi-cohort Study of the Prognostic Significance of Microsatellite Instability or Mismatch Repair Status after Recurrence of Resectable Gastric Cancer. Cancer Res. Treat. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Wang, L.; Ni, S.; Tan, C.; Cai, X.; Huang, D.; Sheng, W. Clinicopathological features and prognostic value of mismatch repair protein deficiency in gastric cancer. Int. J. Clin. Exp. Pathol. 2018, 11, 2579–2587. [Google Scholar] [PubMed]
- Bass, A.J.; Thorsson, V.; Shmulevich, I.; Reynolds, S.M.; Miller, M.; Bernard, B.; Hinoue, T.; Laird, P.W.; Curtis, C.; Shen, H.; et al. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef]
- Puhr, H.C.; Ilhan-Mutlu, A. Molecular profiling in gastroesophageal cancer—Clinical routine and future perspective. Memo Mag. Eur. Med. Oncol. 2019. [Google Scholar] [CrossRef]
- Morales-Sanchez, A.; Fuentes-Panana, E.M. Epstein-Barr Virus-associated Gastric Cancer and Potential Mechanisms of Oncogenesis. Curr. Cancer Drug Targets 2017, 17, 534–554. [Google Scholar] [CrossRef]
- Fusco, N.; Bosari, S. HER2 aberrations and heterogeneity in cancers of the digestive system: Implications for pathologists and gastroenterologists. World J. Gastroenterol. 2016, 22, 7926–7937. [Google Scholar] [CrossRef]
- Patel, T.H.; Cecchini, M. Targeted Therapies in Advanced Gastric Cancer. Curr. Treat Options Oncol. 2020, 21, 70. [Google Scholar] [CrossRef]
- Park, Y.; Koh, J.; Kwak, Y.; Ahn, S.H.; Park, D.J.; Kim, H.H.; Kim, W.H.; Lee, H.S. Clinicopathologic significance of human leukocyte antigen class I expression in patients with stage II and III gastric cancer. Cancer Immunol. Immunother. 2019, 68, 1779–1790. [Google Scholar] [CrossRef]
- Torrejon, D.Y.; Abril-Rodriguez, G.; Champhekar, A.S.; Tsoi, J.; Campbell, K.M.; Kalbasi, A.; Parisi, G.; Zaretsky, J.M.; Garcia-Diaz, A.; Puig-Saus, C.; et al. Overcoming Genetically Based Resistance Mechanisms to PD-1 Blockade. Cancer Discov. 2020, 10, 1140–1157. [Google Scholar] [CrossRef]
- Vrána, D.; Matzenauer, M.; Neoral, Č.; Aujeský, R.; Vrba, R.; Melichar, B.; Rušarová, N.; Bartoušková, M.; Jankowski, J. From Tumor Immunology to Immunotherapy in Gastric and Esophageal Cancer. Int. J. Mol. Sci. 2018, 20, 13. [Google Scholar] [CrossRef]
- Coutzac, C.; Pernot, S.; Chaput, N.; Zaanan, A. Immunotherapy in advanced gastric cancer, is it the future? Crit. Rev. Oncol. Hematol. 2019, 133, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Serra, O.; Smyth, E.C.; Lordick, F. Progress and challenges in gastroesophageal cancer. Curr. Probl. Cancer 2020, 100590. [Google Scholar] [CrossRef]
- Pagni, F.; Guerini-Rocco, E.; Schultheis, A.M.; Grazia, G.; Rijavec, E.; Ghidini, M.; Lopez, G.; Venetis, K.; Croci, G.A.; Malapelle, U.; et al. Targeting Immune-Related Biological Processes in Solid Tumors: We do Need Biomarkers. Int. J. Mol. Sci. 2019, 20, 5452. [Google Scholar] [CrossRef]
- von Loga, K.; Woolston, A.; Punta, M.; Barber, L.J.; Griffiths, B.; Semiannikova, M.; Spain, G.; Challoner, B.; Fenwick, K.; Simon, R.; et al. Extreme intratumour heterogeneity and driver evolution in mismatch repair deficient gastro-oesophageal cancer. Nat. Commun. 2020, 11, 139. [Google Scholar] [CrossRef] [PubMed]
- Cohen, R.; Buhard, O.; Cervera, P.; Hain, E.; Dumont, S.; Bardier, A.; Bachet, J.B.; Gornet, J.M.; Lopez-Trabada, D.; Kaci, R.; et al. Clinical and molecular characterisation of hereditary and sporadic metastatic colorectal cancers harbouring microsatellite instability/DNA mismatch repair deficiency. Eur. J. Cancer 2017, 86, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Eso, Y.; Shimizu, T.; Takeda, H.; Takai, A.; Marusawa, H. Microsatellite instability and immune checkpoint inhibitors: toward precision medicine against gastrointestinal and hepatobiliary cancers. J. Gastroenterol. 2020, 55, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Shi, Y.; Li, A. Association Between hMLH1 Promoter Methylation and Risk of Gastric Cancer: A Meta-Analysis. Front. Physiol. 2018, 9, 368. [Google Scholar] [CrossRef] [PubMed]
- Polom, K.; Marano, L.; Marrelli, D.; De Luca, R.; Roviello, G.; Savelli, V.; Tan, P.; Roviello, F. Meta-analysis of microsatellite instability in relation to clinicopathological characteristics and overall survival in gastric cancer. Br. J. Surg. 2018, 105, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; An, J.Y.; Noh, S.H.; Shin, S.K.; Lee, Y.C. High microsatellite instability predicts good prognosis in intestinal-type gastric cancers. J. Gastroenterol. Hepatol. 2011, 26, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Dulak, A.M.; Stojanov, P.; Peng, S.; Lawrence, M.S.; Fox, C.; Stewart, C.; Bandla, S.; Imamura, Y.; Schumacher, S.E.; Shefler, E.; et al. Exome and whole-genome sequencing of esophageal adenocarcinoma identifies recurrent driver events and mutational complexity. Nat. Genet. 2013, 45, 478–486. [Google Scholar] [CrossRef]
- Broad, I.; Brown, U. Integrated genomic characterization of oesophageal carcinoma. Nature 2017, 541, 169–175. [Google Scholar] [CrossRef]
- Chen, K.; Yang, D.; Li, X.; Sun, B.; Song, F.; Cao, W.; Brat, D.J.; Gao, Z.; Li, H.; Liang, H.; et al. Mutational landscape of gastric adenocarcinoma in Chinese: implications for prognosis and therapy. Proc. Natl. Acad. Sci. USA 2015, 112, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yuen, S.T.; Xu, J.; Lee, S.P.; Yan, H.H.; Shi, S.T.; Siu, H.C.; Deng, S.; Chu, K.M.; Law, S.; et al. Whole-genome sequencing and comprehensive molecular profiling identify new driver mutations in gastric cancer. Nat. Genet. 2014, 46, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Kakiuchi, M.; Nishizawa, T.; Ueda, H.; Gotoh, K.; Tanaka, A.; Hayashi, A.; Yamamoto, S.; Tatsuno, K.; Katoh, H.; Watanabe, Y.; et al. Recurrent gain-of-function mutations of RHOA in diffuse-type gastric carcinoma. Nat. Genet. 2014, 46, 583–587. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Kan, J.; Yuen, S.T.; Shi, S.T.; Chu, K.M.; Law, S.; Chan, T.L.; Kan, Z.; Chan, A.S.; Tsui, W.Y.; et al. Exome sequencing identifies frequent mutation of ARID1A in molecular subtypes of gastric cancer. Nat. Genet. 2011, 43, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Broad Institute Firehose. Stomach Adenocarcinoma (STAD); Broad Institute Firehose: Cambridge, MA, USA, 2020. [Google Scholar]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors From 33 Types of Cancer. Cell 2018, 173. [Google Scholar] [CrossRef]
- van Velzen, M.J.M.; Derks, S.; van Grieken, N.C.T.; Haj Mohammad, N.; van Laarhoven, H.W.M. MSI as a predictive factor for treatment outcome of gastroesophageal adenocarcinoma. Cancer Treat Rev. 2020, 86, 102024. [Google Scholar] [CrossRef]
- Tessier-Cloutier, B.; Cai, E.; Schaeffer, D.F. Off-label use of common predictive biomarkers in gastrointestinal malignancies: a critical appraisal. Diagn. Pathol. 2019, 14, 62. [Google Scholar] [CrossRef]
- Cho, J.; Kang, S.Y.; Kim, K.M. MMR protein immunohistochemistry and microsatellite instability in gastric cancers. Pathology 2019, 51, 110–113. [Google Scholar] [CrossRef]
- McCarthy, A.J.; Capo-Chichi, J.M.; Spence, T.; Grenier, S.; Stockley, T.; Kamel-Reid, S.; Serra, S.; Sabatini, P.; Chetty, R. Heterogenous loss of mismatch repair (MMR) protein expression: a challenge for immunohistochemical interpretation and microsatellite instability (MSI) evaluation. J. Pathol. Clin. Res. 2019, 5, 115–129. [Google Scholar] [CrossRef]
- Venetis, K.; Sajjadi, E.; Haricharan, S.; Fusco, N. Mismatch repair testing in breast cancer: the path to tumor-specific immuno-oncology biomarkers. Transl. Cancer Res. 2020, 9, 4060–4064. [Google Scholar] [CrossRef]
- Ratti, M.; Lampis, A.; Hahne, J.C.; Passalacqua, R.; Valeri, N. Microsatellite instability in gastric cancer: molecular bases, clinical perspectives, and new treatment approaches. Cell Mol. Life Sci. 2018, 75, 4151–4162. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Jang, Y.J.; Lee, E.J.; Kim, J.H.; Park, S.S.; Park, S.H.; Kim, C.S.; Mok, Y.J. The significance of mismatch repair genes in gastric cancer. J. Cancer Res. Ther. 2013, 9, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Frankel, W.L. A practical guide to biomarkers for the evaluation of colorectal cancer. Mod. Pathol. 2019, 32, 1–15. [Google Scholar] [CrossRef]
- Boland, C.R.; Thibodeau, S.N.; Hamilton, S.R.; Sidransky, D.; Eshleman, J.R.; Burt, R.W.; Meltzer, S.J.; Rodriguez-Bigas, M.A.; Fodde, R.; Ranzani, G.N.; et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998, 58, 5248–5257. [Google Scholar]
- Buhard, O.; Suraweera, N.; Lectard, A.; Duval, A.; Hamelin, R. Quasimonomorphic Mononucleotide Repeats for High-Level Microsatellite Instability Analysis. Dis. Markers 2004, 20, 159347. [Google Scholar] [CrossRef]
- Murphy, K.M.; Zhang, S.; Geiger, T.; Hafez, M.J.; Bacher, J.; Berg, K.D.; Eshleman, J.R. Comparison of the microsatellite instability analysis system and the Bethesda panel for the determination of microsatellite instability in colorectal cancers. J. Mol. Diagn. 2006, 8, 305–311. [Google Scholar] [CrossRef]
- Bacani, J.; Zwingerman, R.; Di Nicola, N.; Spencer, S.; Wegrynowski, T.; Mitchell, K.; Hay, K.; Redston, M.; Holowaty, E.; Huntsman, D.; et al. Tumor microsatellite instability in early onset gastric cancer. J. Mol. Diagn. 2005, 7, 465–477. [Google Scholar] [CrossRef]
- Fusco, N.; Lopez, G.; Corti, C.; Pesenti, C.; Colapietro, P.; Ercoli, G.; Gaudioso, G.; Faversani, A.; Gambini, D.; Michelotti, A.; et al. Mismatch Repair Protein Loss as a Prognostic and Predictive Biomarker in Breast Cancers Regardless of Microsatellite Instability. JNCI Cancer Spectr. 2018, 2, pky056. [Google Scholar] [CrossRef]
- Lopez, G.; Noale, M.; Corti, C.; Gaudioso, G.; Sajjadi, E.; Venetis, K.; Gambini, D.; Runza, L.; Costanza, J.; Pesenti, C.; et al. PTEN Expression as a Complementary Biomarker for Mismatch Repair Testing in Breast Cancer. Int. J. Mol. Sci. 2020, 21, 1461. [Google Scholar] [CrossRef]
- Lopez, G.; Fusco, N. RE: Mismatch repair protein loss in breast cancer: clinicopathological associations in a large British Columbia cohort. Breast Cancer Res. Treat 2020. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.L.; Chen, J.Y.; Hu, J.; Chen, Q.; Yu, L.X.; Liu, B.R.; Qian, X.P.; Yang, M. Comparison of microsatellite status detection methods in colorectal carcinoma. Int. J. Clin. Exp. Pathol. 2018, 11, 1431–1438. [Google Scholar] [PubMed]
- Battaglin, F.; Naseem, M.; Puccini, A.; Lenz, H.-J. Molecular biomarkers in gastro-esophageal cancer: recent developments, current trends and future directions. Cancer Cell Int. 2018, 18, 99. [Google Scholar] [CrossRef] [PubMed]
- Leite, M.; Corso, G.; Sousa, S.; Milanezi, F.; Afonso, L.P.; Henrique, R.; Soares, J.M.; Castedo, S.; Carneiro, F.; Roviello, F.; et al. MSI phenotype and MMR alterations in familial and sporadic gastric cancer. Int. J. Cancer 2011, 128, 1606–1613. [Google Scholar] [CrossRef]
- Weinberg, B.A.; Xiu, J.; Hwang, J.J.; Shields, A.F.; Salem, M.E.; Marshall, J.L. Immuno-Oncology Biomarkers for Gastric and Gastroesophageal Junction Adenocarcinoma: Why PD-L1 Testing May Not Be Enough. Oncol. 2018, 23, 1171–1177. [Google Scholar] [CrossRef]
- Lin, E.M.; Gong, J.; Klempner, S.J.; Chao, J. Advances in immuno-oncology biomarkers for gastroesophageal cancer: Programmed death ligand 1, microsatellite instability, and beyond. World J. Gastroenterol. 2018, 24, 2686–2697. [Google Scholar] [CrossRef]
- Haag, G.M.; Czink, E.; Ahadova, A.; Schmidt, T.; Sisic, L.; Blank, S.; Heger, U.; Apostolidis, L.; Berger, A.K.; Springfeld, C.; et al. Prognostic significance of microsatellite-instability in gastric and gastroesophageal junction cancer patients undergoing neoadjuvant chemotherapy. Int. J. Cancer 2019, 144, 1697–1703. [Google Scholar] [CrossRef]
- Prasad, V.; Kaestner, V.; Mailankody, S. Cancer Drugs Approved Based on Biomarkers and Not Tumor Type-FDA Approval of Pembrolizumab for Mismatch Repair-Deficient Solid Cancers. JAMA Oncol. 2018, 4, 157–158. [Google Scholar] [CrossRef]
- du Rusquec, P.; de Calbiac, O.; Robert, M.; Campone, M.; Frenel, J.S. Clinical utility of pembrolizumab in the management of advanced solid tumors: an evidence-based review on the emerging new data. Cancer Manag. Res. 2019, 11, 4297–4312. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. FDA Grants Accelerated Approval to Pembrolizumab for Advanced Gastric Cancer; FDA: Washington, DC, USA, 2017. [Google Scholar]
- Fuchs, C.S.; Doi, T.; Jang, R.W.; Muro, K.; Satoh, T.; Machado, M.; Sun, W.; Jalal, S.I.; Shah, M.A.; Metges, J.P.; et al. Safety and Efficacy of Pembrolizumab Monotherapy in Patients with Previously Treated Advanced Gastric and Gastroesophageal Junction Cancer: Phase 2 Clinical KEYNOTE-059 Trial. JAMA Oncol. 2018, 4, e180013. [Google Scholar] [CrossRef]
- Kim, S.T.; Cristescu, R.; Bass, A.J.; Kim, K.M.; Odegaard, J.I.; Kim, K.; Liu, X.Q.; Sher, X.; Jung, H.; Lee, M.; et al. Comprehensive molecular characterization of clinical responses to PD-1 inhibition in metastatic gastric cancer. Nat. Med. 2018, 24, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Zaanan, A.; Taieb, J. How to better select patients with advanced gastric cancer for immunotherapy. Transl. Gastroenterol. Hepatol. 2019, 4, 6. [Google Scholar] [PubMed]
- Shitara, K.; Van Cutsem, E.; Bang, Y.; Fuchs, C.S.; Wyrwicz, L.; Lee, K.W.; Kudaba, I.; Garrido, M.; Cheol Chung, H.; Castro, H.R.; et al. Pembrolizumab With or Without Chemotherapy vs Chemotherapy in Patients With Advanced G/GEJ Cancer (GC) Including Outcomes According to Microsatellite Instability-High (MSI-H) Status in KEYNOTE-062. Ann Oncol. 2019, 30 (Suppl. 5), v-878–v-879. [Google Scholar] [CrossRef]
- Wrangle, J.M.; Velcheti, V.; Patel, M.R.; Garrett-Mayer, E.; Hill, E.G.; Ravenel, J.G.; Miller, J.S.; Farhad, M.; Anderton, K.; Lindsey, K.; et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: a non-randomised, open-label, phase 1b trial. Lancet Oncol. 2018, 19, 694–704. [Google Scholar] [CrossRef] [PubMed]
- Santuray, R.T.; Johnson, D.E.; Grandis, J.R. New Therapies in Head and Neck Cancer. Trends Cancer 2018, 4, 385–396. [Google Scholar] [CrossRef] [PubMed]
- Cuoco, J.A.; Benko, M.J.; Busch, C.M.; Rogers, C.M.; Prickett, J.T.; Marvin, E.A. Vaccine-Based Immunotherapeutics for the Treatment of Glioblastoma: Advances, Challenges, and Future Perspectives. World Neurosurg. 2018, 120, 302–315. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| dMMR | pMMR | Log Ratio | p-Value | q-Value | ||
|---|---|---|---|---|---|---|
| Highly recurrent | TTN | 84 (82.35%) | 581 (48.86%) | 0.75 | 1.54 × 10−11 | 6.78 × 10−10 |
| ARID1A | 71 (69.61%) | 204 (17.16%) | 2.02 | 3.46 × 10−28 | 2.11 × 10−24 | |
| TP53 | 55 (53.92%) | 647 (54.42%) | −0.01 | 0.502 | 0.59 | |
| KMT2D | 54 (52.94%) | 108 (9.08%) | 2.54 | 1.22 × 10−25 | 3.12 × 10−22 | |
| ACVR2A | 53 (51.96%) | 80 (6.73%) | 2.95 | 1.13 × 10−29 | 2.08 × 10−25 | |
| EGFR family | EGFR | 17 (16.67%) | 33 (2.78%) | 2.59 | 5.57 × 10−8 | 6.81 × 10−7 |
| ERBB2 | 18 (17.65%) | 59 (4.96%) | 1.83 | 1.12 × 10−5 | 5.96 × 10−5 | |
| ERBB3 | 18 (17.65%) | 65 (5.47%) | 1.69 | 3.44 × 10−5 | 1.54 × 10−4 | |
| ERBB4 | 20 (19.61%) | 127 (10.68%) | 0.88 | 7.92 × 10−3 | 0.01 | |
| PI3K family | PIK3CA | 30 (29.41%) | 129 (10.85%) | 1.44 | 1.06 × 10−6 | 8.00 × 10−6 |
| PIK3CB | 8 (7.84%) | 20 (1.68%) | 2.22 | 9.50 × 10−4 | 2.65 × 10−3 | |
| PIK3CG | 17 (16.67%) | 36 (3.03%) | 2.46 | 1.50 × 10−7 | 1.55 × 10−6 | |
| PTEN | 17 (16.67%) | 58 (4.88%) | 1.77 | 3.18 × 10−5 | 1.48 × 10−4 | |
| AKT1 | 1 (0.98%) | 8 (0.67%) | 0.54 | 0.52 | 0.61 | |
| AKT2 | 12 (11.76%) | 7 (0.59%) | 4.32 | 1.00 × 10−9 | 2.31 × 10−8 | |
| AKT3 | 2 (1.96%) | 17 (1.43%) | 0.46 | 0.45 | 0.54 | |
| MTOR | 26 (25.49%) | 40 (3.36%) | 2.92 | 1.27 × 10−13 | 1.03 × 10−11 | |
| IC | CD274 (PD-L1) | 3 (2.94%) | 5 (0.42%) | 2.81 | 0.02 | 0.04 |
| PDCD1 (PD-1) | 8 (7.84%) | 5 (0.42%) | 4.22 | 1.08 × 10−6 | 8.00 × 10−6 |
| Drug | Phase | Setting | Status | Patients | Basket Trial | Primary Outcome | Secondary Outcome | NCT Number |
|---|---|---|---|---|---|---|---|---|
| Pembro | I | A, R | Ac(nr) | 297 | Yes | AEs, ORR | - | NCT01848834 |
| Pembro | II | E | C | 113 | Yes | irPFS, iORR, PFS | OS, irPFS, ORR, PFS | NCT01876511 |
| Pembro | I | A | Ac(nr) | 477 | Yes | BRR | PFS, OS, DOR | NCT02054806 |
| Pembro | II | A | Ac | 1395 | Yes | ORR | - | NCT02628067 |
| Pembro + CT | II | A, R | Ac(nr) | 315 | No | AEs, ORR | - | NCT02335411 |
| Pembro + CT | III | A | Ac(nr) | 763 | No | PFS, OS | ORR, DOR, PFS, AEs | NCT02494583 |
| Durva/Treme + CT | II | A | Ac | 105 | No | PFS | OS, AEs, QoL, TTP, PFS, BRR, DCR | NCT03959293 |
| Camre + CT | II | Stage III | Ac | 20 | No | DFS | - | NCT04152889 |
| ALT-803 + Pembro/Nivo/Atezo/Ave | II | A | Ac | 611 | Yes | ORR | DFS, OS, AEs, QoL, PFS | NCT03228667 |
| anti-GITR + Nivo/Ipili | I/II | A | Ac | 285 | Yes | AEs, ORR | PFS, OS | NCT03126110 |
| DSP-7888/Nivo/Pembro | I/II | A | Ac | 84 | Yes | AEs, DLTs, ORR | ORR, DCR, DOR, PFS, OS | NCT03311334 |
| Regorafenib + Ave | I/II | A | Ac | 362 | Yes | RP2D | MTD, DLT, AEs, BRR, ORR, PFS, OS | NCT03475953 |
| Pembro | II | A | Ac | 40 | No | RR | - | NCT02589496 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopez, G.; Venetis, K.; Sajjadi, E.; Fusco, N. Mismatch Repair System Genomic Scars in Gastroesophageal Cancers: Biology and Clinical Testing. Gastrointest. Disord. 2020, 2, 341-352. https://doi.org/10.3390/gidisord2040031
Lopez G, Venetis K, Sajjadi E, Fusco N. Mismatch Repair System Genomic Scars in Gastroesophageal Cancers: Biology and Clinical Testing. Gastrointestinal Disorders. 2020; 2(4):341-352. https://doi.org/10.3390/gidisord2040031
Chicago/Turabian StyleLopez, Gianluca, Konstantinos Venetis, Elham Sajjadi, and Nicola Fusco. 2020. "Mismatch Repair System Genomic Scars in Gastroesophageal Cancers: Biology and Clinical Testing" Gastrointestinal Disorders 2, no. 4: 341-352. https://doi.org/10.3390/gidisord2040031
APA StyleLopez, G., Venetis, K., Sajjadi, E., & Fusco, N. (2020). Mismatch Repair System Genomic Scars in Gastroesophageal Cancers: Biology and Clinical Testing. Gastrointestinal Disorders, 2(4), 341-352. https://doi.org/10.3390/gidisord2040031