
Hydrogen Absorption and Self-Corrosion of Mg Anode: Influence of Aqueous Electrolyte Species


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Correspondence: Mechanisms of Self-Corrosion of Mg Anode
Mg+ + H2O→Mg2+ + ½ H2 g + OH−
1.2. The Effect of Surface Impurities and Electrolyte Nature on Anomalous HE
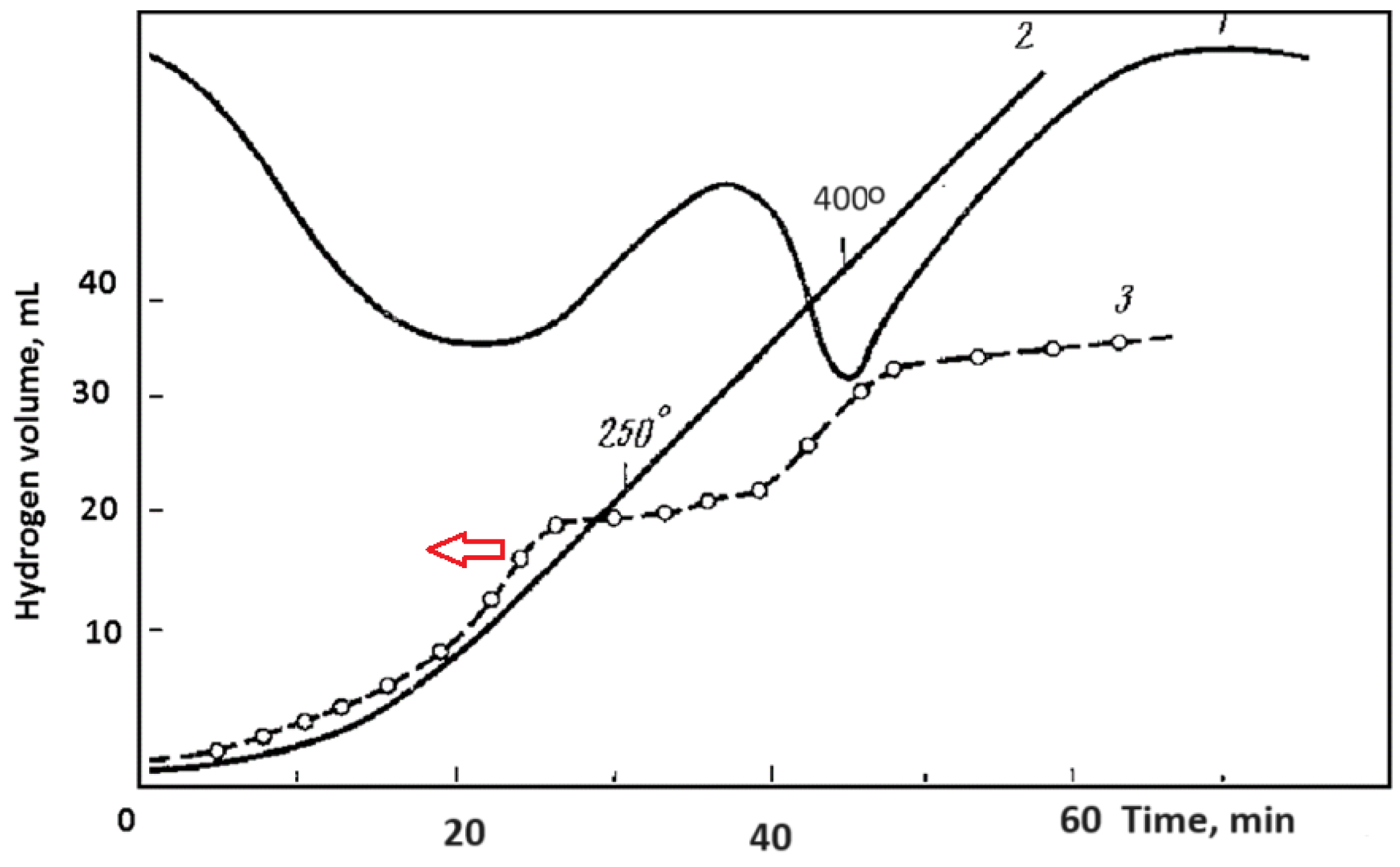
1.3. Hydride Formation and the Anomalous HE on the Mg Anode
E = −1.684 − 0.059 pH + 0.0291 log pH2
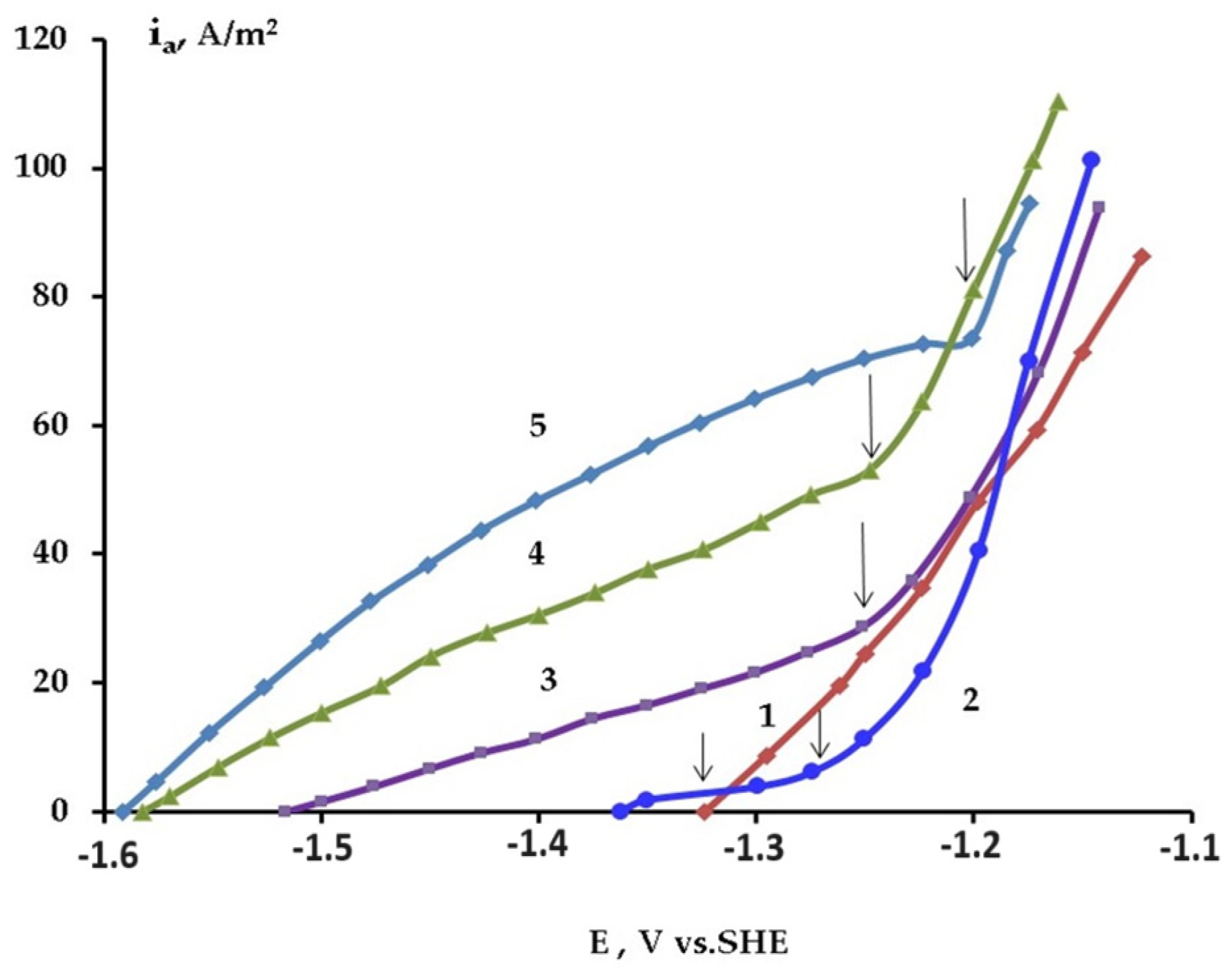
2. Self-Corrosion and Hydrogen Absorption in Halide Containing Aqueous Electrolytes
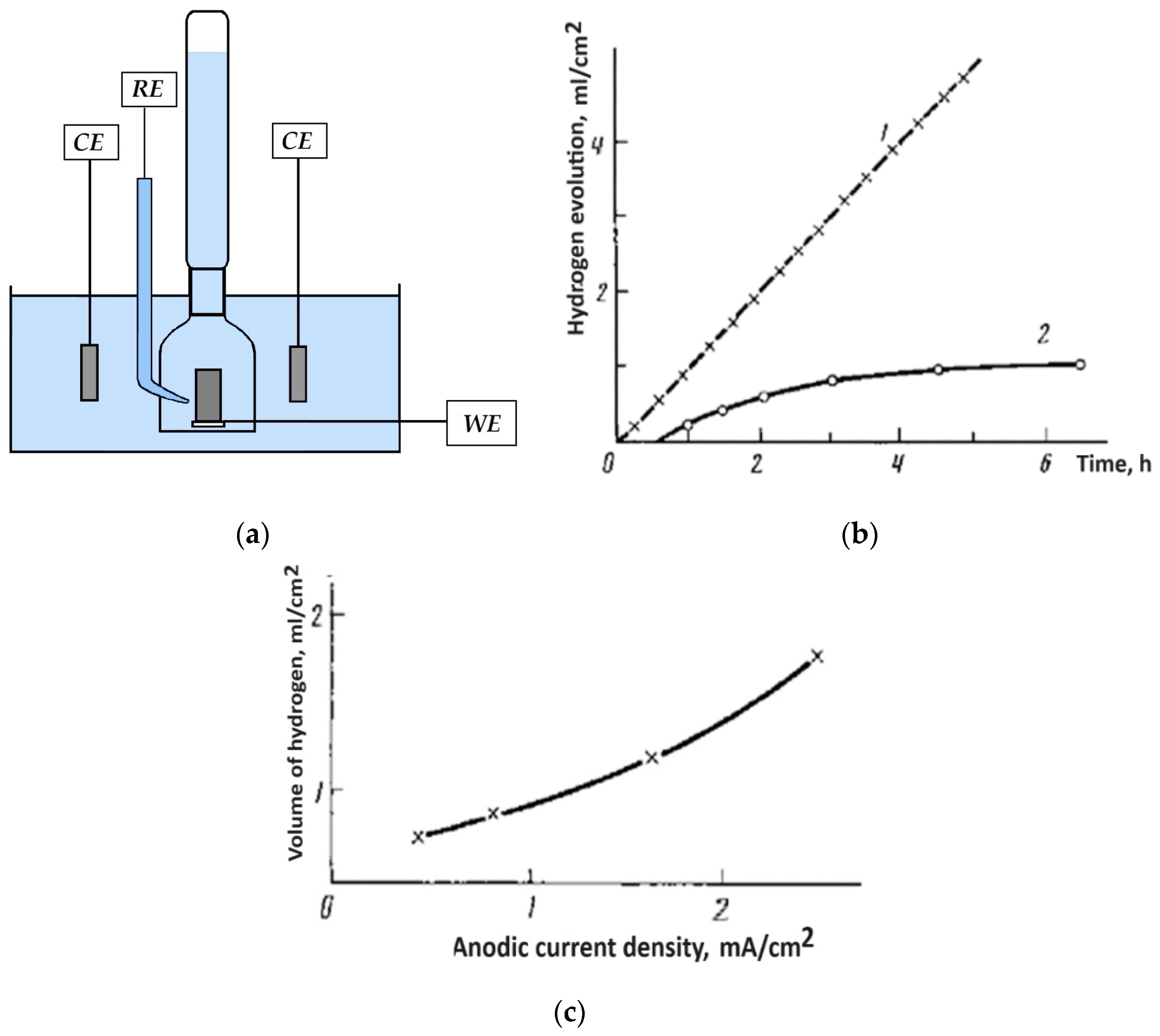
2.1. Electrochemical Detection of Hydride Ions on the Surface of Mg Anode
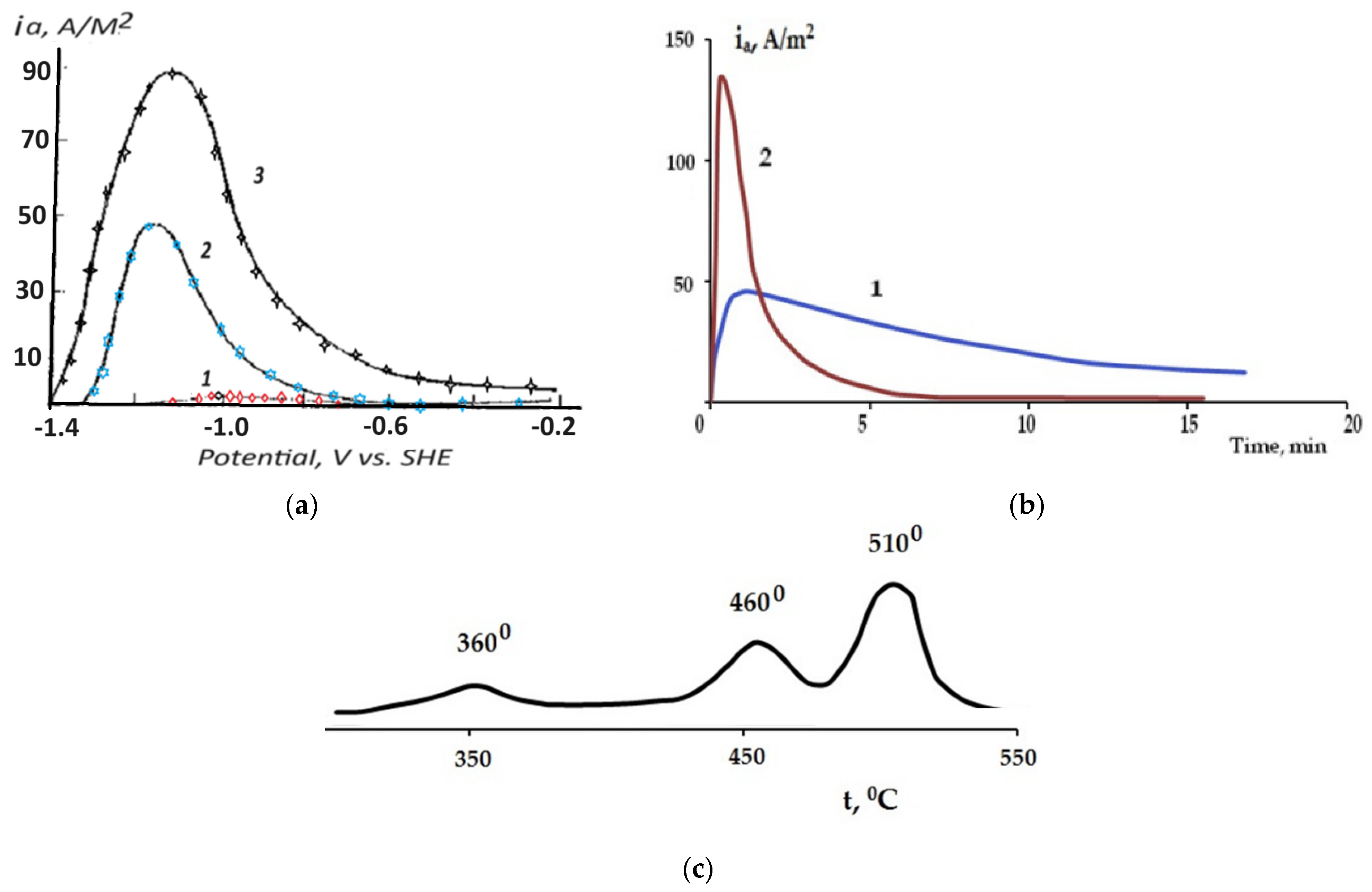
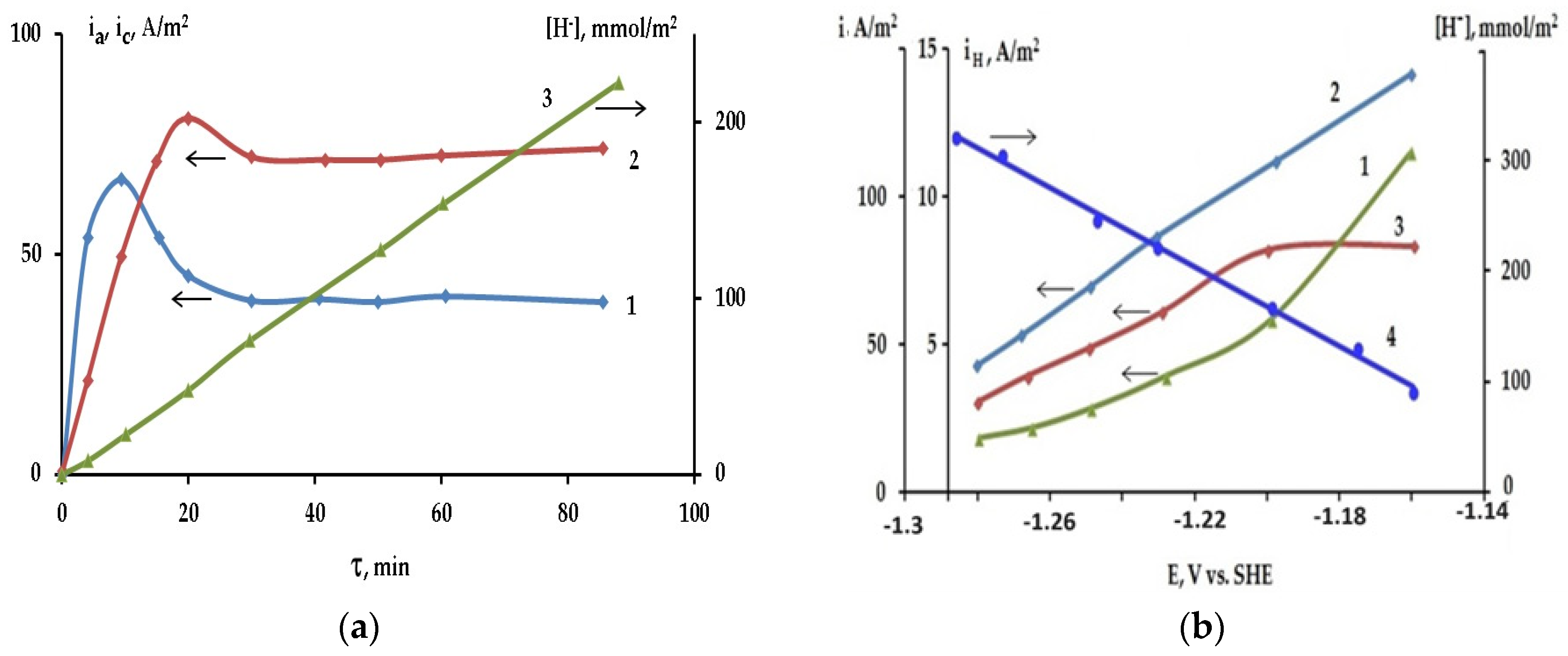
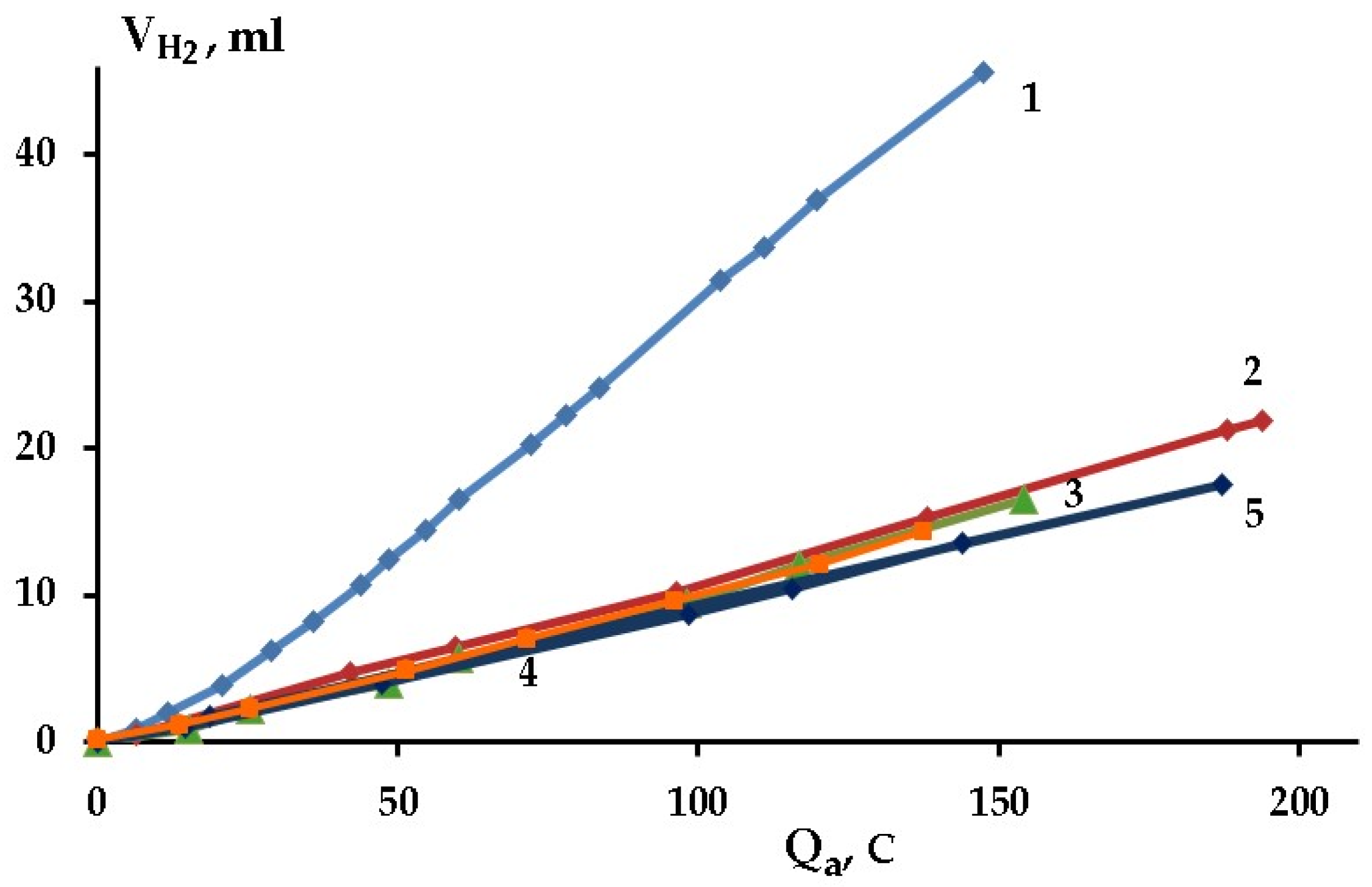
2.2. Kinetics of Self-Corrosion and Hydride Collection on Mg Anode in 0.1 M NaCl Aqueous Electrolyte
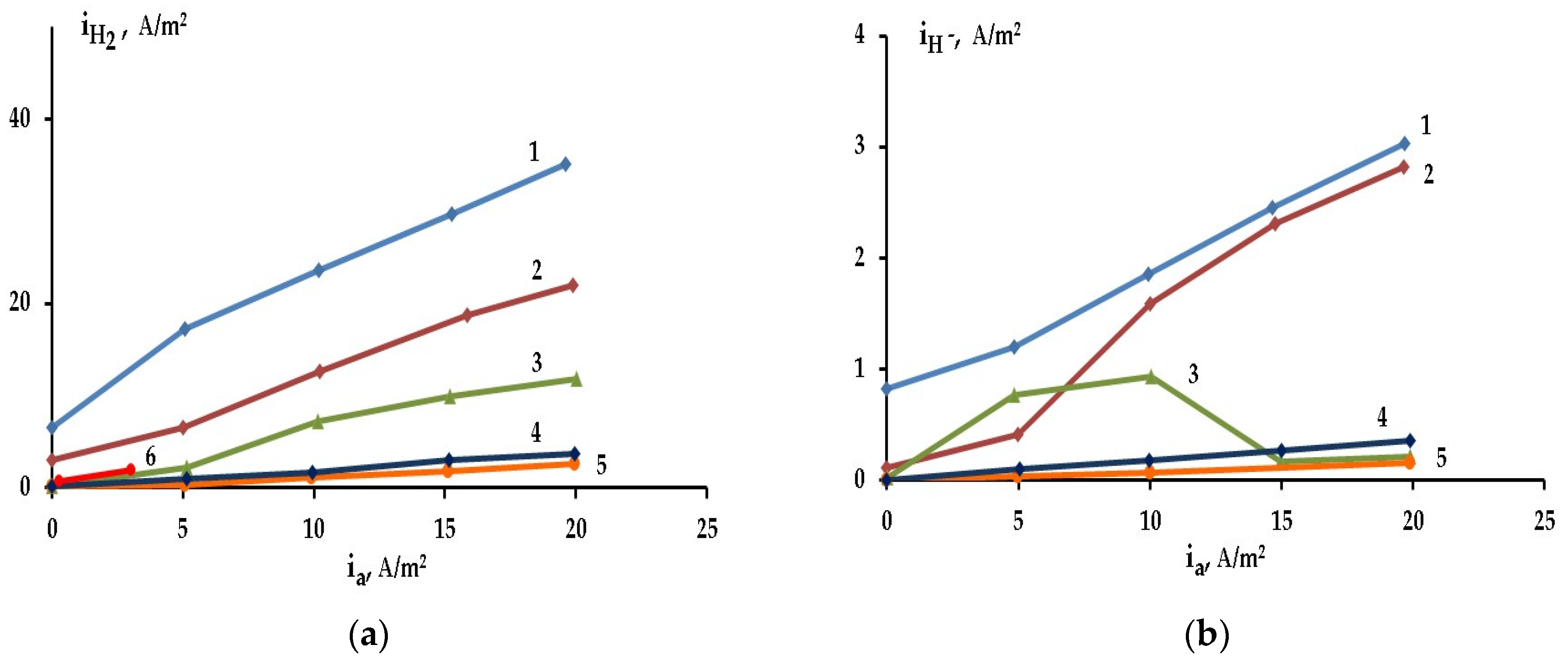
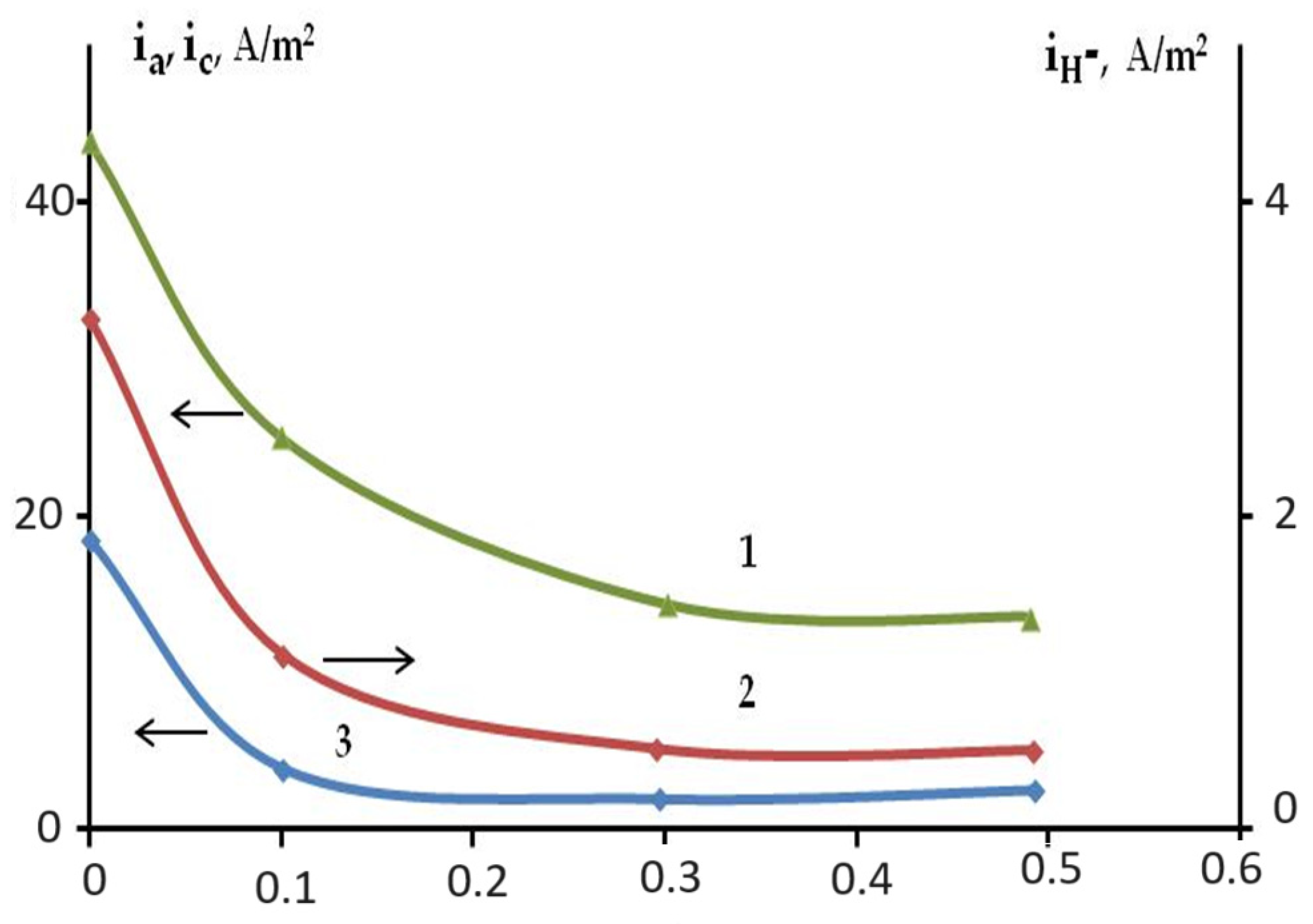
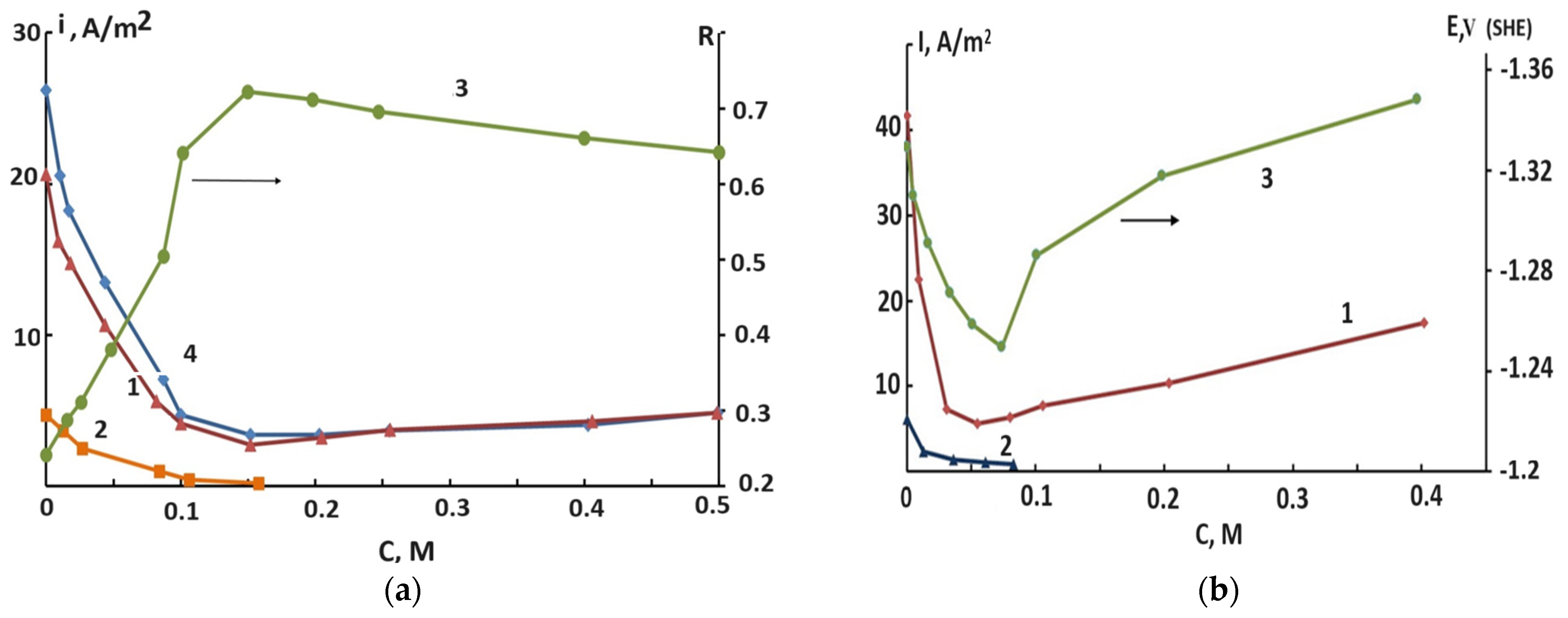
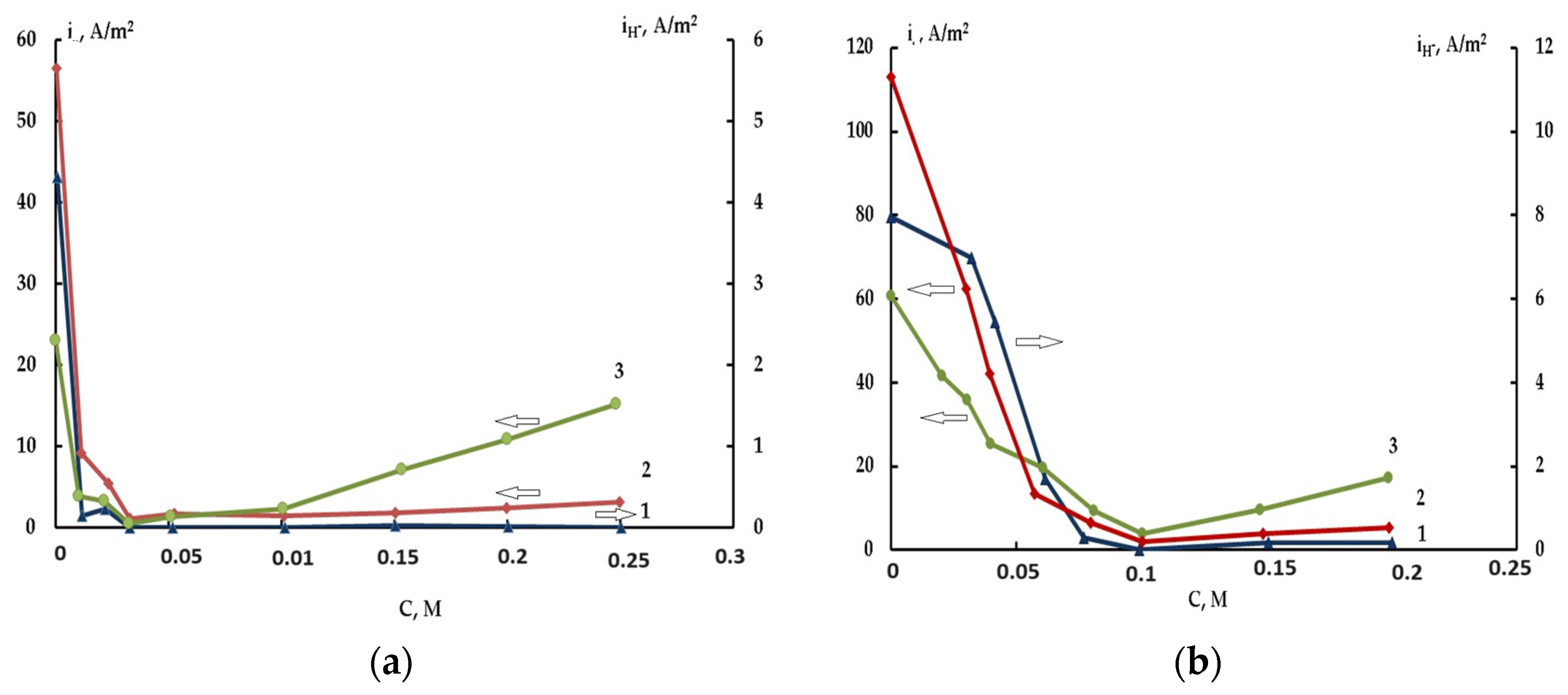
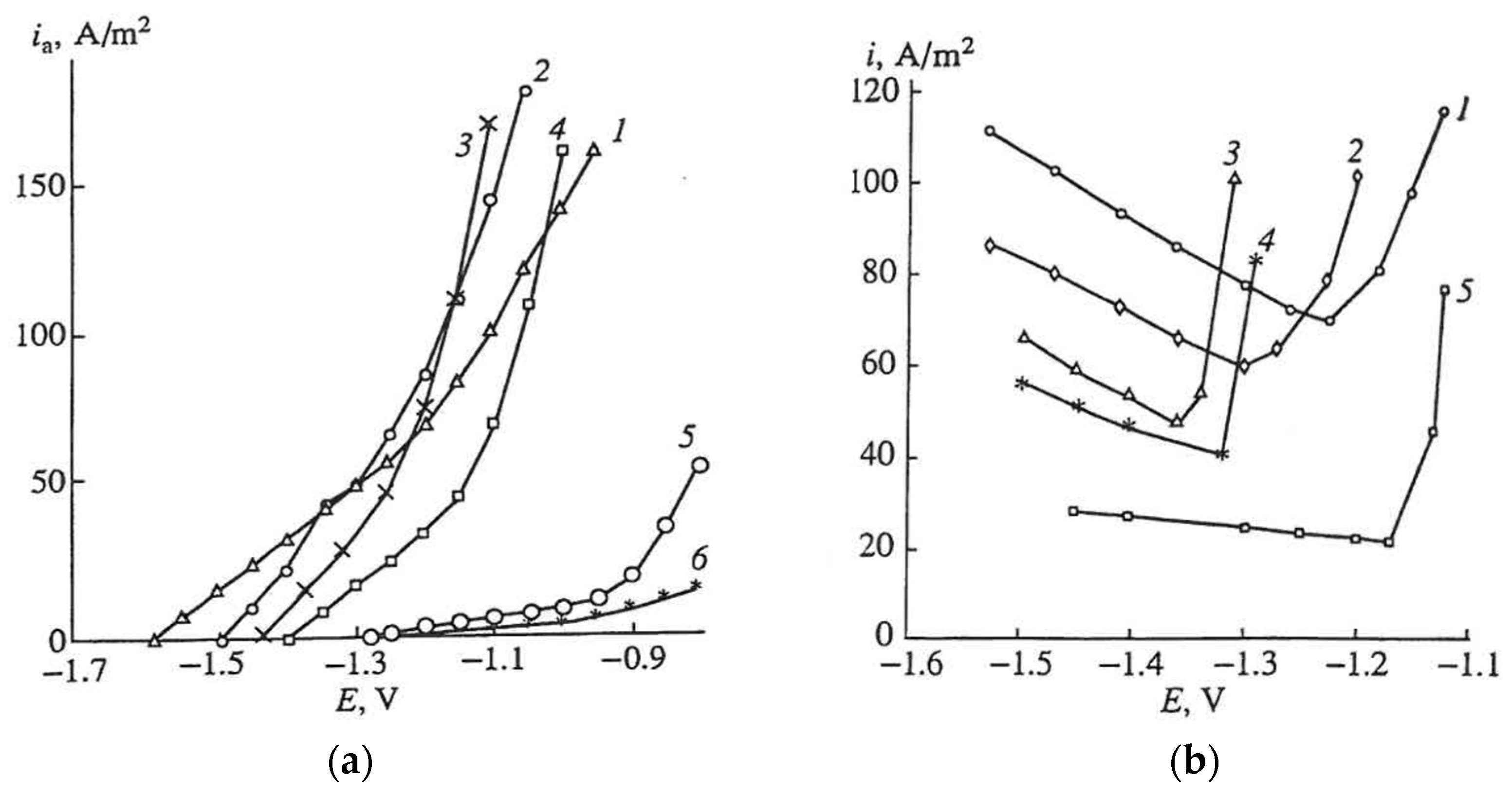
2.3. The Effect of Halide Ions on the Kinetics of Self-Corrosion and Hydride Formation
E = −1.862 − 0.059 pH
E = −1.72 − 0.029 log [Cl−]2/[MgCl2]
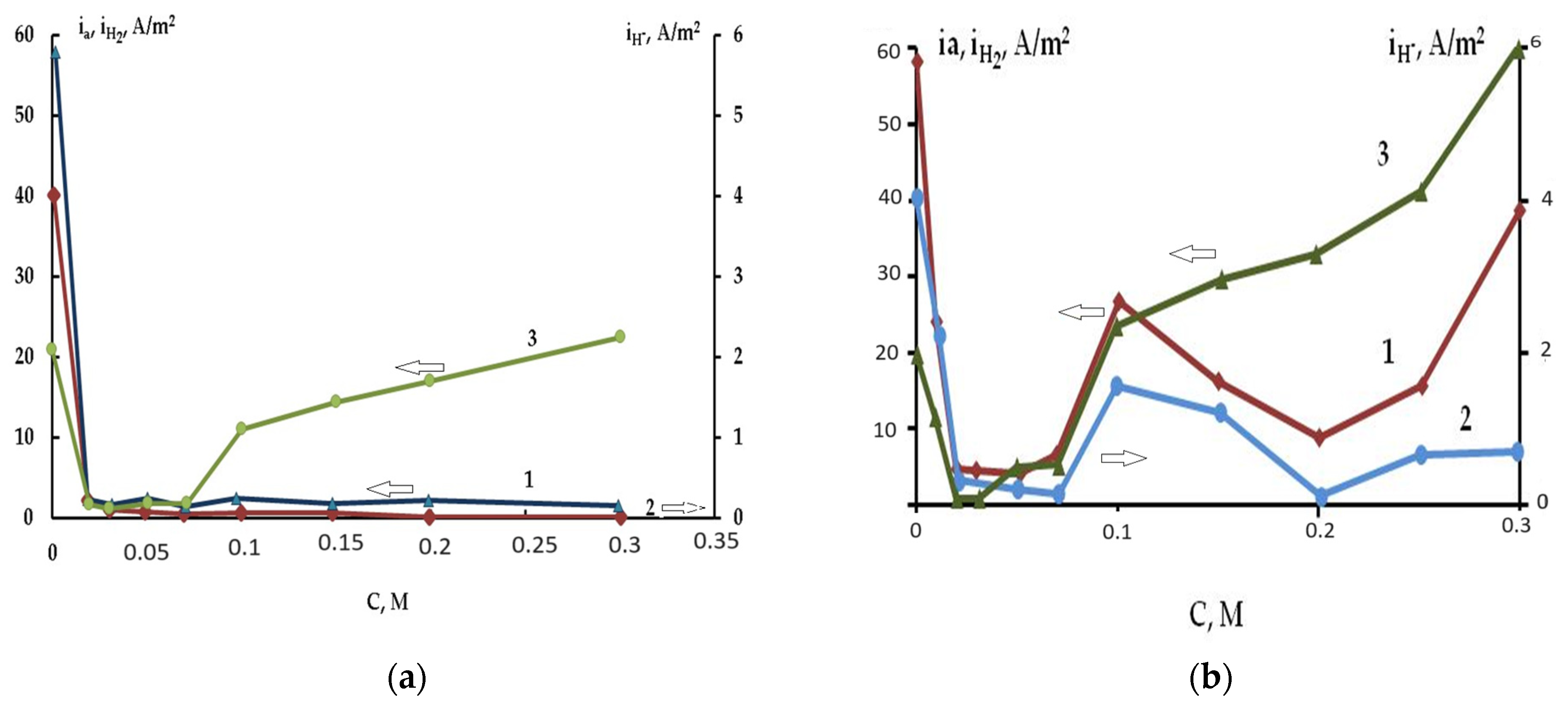
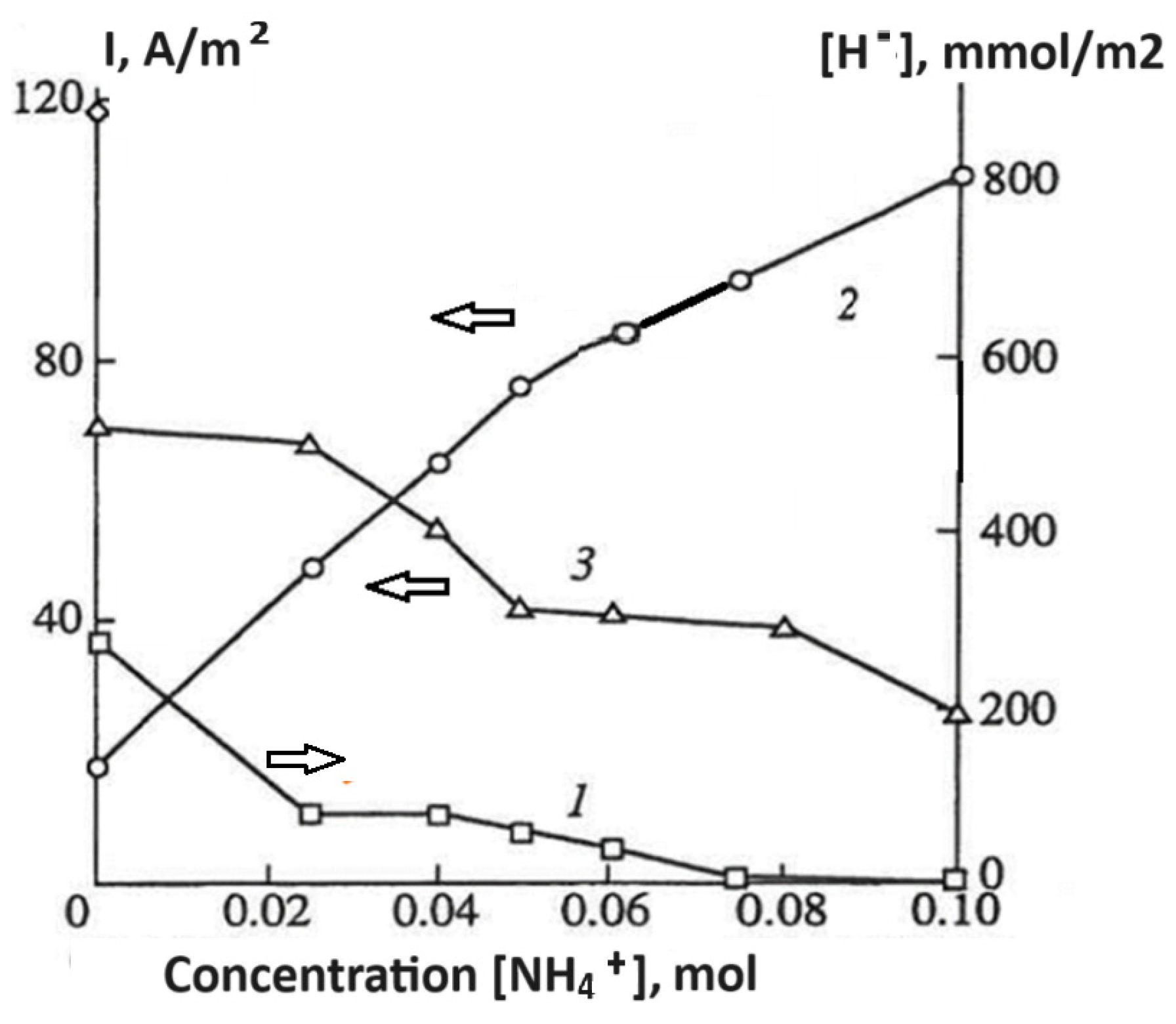
3. Impact of Addition of Complex Formed Ligands on the Self-Corrosion and Hydride Formation Rates
E = −2.94 − 0.059 log [C2O42−]
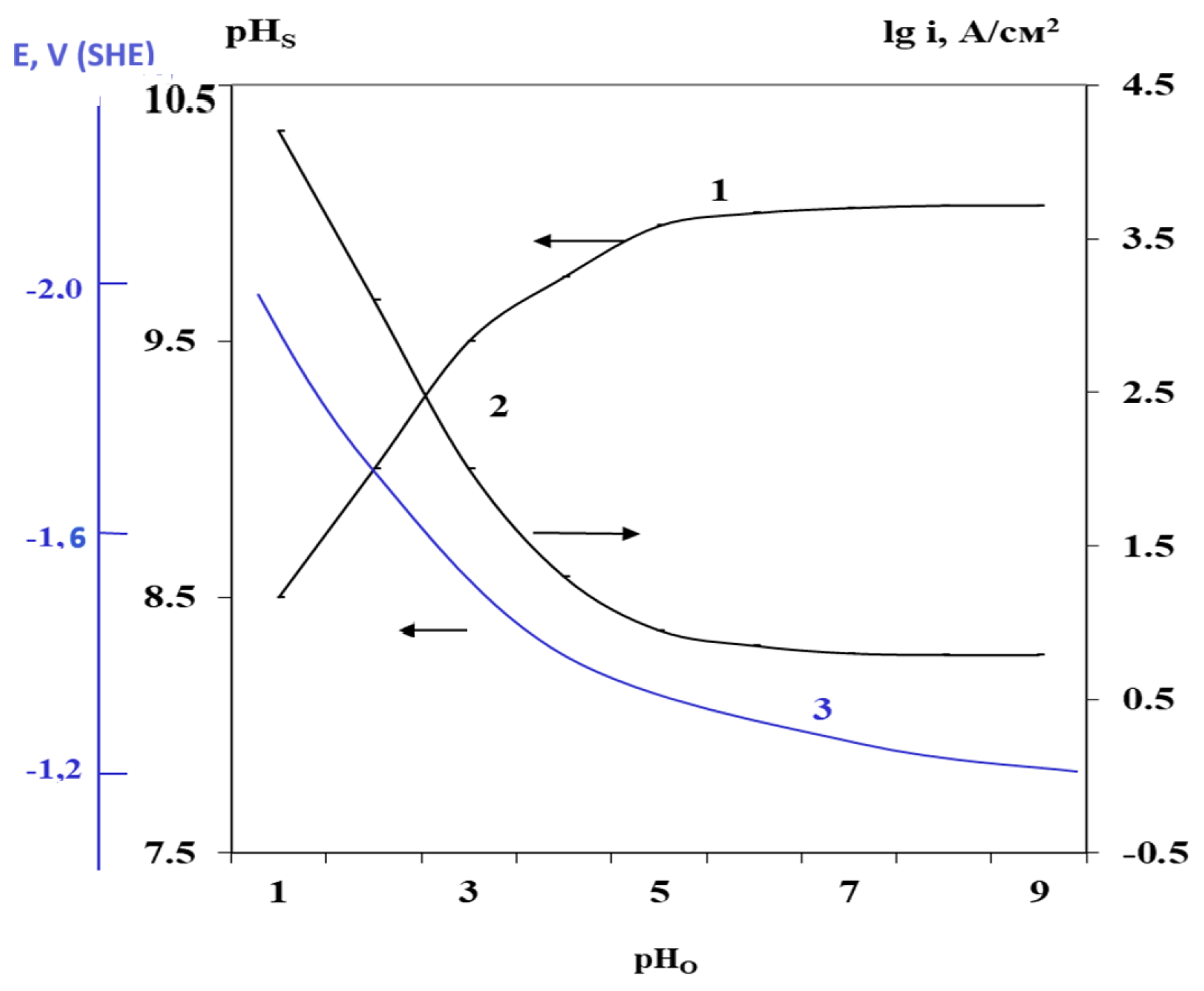
4. Influence of Buffering the Acidity of Aqueous Electrolyte on Hydrogen Absorption and Mg Self-Corrosion
5. Summary and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Prasada Satya, S.V.; Prasada, S.B.; Verma, K.; Mishrac, R.K.; Kumar, V.; Singha, S. The role and significance of Magnesium in modern day research—A review. J. Magnes. Alloys 2022, 10, 1–61. [Google Scholar] [CrossRef]
- Joost, W.J.; Krajewski, P.E. Towards magnesium alloys for high volume automotive applications. Scr. Mater. 2017, 128, 107–112. [Google Scholar] [CrossRef]
- Trang, T.T.T.; Zhang, J.H.; Kim, J.H.; Zargaran, A.; Hwang, J.H.; Suh, B.C.; Kim, N.J. Designing a magnesium alloy with high strength and high formability. Nat. Commun. 2018, 9, 2522–2527. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, N.T.; Birbilis, N.; Walker, J.; Woodfield, T.; Dias, G.J.; Staiger, M.P. In-vitro dissolution of magnesium-calcium binary alloys: Clarifying the unique role of calcium additions in bioresorbable magnesium implant alloys. J. Biomed. Mater. Res. Part B 2010, 95B, 91–100. [Google Scholar] [CrossRef]
- Zheng, Y.F.; Gu, X.N.; Witte, F. Biodegradable metals. Mater. Sci. Eng. R 2014, 77, 1–34. [Google Scholar] [CrossRef]
- Gu, X.; Zheng, Y.; Cheng, Y.; Zhong, S.; Xi, T. In vitro corrosion and biocompatibility of binary magnesium alloys. Biomaterials 2009, 30, 484–498. [Google Scholar] [CrossRef]
- Zhang, T.; Tao, Z.; Chen, J. Magnesium-air batteries: From principles to applications. Mater. Horiz. 2014, 1, 196–206. [Google Scholar] [CrossRef]
- Peng, B.; Chen, J. Functional materials with high-efficiency energy storage and conversion for batteries and fuel cells. Coord. Chem. Rev. 2009, 253, 2805–2813. [Google Scholar] [CrossRef]
- Chen, J.; Cheng, F. Combination of lightweight elements and nanostructured materials for batteries. Acc. Chem. Res. 2009, 42, 713–723. [Google Scholar] [CrossRef]
- Esmaily, M.; Svensson, J.E.; Fajardo, S.; Birbilis, N.; Frankel, G.S.; Virtanen, S.; Arrabal, R.; Thomas, S.; Johansson, L.G. Fundamentals and advances in magnesium alloy corrosion. Prog. Mater. Sci. 2017, 89, 92–193. [Google Scholar] [CrossRef]
- Huanga, J.; Songa, G.-L.; Atrens, A.; Dargusch, M. What activates the Mg surface—A comparison of Mg dissolution mechanisms. J. Mater. Sci. Technol. 2020, 57, 204–220. [Google Scholar] [CrossRef]
- Kappes, M.; Iannuzzi, M.; Carranza, R.M. Hydrogen embrittlement of magnesium and magnesium alloys: A Review. J. Electrochem. Soc. 2013, 160, C168–C178. [Google Scholar] [CrossRef]
- Atrens, A.; Chen, X.; Shi, Z. Review Mg corrosion—Recent progress. MDPI Corros. Mater. Degrad. 2022, 3, 566–597. [Google Scholar] [CrossRef]
- Tomashov, N.D.; Komisarova, V.S.; Timonova, M.A. Investigation of electrochemical corrosion of magnesium. In Trudy of Institute Physical Chemistry AN SSSR; Publication of Academy of Sciences: Moscow, Russia, 1955; Volume 5, pp. 172–197. [Google Scholar]
- Straumanis, M.E.; Bhatia, B.K. Disintegration of magnesium while dissolving anodically in neutral and acidic solutions. J. Electrochem. Soc. 1963, 110, 357–360. [Google Scholar] [CrossRef]
- James, W.J.; Straumanis, M.E.; Bhatia, D.K.; Johnson, J.W. The difference effect on magnesium dissolving in acids. J. Electrochem. Soc. 1962, 110, 1117–1121. [Google Scholar] [CrossRef]
- Petty, R.L.; Davidson, A.W.; Kleinberg, J. The anodic oxidation of magnesium metal: Evidence for the existence of unipositive Mg. J. Am. Chem. Soc. 1954, 76, 363–366. [Google Scholar] [CrossRef]
- Shi, Z.; Cao, F.; Song, G.-L.; Atrens, A. Low apparent valence of Mg during corrosion. Corros. Sci. 2014, 88, 434–443. [Google Scholar] [CrossRef]
- Atrens, A.; Song, G.-L.; Liu, M.; Shi, Z.; Cao, F.; Dargusch, M.S. Review of recent developments in the field of magnesium corrosion. Adv. Eng. Mater. 2015, 17, 400–453. [Google Scholar] [CrossRef]
- Samaniego, A.; Hurley, B.L.; Frankel, G.S. On the evidence for univalent Mg. J. Electroanal. Chem. 2015, 737, 123–128. [Google Scholar] [CrossRef]
- Birbilis, N.; King, A.D.; Thomas, S.; Frankel, G.S.; Scully, J.R. Evidence for enhanced catalytic activity of magnesium arising from anodic dissolution. Electrochim. Acta 2014, 132, 277–283. [Google Scholar] [CrossRef]
- Williams, G.; Birbilis, N.; McMurray, H.N. The source of hydrogen evolved from a magnesium anode. Electrochem. Com. 2013, 36, 2–5. [Google Scholar] [CrossRef]
- Taheri, M.; Kish, J.R.; Birbilis, N.; Danaie, M.; McNally, E.A.; McDermid, J.R. Towards a physical description for the origin of enhanced catalytic activity of corroding magnesium surfaces. Electrochim. Acta 2014, 116, 396–403. [Google Scholar] [CrossRef]
- Frankel, G.S.; Samaniego, A.; Birbilis, N. Evolution of hydrogen at dissolving magnesium surfaces. Corr. Sci. 2013, 70, 104–111. [Google Scholar] [CrossRef]
- Thomas, S.; Gharbi, O.; Salleha, S.H.; Volovitch, P.; Ogle, K.; Birbilis, N. On the effect of Fe concentration on Mg dissolution and activation studied using atomic emission spectroelectrochemistry and scanning electrochemical microscopy. Electrochim. Acta 2016, 210, 271–284. [Google Scholar] [CrossRef]
- Curioni, M. The behaviour of magnesium during free corrosion and potentiodynamic polarisation investigated by real-time hydrogen measurement and optical imaging. Electrochim. Acta. 2014, 120, 284–292. [Google Scholar] [CrossRef]
- Cain, T.; Madden, S.B.; Birbilis, N.; Scully, J.R. Evidence of the enrichment of transition metal elements on corroding magnesium surfaces using Rutherford backscattering spectrometry. J. Electrochem. Soc. 2015, 162, C228–C237. [Google Scholar] [CrossRef]
- Bender, S.; Goellner, J.; Heyn, A.; Schmigalla, S. A new theory for the negative difference effect in magnesium corrosion. Mater. Corros. 2013, 63, 707–712. [Google Scholar] [CrossRef]
- Rossrucker, L.; Samaniego, A.; Grote, J.-P.; Mingers, A.M.; Laska, C.A.; Birbilis, N. The pH dependence of magnesium dissolution and hydrogen evolution during anodic polarization. J. Electrochem. Soc. 2015, 162, C333–C339. [Google Scholar] [CrossRef]
- Rossrucker, L.; Mayrhofer, K.J.; Frankel, G.S.; Birbilis, N. Investigating the real-time dissolution of magnesium using online analysis by ICP-MS. J. Electrochem. Soc. 2014, 161, C115–C119. [Google Scholar] [CrossRef]
- Lebouil, S.; Gharbi, O.; Volovitch, P.; Ogle, K. Mg dissolution in phosphate and chloride electrolytes: Insight into the mechanism of the negative difference effect. Corrosion 2015, 71, 234–241. [Google Scholar] [CrossRef]
- Swiatowska, J.; Volovitch, P.; Ogle, K. The anodic dissolution of Mg in NaCl and Na2SO4 electrolytes by atomic emission spectroelectrochemistry. Corros. Sci. 2010, 52, 2372–2378. [Google Scholar] [CrossRef]
- Salleh, S.H.; Thomas, S.; Yuwono, J.A.; Venkatesan, K.; Birbilis, N. Enhanced hydrogen evolution on Mg(OH)2 covered Mg surfaces. Electrochim. Acta 2015, 161, 144–152. [Google Scholar] [CrossRef]
- Lysne, D.; Thomas, S.; Hurley, M.F.; Birbilis, N. On the Fe enrichment during anodic polarization of magnesium and its impact on hydrogen evolution. J. Electrochem. Soc. 2015, 162, C396. [Google Scholar] [CrossRef]
- Fajardo, S.; Glover, C.F.; Williams, G.; Frankel, G.S. The evolution of anodic hydrogen on high-purity magnesium in acidic buffer solution. Corrosion 2017, 73, 482–493. [Google Scholar] [CrossRef]
- Lynch, S.P.; Trevena, P. Stress Corrosion Cracking and Liquid Metal Embrittlement in Pure Magnesium. Corrosion 1988, 44, 113–120. [Google Scholar] [CrossRef]
- Kamilyan, M.; Silverstein, R.; Eliezer, D. Hydrogen trapping and hydrogen embrittlement of Mg alloys. J. Mater. Sci. 2017, 52, 11091–11100. [Google Scholar] [CrossRef]
- Perrault, G. Magnesium. In Encyclopedia of Electrochemistry of the Elements; Bard, A.J., Ed.; Marcel Dekker: New York, NY, USA, 1978; pp. 263–319. [Google Scholar]
- Perrault, G. The potential–pH diagram of the magnesium–water system. J. Electroan. Chem. 1974, 51, 107–119. [Google Scholar] [CrossRef]
- Nazarov, A.; Lisovskii, A.; Mikhailovskii, Y.N. Formation of MgH2 on electrochemical dissolution of magnesium in aqueous-electrolytes. Prot. Met. 1989, 25, 606–610. [Google Scholar]
- Nazarov, A.; Yurasova, T. Influence of the Electrolyte Nature on Mg Hydrogenation and Corrosion. In Proceedings of the Eurocorr’91, Budapest, Hungary, 21–25 October 1991; Karl, I., Bod, M., Eds.; European Federation of Corrosion: Bruxelles, Belgium, 1991; Volume 1, pp. 124–129. [Google Scholar]
- Gulbrandsen, E. Anodic behavior of Mg in HCO−3/CO2−3 buffer solutions. Quasi-steady measurements. Electrochim. Acta 1992, 37, 1403–1412. [Google Scholar] [CrossRef]
- Nazarov, A.; Yurasova, T.; Gubin, V.V.; Buriak, A.K.; Glazunov, M.P. On hydrogenation of magnesium at the free corrosion and anodic corrosion in chloride electrolyte. Prot. Met. 1993, 29, 392–397. [Google Scholar]
- Chen, J.; Wang, E.; Han, J.; Dong, W.K.; Shoesmith, D.W. Effect of Hydrogen on Corrosion and Stress Corrosion Cracking of AZ91 Alloy in Aqueous Solutions. J. Acta Metallurg. Sin. (Engl. Lett.) 2016, 29, 1–7. [Google Scholar] [CrossRef]
- Seyeux, A.; Liu, M.; Schmutz, P.; Song, G.; Atrens, A.; Marcus, P. ToF-SIMS depth profile of the surface film on pure magnesium formed by immersion in pure water and the identification of magnesium hydride. Corros. Sci. 2009, 51, 1883–1886. [Google Scholar] [CrossRef]
- Unocic, K.A.; Elsentriecy, H.H.; Brady, M.P.; Meyer, H.M.; Song, G.; Fayek, M.; Meisner, R.A.; Davis, B. Transmission Electron Microscopy Study of Aqueous Film Formation and Evolution on Magnesium Alloys. J. Electrochem. Soc. 2014, 161, C30–C38. [Google Scholar] [CrossRef]
- Binns, W.J.; Zargarzadah, F.; Dehnavi, V.; Chen, J.; Noël, J.J.; Shoesmith, D.W. Physical and Electrochemical Evidence for the Role of a Mg Hydride Species in Mg Alloy Corrosion. Corrosion 2018, 75, 58–68. [Google Scholar] [CrossRef]
- Cain, T.W.; Gonzalez-Afanador, I.; Birbilis, N.; Scully, J.R. The Role of Surface Films and Dissolution Products on the Negative Difference Effect for Magnesium: Comparison of Cl− versus Cl− Free Solutions. J. Electrochem. Soc. 2017, 164, C300–C311. [Google Scholar] [CrossRef]
- Nazarov, A.P.; Mikhailovski, Y.N. Influence of complex formation on the self-dissolution of Mg-anode. Prot. Met. 1990, 26, 13–18. [Google Scholar]
- Nazarov, A.P.; Yurasova, T.A. Anodic dissolution and self-dissolution of magnesium in the presence de-passivating ions. Prot. Met. 1993, 29, 381–391. [Google Scholar]
- Nazarov, A.P.; Yurasova, T.A. Hydrogen release, hydride formation and self-dissolution of magnesium in presence of complex formed reagents. Prot. Met. 1995, 31, 139–144. [Google Scholar]
- Xu, S.; Dong, X.; Ke, W. Effect of Magnesium Hydride on the Corrosion Behavior of Pure Magnesium in 0.1MNaCl Solution, Hindawi. Int. J. Corros. 2010, 2010, 934867. [Google Scholar] [CrossRef]
- Yasakau, K.A.; Maltseva, A.; Lamaka, S.V.; Mei, D.; Orvi, H.; Volovitch, P.; Ferreira, M.G.S.; Zheludkevich, M.I. The effect of carboxylate compounds on Volta potential and corrosion inhibition of Mg containing different levels of iron. Corros. Sci. 2022, 194, 109937. [Google Scholar] [CrossRef]
- Fockaert, L.I.; Würger, T.; Unbehau, R.; Boelen, B.; Meißner, R.H.; Lamaka, S.V.; Zheludkevich, M.L.; Terryn, H.; Mol, J.M.C. ATR-FTIR in Kretschmann configuration integrated with electrochemical cell as in situ interfacial sensitive tool to study corrosion inhibitors for magnesium substrates. Electrochim. Acta 2020, 345, 136166. [Google Scholar] [CrossRef]
- Prince, L.; Rousseau, M.-A.; Noirfalise, X.; Dangreau, L.; Coelho, L.B.; Olivier, M.-G. Inhibitive effect of sodium carbonate on corrosion of AZ31 magnesium alloy in NaCl solution. Corros. Sci. 2021, 179, 109131. [Google Scholar] [CrossRef]
- Atrens, A.D.; Gentle, I.; Atrens, A. Possible dissolution pathways participating in the Mg corrosion reaction. Corros. Sci. 2015, 92, 173–181. [Google Scholar] [CrossRef]
- Roald, R.; Beck, W. The Dissolution of Magnesium in Hydrochloric Acid. J. Electrochem. Soc. 1951, 98, 277–298. [Google Scholar] [CrossRef]
- Maiangoris, J. Corrosion and Mass Transfer with Chemical Reaction the Dissolution of Mg in HCl. Corrosion 1968, 24, 255–259. [Google Scholar]
- Simaranov, A.I.; Sokolova, T.I.; Marshakov, A.I.; Mikhailovskii, Y.N. Corrosion and electrochemistry of magnesium in acidic media contained the oxidizers. Prot. Met. 1991, 27, 403–409. [Google Scholar]
- Nazarov, A.P.; Yurasova, T.A. Anodic dissolution of Magnesium under positive and negative difference effect. Prot. Met. 1996, 32, 33–37. [Google Scholar]
- Buggio, D.; Trueba, M.; Trasatti, S.P. Corrosion of Mg Alloy in the Presence of Ammonium Ion. Evidence of Hydride Sub-Products. Corros. Sci. 2016, 104, 173–186. [Google Scholar] [CrossRef]
- Taylor, C.D. A first-principles surface reaction kinetic model for hydrogen evolution under cathodic and anodic conditions on magnesium. J. Electrochem. Soc. 2016, 163, C602–C608. [Google Scholar] [CrossRef]
- Xia, X.; Nie, J.F.; Davies, C.H.J.; Tang, W.N.; Xu, S.W.; Birbilis, N. The Influence of Low Levels of Zinc, Calcium, Gadolinium, Strontium, and Zirconium on the Corrosion of Magnesium for Wrought Applications. Corrosion 2015, 71, 1370–1386. [Google Scholar] [CrossRef]
- Lynch, S.P. Chapter 2—Hydrogen Embrittlement (HE) Phenomena and Mechanisms. In Stress Corrosion Cracking: Theory and Practice; Raja, V.S., Shoji, T., Eds.; Woodhead Publishing: Oxford, UK, 2011; pp. 90–130. [Google Scholar]
- Stampella, R.P.; Procter, M.; Ashworth, V. Environmentally-induced cracking of magnesium. Corros. Sci. 1984, 24, 325–341. [Google Scholar] [CrossRef]
- Bobby Kannan, M.; Dietzel, W. Pitting-induced hydrogen embrittlement of magnesium–aluminum alloy. Mater. Des. 2012, 42, 321–326. [Google Scholar] [CrossRef]
- Song, R.G.; Blawert, C.; Dietzel, W.; Atrens, A. A study of stress corrosion cracking and hydrogen embrittlement of AZ31 magnesium alloy. Mater. Sci. Eng. A 2005, 399, 308–317. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nazarov, A.; Yurasova, T.; Marshakov, A. Hydrogen Absorption and Self-Corrosion of Mg Anode: Influence of Aqueous Electrolyte Species. Corros. Mater. Degrad. 2024, 5, 350-369. https://doi.org/10.3390/cmd5030015
Nazarov A, Yurasova T, Marshakov A. Hydrogen Absorption and Self-Corrosion of Mg Anode: Influence of Aqueous Electrolyte Species. Corrosion and Materials Degradation. 2024; 5(3):350-369. https://doi.org/10.3390/cmd5030015
Chicago/Turabian StyleNazarov, Andrei, Tatiana Yurasova, and Andrey Marshakov. 2024. "Hydrogen Absorption and Self-Corrosion of Mg Anode: Influence of Aqueous Electrolyte Species" Corrosion and Materials Degradation 5, no. 3: 350-369. https://doi.org/10.3390/cmd5030015
APA StyleNazarov, A., Yurasova, T., & Marshakov, A. (2024). Hydrogen Absorption and Self-Corrosion of Mg Anode: Influence of Aqueous Electrolyte Species. Corrosion and Materials Degradation, 5(3), 350-369. https://doi.org/10.3390/cmd5030015