
The AA7075–CS1018 Galvanic Couple under Evaporating Droplets


Abstract
1. Introduction
2. Materials and Methods
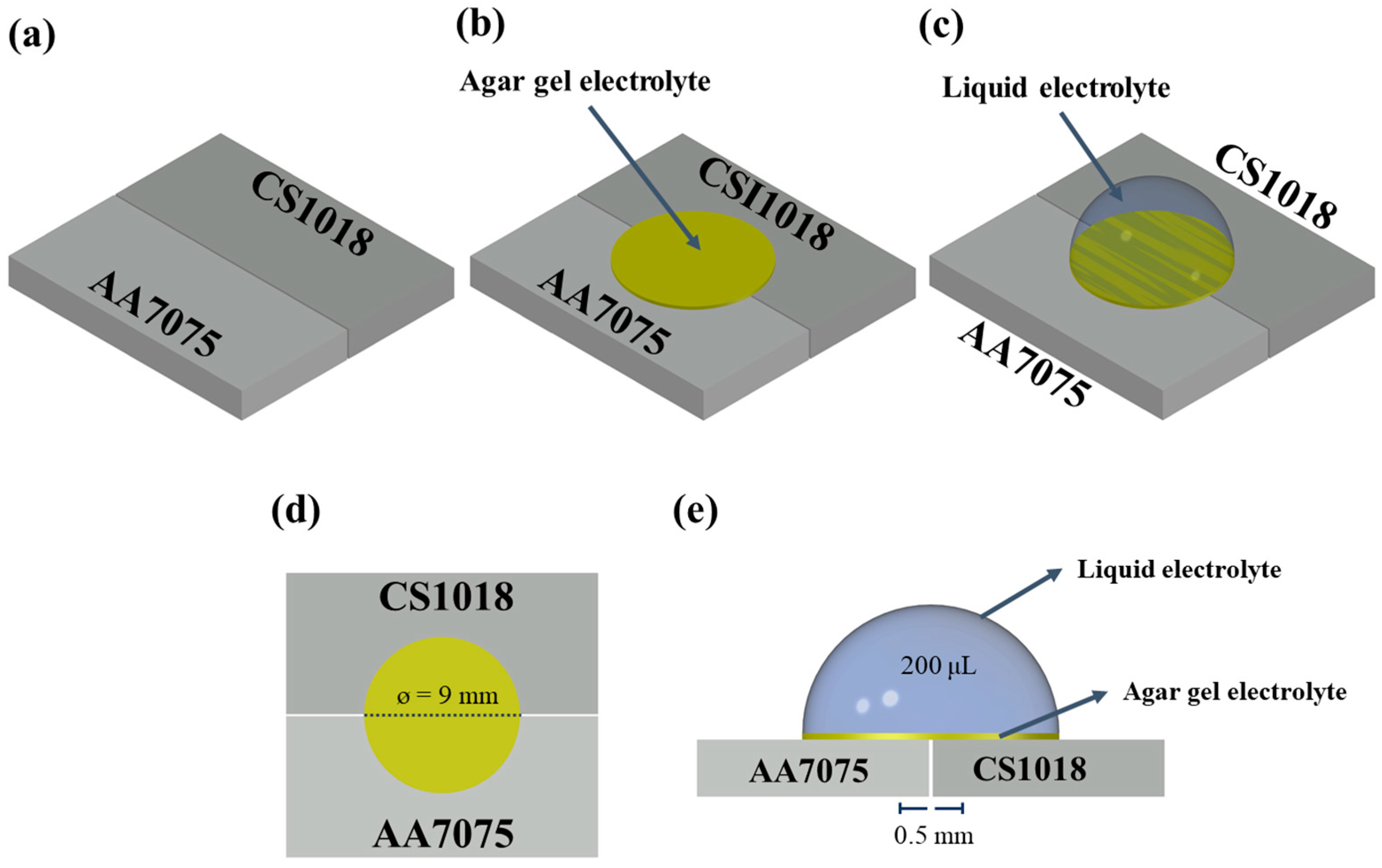
2.1. Galvanic Couple Specimens
2.2. Preparation of the Electrolytes
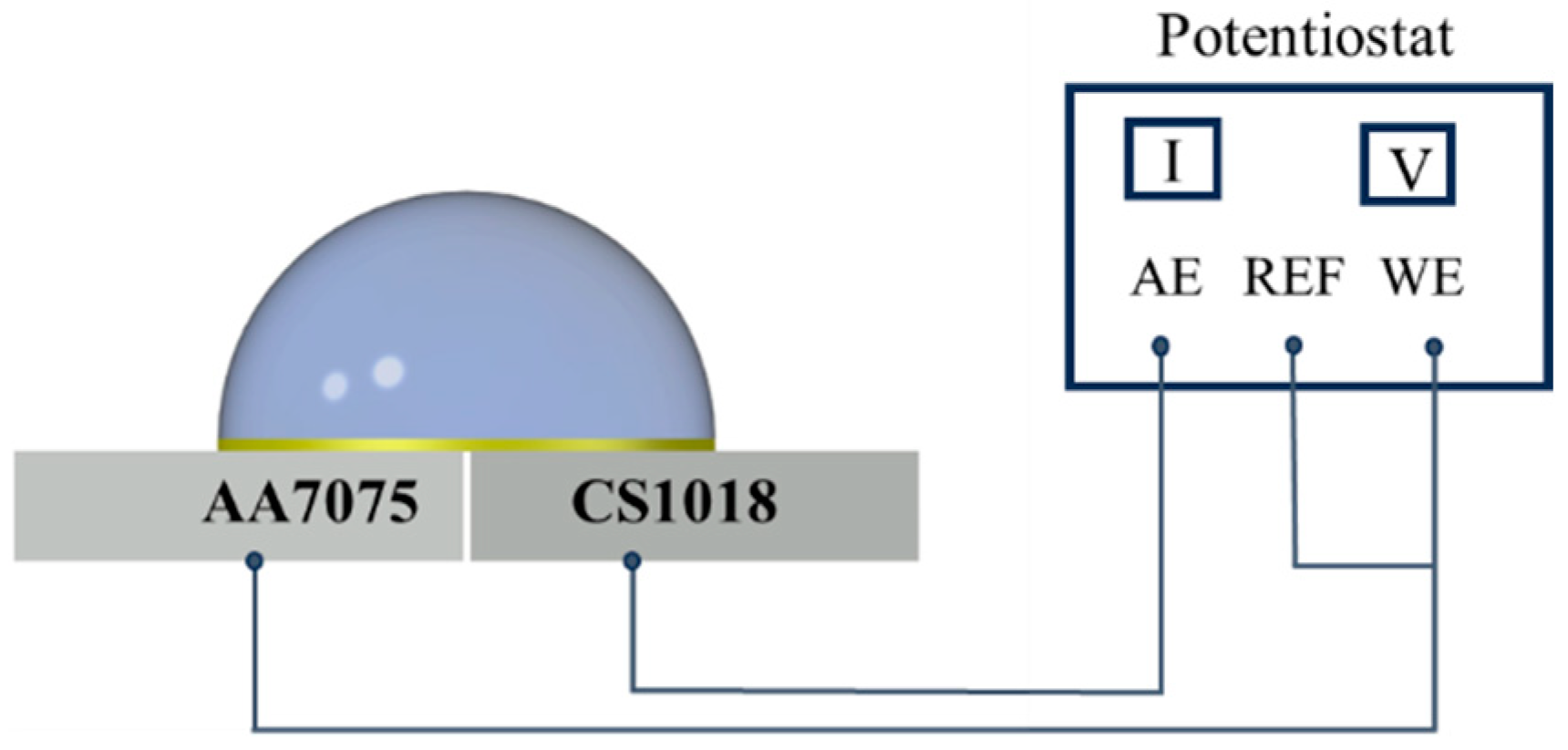
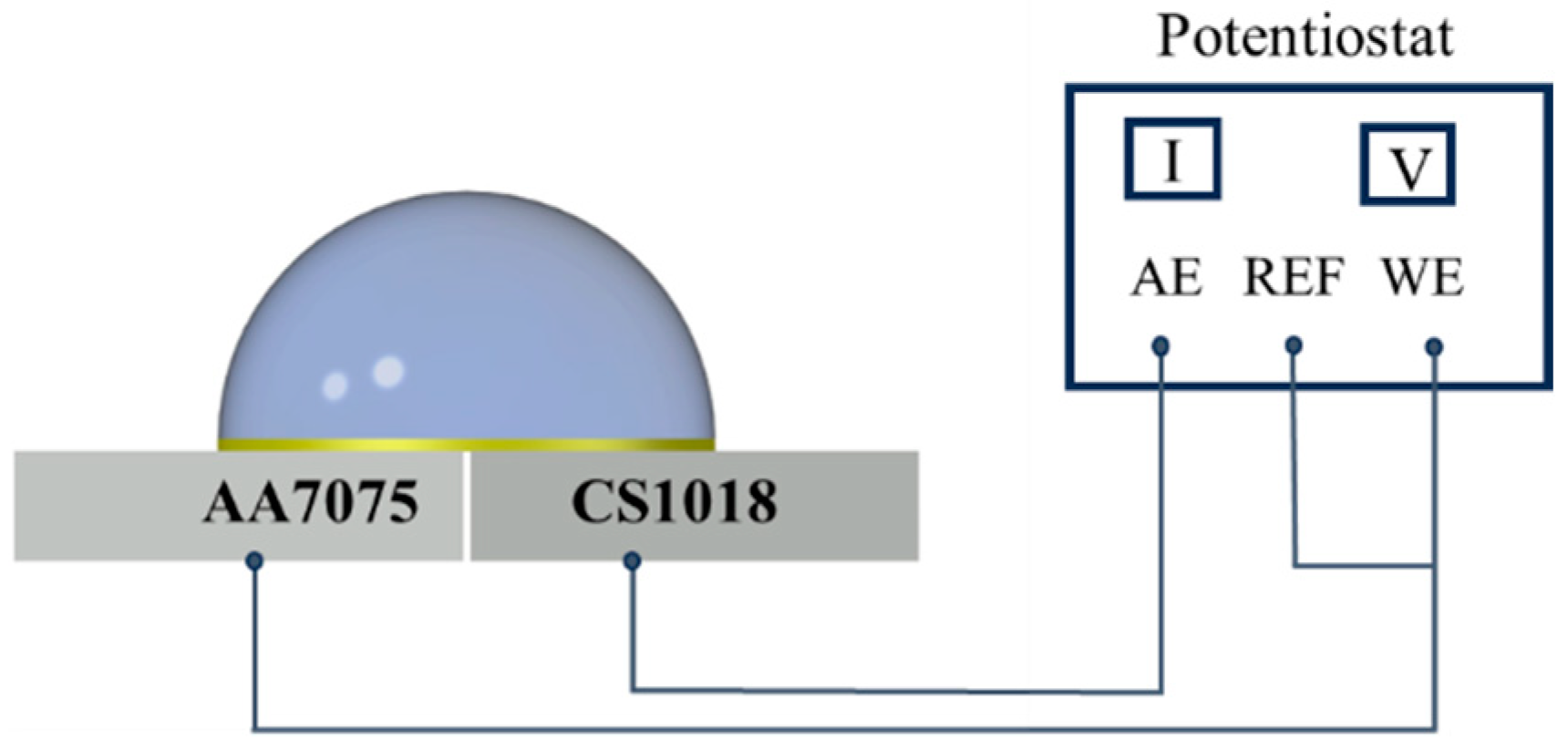
2.3. Experimental Setup of the Dynamic Droplet Process
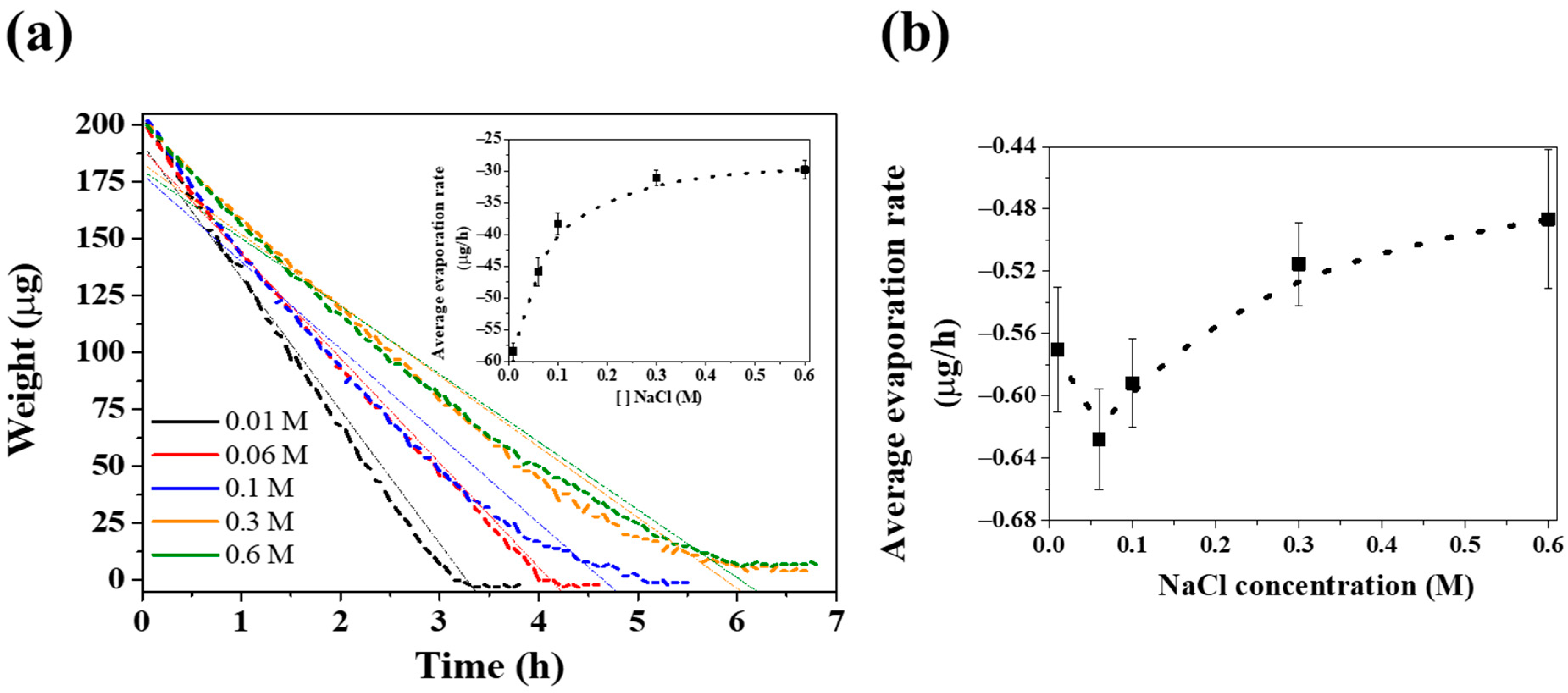
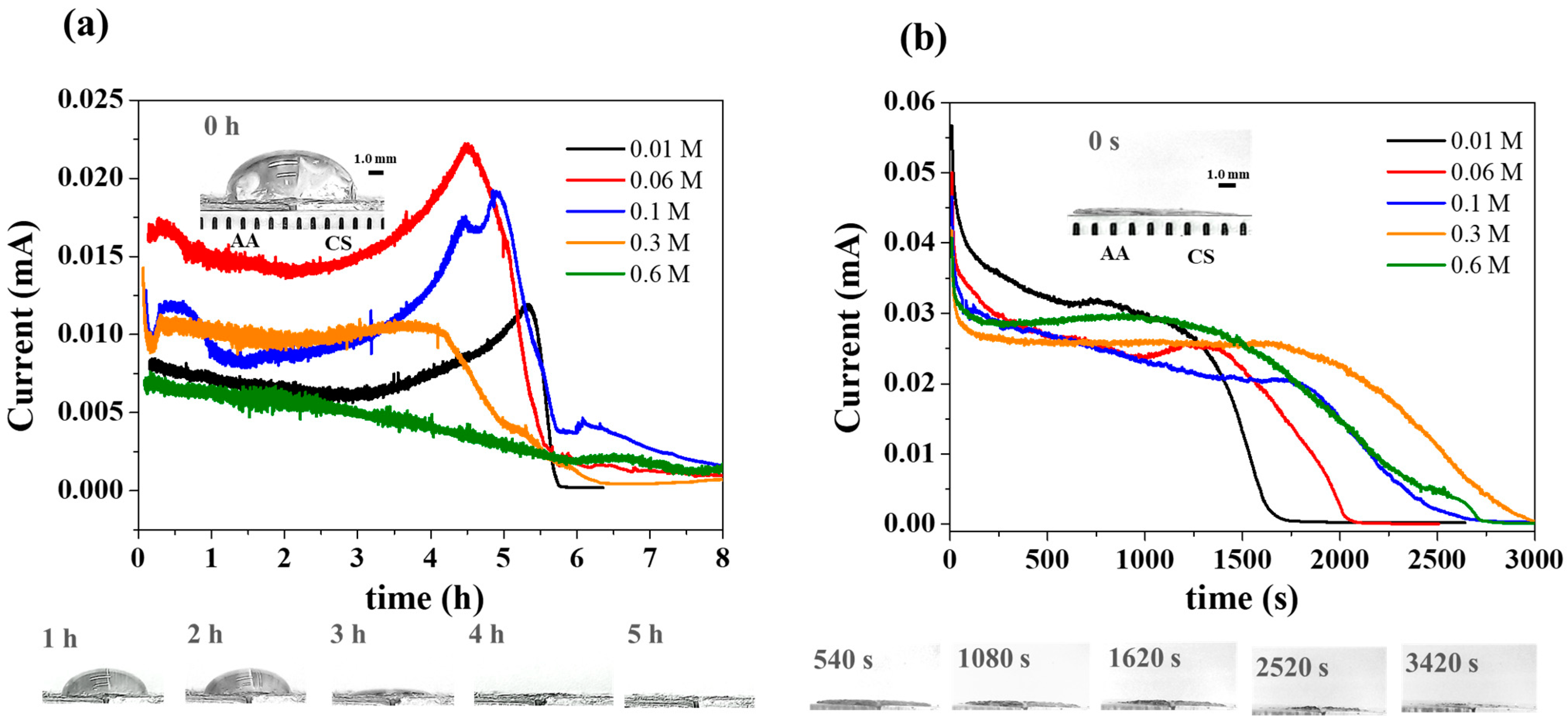
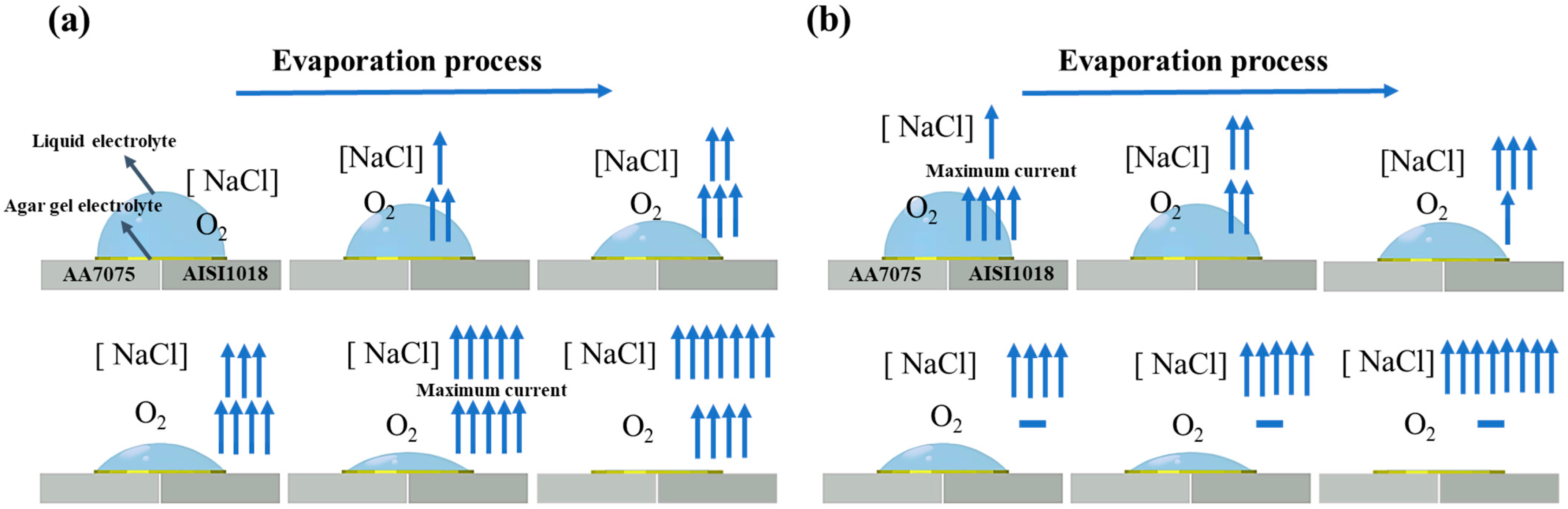
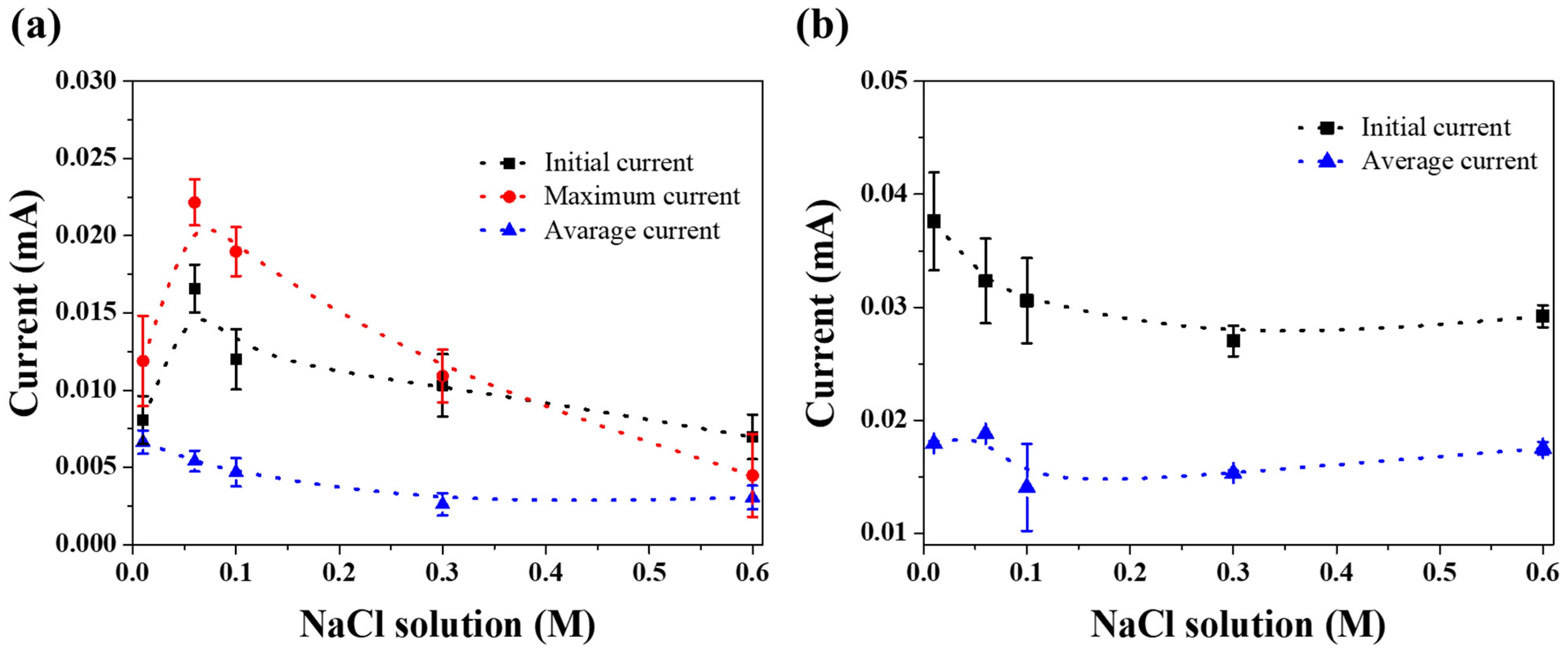
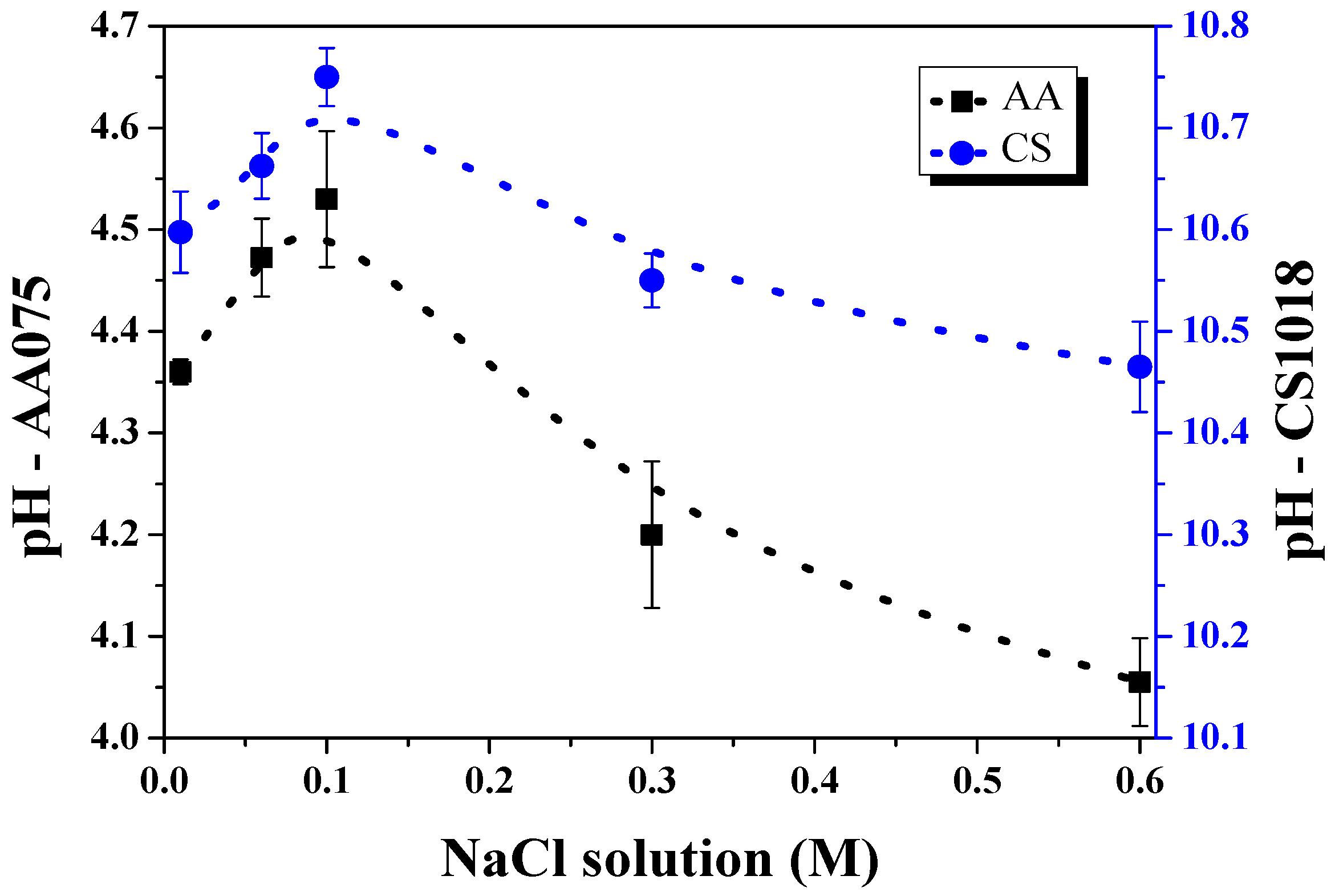
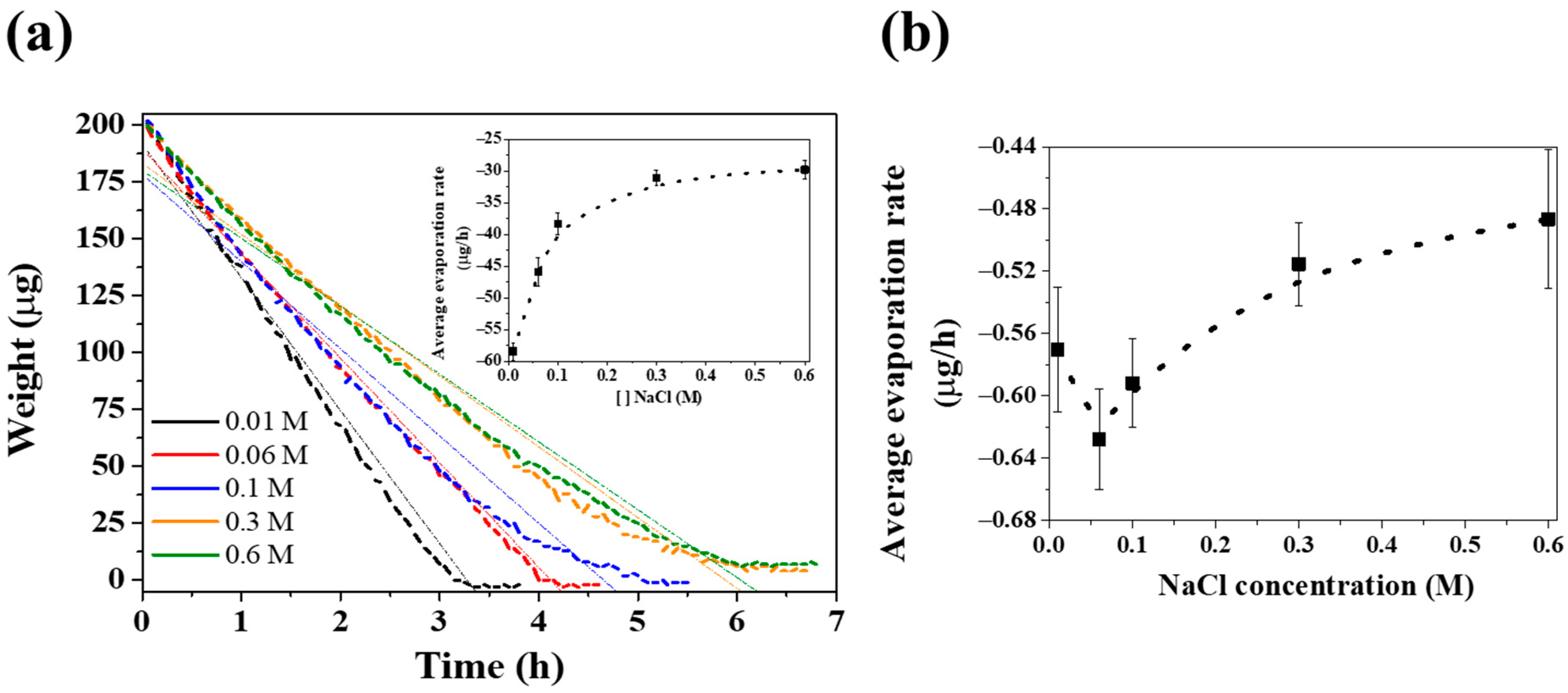
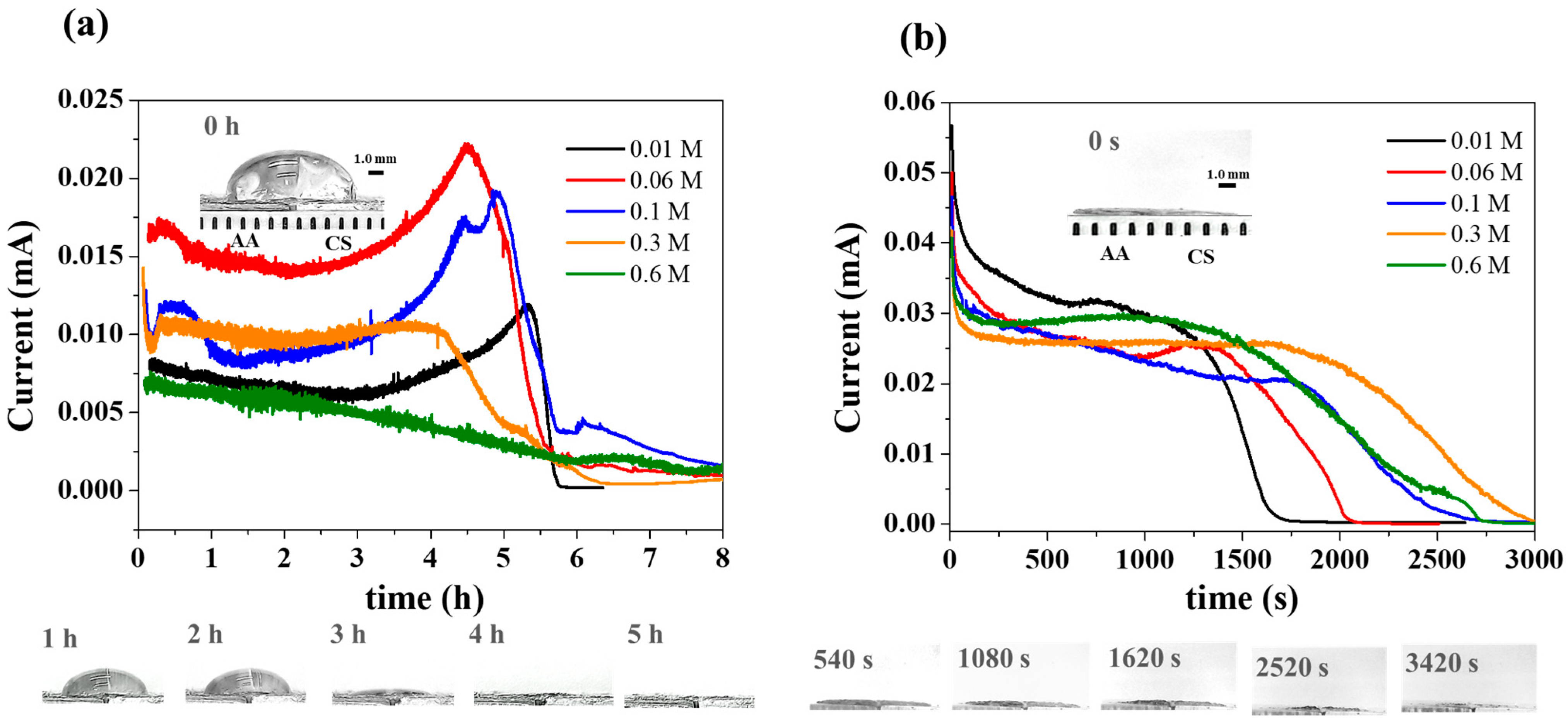
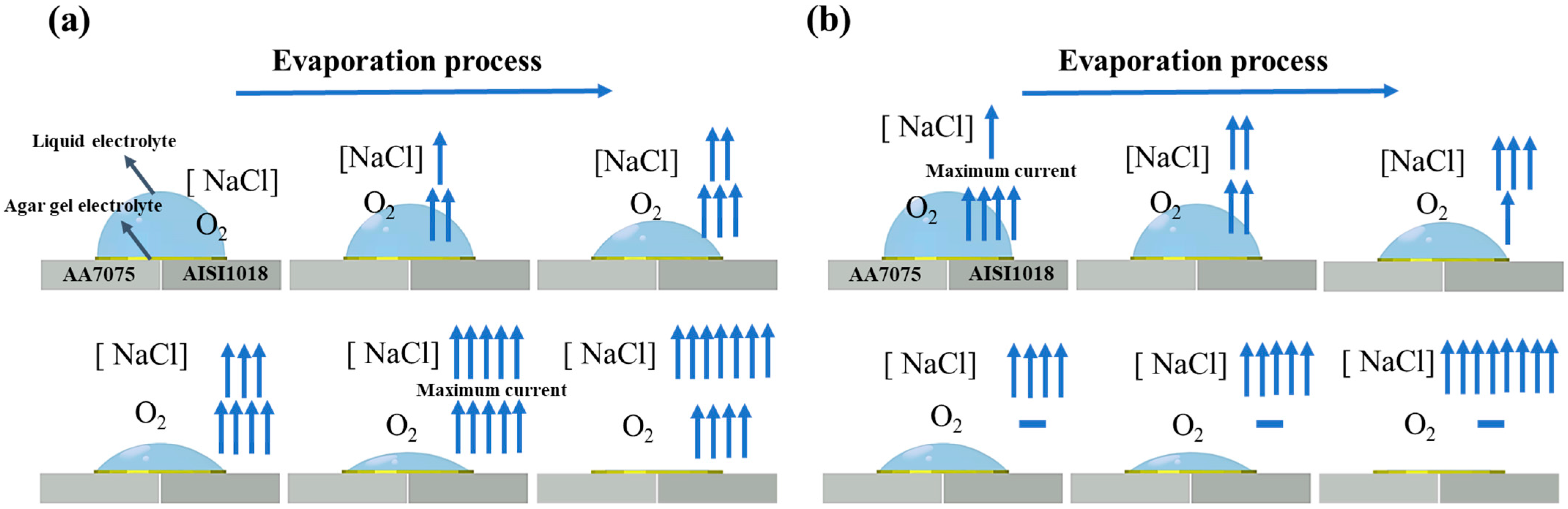
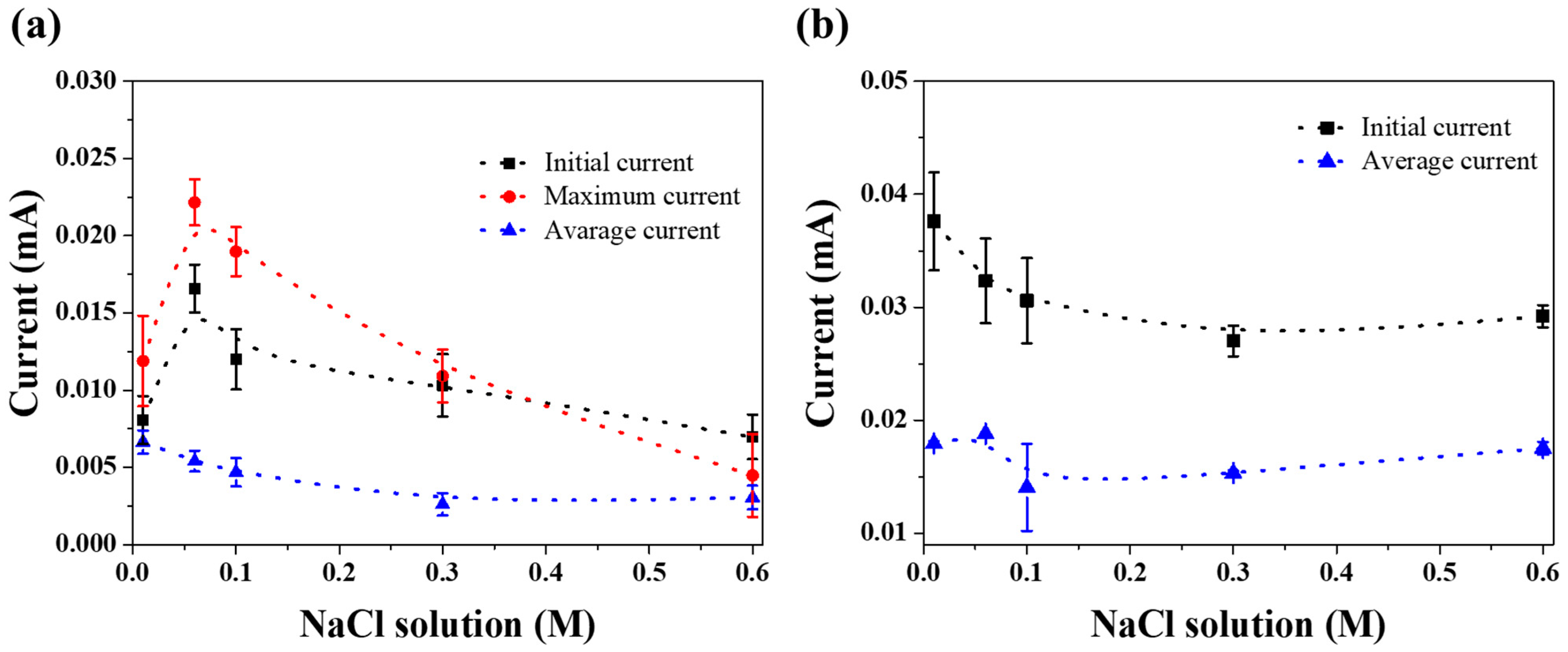
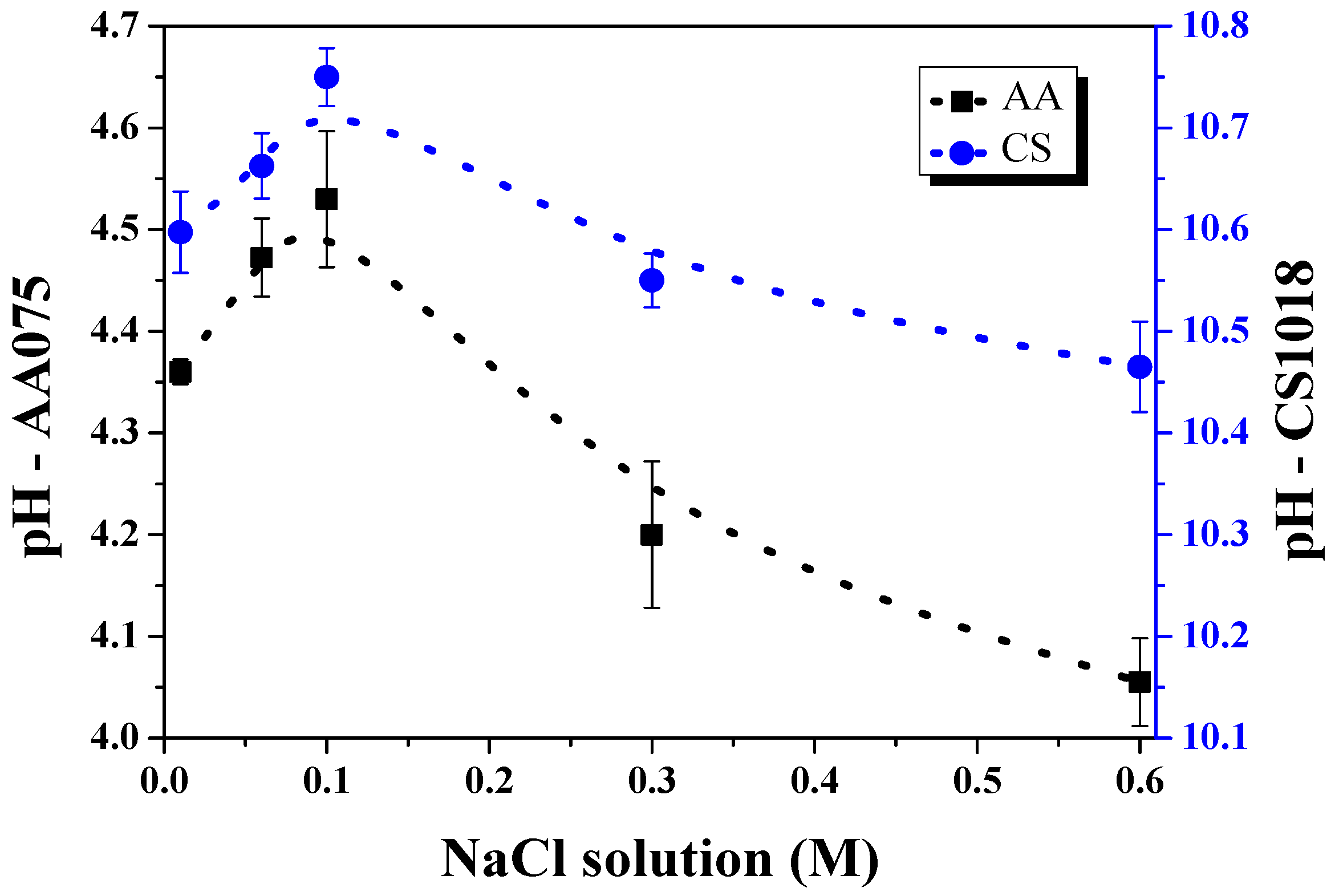
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahmad, Z. Chapter 10—Atmospheric Corrosion. In Principles of Corrosion Engineering and Corrosion Control; Ahmad, Z., Ed.; Butterworth-Heinemann: Oxford, UK, 2006; pp. 550–575. [Google Scholar] [CrossRef]
- Alcántara, J.; de la Fuente, D.; Chico, B.; Simancas, J.; Díaz, I.; Morcillo, M. Marine Atmospheric Corrosion of Carbon Steel: A Review. Materials 2017, 10, 406. [Google Scholar] [CrossRef]
- Popov, B.N. Chapter 10—Atmospheric Corrosion. In Corrosion Engineering; Popov, B.N., Ed.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 451–480. [Google Scholar] [CrossRef]
- Tomashov, N.D. Development of the Electrochemical Theory of Metallic Corrosion. Corrosion 1964, 20, 7t–14t. [Google Scholar] [CrossRef]
- Adams, F.V.; Akinwamide, S.O.; Obadele, B.; Olubambi, P.A. Comparison Study on the Corrosion Behavior of Aluminum Alloys in Different Acidic Media. Mater. Today Proc. 2021, 38, 1040–1043. [Google Scholar] [CrossRef]
- Frankel, G.S. Pitting Corrosion. In Corrosion: Fundamentals, Testing, and Protection; ASM International: Almere, The Netherlands, 2003; pp. 236–241. [Google Scholar] [CrossRef]
- Jinlong, L.; Tongxiang, L.; Chen, W.; Ting, G. The Passive Film Characteristics of Several Plastic Deformation 2099 Al–Li Alloy. J. Alloys Compd. 2016, 662, 143–149. [Google Scholar] [CrossRef]
- Liu, Y.; Meng, G.Z.; Cheng, Y.F. Electronic Structure and Pitting Behavior of 3003 Aluminum Alloy Passivated under Various Conditions. Electrochim. Acta 2009, 54, 4155–4163. [Google Scholar] [CrossRef]
- Vargel, C. Chapter H.5—Resistance of aluminium to atmospheric corrosion. In Corrosion of Aluminium, 2nd ed.; Vargel, C., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 575–589. [Google Scholar] [CrossRef]
- Wang, B.B.; Wang, Z.Y.; Han, W.; Ke, W. Atmospheric Corrosion of Aluminium Alloy 2024-T3 Exposed to Salt Lake Environment in Western China. Corros. Sci. 2012, 59, 63–70. [Google Scholar] [CrossRef]
- Akid, R.; Mills, D.J. A Comparison between Conventional Macroscopic and Novel Microscopic Scanning Electrochemical Methods to Evaluate Galvanic Corrosion. Corros. Sci. 2001, 43, 1203–1216. [Google Scholar] [CrossRef]
- Mao, Y.; Zhu, Y.; Deng, C.M.; Sun, S.; Xia, D.H. Analysis of Localized Corrosion Mechanism of 2024 Aluminum Alloy at a Simulated Marine Splash Zone. Eng. Fail. Anal. 2022, 142, 106759. [Google Scholar] [CrossRef]
- Gao, M.; Sun, Z.H.; Yan, W.; Tang, Z.H.; Zhang, Y.N. Influence of Solid Particles Deposition on the Initial Atmospheric Corrosion of 7B04 Aluminum Alloy. Mater. Today Commun. 2022, 31, 103562. [Google Scholar] [CrossRef]
- Dan, Z.; Takigawa, S.; Muto, I.; Hara, N. Applicability of Constant Dew Point Corrosion Tests for Evaluating Atmospheric Corrosion of Aluminium Alloys. Corros. Sci. 2011, 53, 2006–2014. [Google Scholar] [CrossRef]
- Zhang, S.; Zhang, T.; He, Y.; Liu, D.; Wang, J.; Du, X.; Ma, B. Long-Term Atmospheric Corrosion of Aluminum Alloy 2024-T4 in Coastal Environment: Surface and Sectional Corrosion Behavior. J. Alloys Compd. 2019, 789, 460–471. [Google Scholar] [CrossRef]
- Vera, R.; Delgado, D.; Rosales, B.M. Effect of Atmospheric Pollutants on the Corrosion of High Power Electrical Conductors: Part 1. Aluminium and AA6201 Alloy. Corros. Sci. 2006, 48, 2882–2900. [Google Scholar] [CrossRef]
- Cao, M.; Liu, L.; Yu, Z.; Fan, L.; Li, Y.; Wang, F. Electrochemical Corrosion Behavior of 2A02 Al Alloy under an Accelerated Simulation Marine Atmospheric Environment. J. Mater. Sci. Technol. 2019, 35, 651–659. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Z.; Ke, W. Study on Influence of Native Oxide and Corrosion Products on Atmospheric Corrosion of Pure Al. Corros. Sci. 2014, 80, 169–176. [Google Scholar] [CrossRef]
- Musa, A.Y.; Mohamad, A.B.; Al-Amiery, A.A.; Tien, L.T. Galvanic Corrosion of Aluminum Alloy (Al2024) and Copper in 1.0M Hydrochloric Acid Solution. Korean J. Chem. Eng. 2012, 29, 818–822. [Google Scholar] [CrossRef]
- Zou, S.; Zhang, Y.; Xu, W.; Wan, Y.; He, C.; Dong, C.; Li, X. Galvanic Corrosion Behavior of Aluminum Alloy (2219 and ZL205A) Coupled to Carbon Fiber-Reinforced Epoxy Composites. Int. J. Electrochem. Sci. 2016, 11, 9625–9633. [Google Scholar] [CrossRef]
- Liu, J.; Huang, X.; Ren, Y.; Wong, L.M.; Liu, H.; Wang, S. Galvanic Corrosion Protection of Al-Alloy in Contact with Carbon Fibre Reinforced Polymer through Plasma Electrolytic Oxidation Treatment. Sci. Rep. 2022, 12, 1–10. [Google Scholar] [CrossRef]
- Mansfield, F.; Hengstenberg, D.H.; Kenkel, J.V. Galvanic Corrosion of Al Alloys I. Effect of Dissimilar Metal. Corrosion 1974, 30, 343–353. [Google Scholar] [CrossRef]
- Kosaba, T.; Muto, I.; Sugawara, Y. Galvanic Corrosion of AA5083/Fe in Diluted Synthetic Seawater: Effect of Anodizing on Local Electrochemistry on and around Al6(Fe,Mn) on Al-Matrix. J. Electrochem. Soc. 2022, 169, 020550. [Google Scholar] [CrossRef]
- Kosaba, T.; Muto, I.; Sugawara, Y. Effect of Anodizing on Galvanic Corrosion Resistance of Al Coupled to Fe or Type 430 Stainless Steel in Diluted Synthetic Seawater. Corros. Sci. 2021, 179, 109145. [Google Scholar] [CrossRef]
- Song, G.; Johannesson, B.; Hapugoda, S.; StJohn, D. Galvanic Corrosion of Magnesium Alloy AZ91D in Contact with an Aluminium Alloy, Steel and Zinc. Corros. Sci. 2004, 46, 955–977. [Google Scholar] [CrossRef]
- Van Den Steen, N.; Simillion, H.; Dolgikh, O.; Terryn, H.; Deconinck, J. An Integrated Modeling Approach for Atmospheric Corrosion in Presence of a Varying Electrolyte Film. Electrochim. Acta 2016, 187, 714–723. [Google Scholar] [CrossRef]
- Street, S.R.; Mi, N.; Cook, A.J.M.C.; Mohammed-Ali, H.B.; Guo, L.; Rayment, T.; Davenport, A.J. Atmospheric Pitting Corrosion of 304L Stainless Steel: The Role of Highly Concentrated Chloride Solutions. Faraday Discuss. 2015, 180, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Tsuru, T.; Tamiya, K.I.; Nishikata, A. Formation and Growth of Micro-Droplets during the Initial Stage of Atmospheric Corrosion. Electrochim. Acta 2004, 49, 2709–2715. [Google Scholar] [CrossRef]
- Azmat, N.S.; Ralston, K.D.; Muddle, B.C.; Cole, I.S. Corrosion of Zn under Acidified Marine Droplets. Corros. Sci. 2011, 53, 1604–1615. [Google Scholar] [CrossRef]
- Tsutsumi, Y.; Nishikata, A.; Tsuru, T. Pitting Corrosion Mechanism of Type 304 Stainless Steel under a Droplet of Chloride Solutions. Corros. Sci. 2007, 49, 1394–1407. [Google Scholar] [CrossRef]
- Nishikata, A.; Ichihara, Y.; Hayashi, Y.; Tsuru, T. Influence of Electrolyte Layer Thickness and PH on the Initial Stage of the Atmospheric Corrosion of Iron. J. Electrochem. Soc. 1997, 144, 1244–1252. [Google Scholar] [CrossRef]
- Tang, X.; Ma, C.; Zhou, X.; Lyu, X.; Li, Q.; Li, Y. Atmospheric Corrosion Local Electrochemical Response to a Dynamic Saline Droplet on Pure Iron. Electrochem. Commun. 2019, 101, 28–34. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, J.; Wang, Y. Micro-Droplets Formation during the Deliquescence of Salt Particles in Atmosphere. Corrosion 2005, 61, 1167–1172. [Google Scholar] [CrossRef]
- Wang, J.; Liang, L.; Jiang, J. The Role of Electrochemical Polarization in Micro-Droplets Formation. Electrochem. Commun. 2008, 10, 1788–1791. [Google Scholar] [CrossRef]
- Tang, X.; Li, J.; Wu, Y.; Hu, H.; Ma, C.R.; Li, Y.; Fan, H. Electrochemical Formation Mechanism of Microdroplets on Pure Iron. Front. Chem. 2021, 9, 610738. [Google Scholar] [CrossRef]
- Bian, L.; Weng, Y.; Li, X. Observation of Micro-Droplets on Metal Surface in Early Atmospheric Corrosion. Electrochem. Commun. 2005, 7, 1033–1038. [Google Scholar] [CrossRef]
- Koushik, B.G.; Van den Steen, N.; Mamme, M.H.; Van Ingelgem, Y.; Terryn, H. Review on Modelling of Corrosion under Droplet Electrolyte for Predicting Atmospheric Corrosion Rate. J. Mater. Sci. Technol. 2021, 62, 254–267. [Google Scholar] [CrossRef]
- Van den Steen, N.; Gonzalez-Garcia, Y.; Mol, J.M.C.; Terryn, H.; Van Ingelgem, Y. Predicting the Effect of Droplet Geometry and Size Distribution on Atmospheric Corrosion. Corros. Sci. 2022, 202, 110308. [Google Scholar] [CrossRef]
- Dolgikh, O.; Bastos, A.C.; Oliveira, A.; Dan, C.; Deconinck, J. Influence of the Electrolyte Film Thickness and NaCl Concentration on the Oxygen Reduction Current on Platinum. Corros. Sci. 2016, 102, 338–347. [Google Scholar] [CrossRef]
- Lyon, S.B.; Wong, C.W.; Ajiboye, P. An Approach to the Modelling of Atmospheric Corrosion; ASTM Technical Publication: Almere, The Netherlands, 1995; pp. 26–37. [Google Scholar] [CrossRef]
- Kadowaki, M.; Katayama, H.; Yamamoto, M. Corrosion Behavior of AA6016/SM490 Galvanic Couple in NaCl-Containing Droplets: Effect of Fe Species on Galvanic Corrosion Acceleration. Corros. Sci. 2023, 218, 111190. [Google Scholar] [CrossRef]
- Tran, V.N.; Ooi, A.; Tada, E.; Nishikata, A. EIS Characteristics of Galvanic Couple of Aluminum Alloy and High-Strength Steel under Thin Solution Films. J. Electrochem. Soc. 2020, 167, 131507. [Google Scholar] [CrossRef]
- Montoya, R.; Ruiz-García, A.G.; Ortiz-Ozuna, A.; Ramírez-Barat, B.; Genesca, J. Acidification of the Electrolyte during the Galvanic Corrosion of AA7075: A Numerical and Experimental Study. Mater. Corros. 2021, 72, 1259–1269. [Google Scholar] [CrossRef]
- Ruiz, A.; Esquivel-Peña, V.; Genesca, J.; Montoya, R. Application of Gel Electrolytes to Validate Transient Models of Galvanic Corrosion Phenomenon. ECS Trans. 2022, 106, 229–234. [Google Scholar] [CrossRef]
- Ruiz-Garcia, A.; Esquivel-Peña, V.; Genesca, J.; Montoya, R. Advances in Galvanic Corrosion of Aluminum Alloys. Electrochim. Acta 2023, 449, 142227. [Google Scholar] [CrossRef]
- Kang, H.; Hwang, S. Short Communication: Electrochemical Characterization of the Hydrophobic Interaction and the Natural Convection within Agarose Gel. Int. J. Electrochem. Sci. 2015, 10, 9706–9713. [Google Scholar] [CrossRef]
- Bergstad, M.; Or, D.; Withers, P.J.; Shokri, N. Retracted: The Influence of NaCl Concentration on Salt Precipitation in Heterogeneous Porous Media. Water Resour. Res. 2017, 53, 1702–1712. [Google Scholar] [CrossRef]
- Sghaier, N.; Prat, M.; Nasrallah, S. Ben on Ions Transport during Drying in a Porous Medium. Transp. Porous Media 2007, 67, 243–274. [Google Scholar] [CrossRef]
- Misyura, S.Y. Evaporation of a Sessile Water Drop and a Drop of Aqueous Salt Solution. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Orlova, E.; Kuznetsov, G.; Feoktistov, D. The Evaporation of the Water-Sodium Chlorides Solution Droplets on the Heated Substrate. EPJ Web Conf. 2014, 76, 01039. [Google Scholar] [CrossRef]
- Asyakina, L.K.; Dyshlyuk, L.S. Study of viscosity of aqueous solutions of natural polysaccharides. Sci. Evol. 2016, 1, 11–19. [Google Scholar] [CrossRef]
- Cui, K.; Zhao, X.; Hu, M.; Yang, C.; Xie, G. Effect of Salt Content on the Evaporation and Cracking of Soil from Heritage Structures. Adv. Mater. Sci. Eng. 2021, 2021, 3213703. [Google Scholar] [CrossRef]
- Pradhan, T.K.; Panigrahi, P.K. Influence of an Adjacent Droplet on Fluid Convection inside an Evaporating Droplet of Binary Mixture. Colloids Surf. A Physicochem. Eng. Asp. 2016, 500, 154–165. [Google Scholar] [CrossRef]
- Stratmann, M.; Streckel, H. On the Atmospheric Corrosion of Metals Which Are Covered with Thin Electrolyte Layers—II. Exp. Results Corros. Sci. 1990, 30, 697–714. [Google Scholar] [CrossRef]
- Monrrabal, G.; Huet, F.; Bautista, A. Electrochemical Noise Measurements on Stainless Steel Using a Gelled Electrolyte. Corros. Sci. 2019, 148, 48–56. [Google Scholar] [CrossRef]
- Qi, R.; Tang, W.; Shi, Y.; Teng, K.; Deng, Y.; Zhang, L.; Zhang, J.; Liu, R. Gel Polymer Electrolyte toward Large-Scale Application of Aqueous Zinc Batteries. Adv. Funct. Mater. 2023, 33, 2306052. [Google Scholar] [CrossRef]
- Dubuisson, E.; Lavie, P.; Dalard, F.; Caire, J.P.; Szunerits, S. Study of the Atmospheric Corrosion of Galvanised Steel in a Micrometric Electrolytic Droplet. Electrochem. Commun. 2006, 8, 911–915. [Google Scholar] [CrossRef]
- Chung, K.W.; Kim, K.B. A Study of the Effect of Concentration Build-up of Electrolyte on the Atmospheric Corrosion of Carbon Steel during Drying. Corros. Sci. 2000, 42, 517–531. [Google Scholar] [CrossRef]
- Ma, Y.; Dong, H.; Li, P.; Wu, B.; Huang, L.; Zhang, L.; Li, C.; Li, J. Galvanic Corrosion of AA5052/304SS Welded Joint with Zn-Based Filler Metal in Marine Engineering. Corros. Sci. 2023, 211, 110912. [Google Scholar] [CrossRef]
- Okonkwo, B.O.; Ming, H.; Wang, J.; Han, E.H.; Rahimi, E.; Davoodi, A.; Hosseinpour, S. A New Method to Determine the Synergistic Effects of Area Ratio and Microstructure on the Galvanic Corrosion of LAS A508/309 L/308 L SS Dissimilar Metals Weld. J. Mater. Sci. Technol. 2021, 78, 38–50. [Google Scholar] [CrossRef]
- Mansfeld, F.; Xiao, H. Electrochemical Noise and Impedance Analysis of Iron in Chloride Media; ASTM Technical Publication: Almere, The Netherlands, 1996; Volume 1277, pp. 59–78. [Google Scholar] [CrossRef]
- Pistorius, P.C. The Effect of Some Fundamental Aspects of the Pitting Corrosion of Stainless Steel on Electrochemical Noise Measurements; ASTM Technical Publication: Almere, The Netherlands, 1996; Volume 1277, pp. 343–358. [Google Scholar] [CrossRef]
- Blasco-Tamarit, E.; Igual-Muñoz, A.; Antón, J.G. Effect of Temperature on the Galvanic Corrosion of a High Alloyed Austenitic Stainless Steel in Its Welded and Non-Welded Condition in LiBr Solutions. Corros. Sci. 2007, 49, 4472–4490. [Google Scholar] [CrossRef]
- Maier, B.; Frankel, G.S. Pitting Corrosion of Bare Stainless Steel 304 under Chloride Solution Droplets. J. Electrochem. Soc. 2010, 157, C302. [Google Scholar] [CrossRef]
- Kadowaki, M.; Katayama, H.; Yamamoto, M. Galvanic Current Analysis of AA6016/SM490 Couple Using Experimental and Numerical Simulation Data in Various NaCl Solutions. Corros. Sci. 2023, 211, 110918. [Google Scholar] [CrossRef]
- Xie, J.; Zhang, J.; You, Z.; Liu, S.; Guan, K.; Wu, R.; Wang, J.; Feng, J. Towards Developing Mg Alloys with Simultaneously Improved Strength and Corrosion Resistance via RE Alloying. J. Magnes. Alloys 2021, 9, 41–56. [Google Scholar] [CrossRef]
- Lebouil, S.; Duboin, A.; Monti, F.; Tabeling, P.; Volovitch, P.; Ogle, K. A Novel Approach to On-Line Measurement of Gas Evolution Kinetics: Application to the Negative Difference Effect of Mg in Chloride Solution. Electrochim. Acta 2014, 124, 176–182. [Google Scholar] [CrossRef]
- Yu, Y.; Li, Y. New Insight into the Negative Difference Effect in Aluminium Corrosion Using In-Situ Electrochemical ICP-OES. Corros. Sci. 2020, 168, 108568. [Google Scholar] [CrossRef]
- Laurent, C.; Scenini, F.; Monetta, T.; Bellucci, F.; Curioni, M. The Contribution of Hydrogen Evolution Processes during Corrosion of Aluminium and Aluminium Alloys Investigated by Potentiodynamic Polarisation Coupled with Real-Time Hydrogen Measurement. NPJ Mater. Degrad. 2017, 1, 1–7. [Google Scholar] [CrossRef]
- Wong, D.; Swette, L.; Cocks, F.H. Aluminum Corrosion in Uninhibited Ethylene Glycol-Water Solutions. J. Electrochem. Soc. 1979, 126, 11–15. [Google Scholar] [CrossRef]
- Seri, O.; Furumata, K. Effect of Al-Fe-Si Intermetallic Compound Phases on Initiation and Propagation of Pitting Attacks for Aluminum 1100. Mater. Corros. 2002, 53, 111–120. [Google Scholar] [CrossRef]
- Khedr, M.G.A.; Lashien, A.M.S. The Role of Metal Cations in the Corrosion and Corrosion Inhibition of Aluminium in Aqueous Solutions. Corros. Sci. 1992, 33, 137–151. [Google Scholar] [CrossRef]
- Shinozaki, I.; Sakakibara, Y.; Nakayama, G.; Tada, E.; Ooi, A.; Nishikata, A. Effects of Fe3+ on the Corrosion Behavior of High-Purity Aluminum in Neutral Solutions Containing Cl−. Mater. Trans. 2021, 62, 492–497. [Google Scholar] [CrossRef]
- Tahara, A.; Kodama, T. Potential Distribution Measurement in Galvanic Corrosion of Zn/Fe Couple by Means of Kelvin Probe. Corros. Sci. 2000, 42, 655–673. [Google Scholar] [CrossRef]
- Felde, M. Atmospheric Corrosion of Galvanically Coupled Aluminum Alloys and Carbon Steel; Williams College, Honors Research Projects; The University of Akron: Akron, OH, USA, 2021; p. 1378. Available online: https://ideaexchange.uakron.edu/honors_research_projects/1378 (accessed on 24 January 2024).
- Cui, T.F.; Liu, D.X.; Shi, P.A.; Liu, J.J.; Yi, Y.H.; Zhou, H.L. Effect of NaCl Concentration, PH Value and Tensile Stress on the Galvanic Corrosion Behavior of 5050 Aluminum Alloy. Mater. Corros. 2016, 67, 72–83. [Google Scholar] [CrossRef]
- Snihirova, D.; Lamaka, S.V.; Taryba, M.; Salak, A.N.; Kallip, S.; Zheludkevich, M.L.; Ferreira, M.G.S.; Montemor, M.F. Hydroxyapatite Microparticles as Feedback-Active Reservoirs of Corrosion Inhibitors. ACS Appl. Mater. Interfaces 2010, 2, 3011–3022. [Google Scholar] [CrossRef]
- Esquivel-Peña, V.; Ruiz-García, A.; Genescá, J.; Montoya, R. Easy and Low-Cost Transient Detection of Al(III) Evolution during the Galvanic Corrosion of Aluminum Alloys. J. Solid State Electrochem. 2023, 1, 1–8. [Google Scholar] [CrossRef]
- Guseva, O.; Derose, J.A.; Schmutz, P. Modelling the Early Stage Time Dependence of Localised Corrosion in Aluminium Alloys. Electrochim. Acta 2013, 88, 821–831. [Google Scholar] [CrossRef]
- Foley, R.T.; Nguyen, T.H. The Chemical Nature of Aluminum Corrosion: V. Energy Transfer in Aluminum Dissolution. Proc.—Electrochem. Soc. 1981, 81–88, 27–36. [Google Scholar] [CrossRef]
- Maulana, F.R.; Ningrum, A.S.; Handayani, W.; Ain, C.A.N.; Taufiqulkhakim, M.; Risanti, D.D. Enhancement of Hydrogen Production by Aluminum Corrosion Using HCl and Promoted by NaOH Solution. Key Eng. Mater. 2023, 965, 107–112. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | C | Si | Mn | Cu | Cr | Ni | Mo | N | P | S | Al | Sn | V | Fe |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| wt% | 0.18 | 0.12 | 0.65 | 0.07 | 0.03 | 0.031 | 0.007 | 0.0067 | 0.005 | 0.005 | 0.005 | 0.005 | 0.001 | Bal. |
| Element | Zn | Mg | Cu | Si | Fe | Cr | Mn | Al |
|---|---|---|---|---|---|---|---|---|
| wt% | 6.67 | 2.89 | 0.91 | 0.89 | 0.35 | 0.33 | 0.13 | Bal. |
| NaCl Solution (M) | 0.01 | 0.06 | 0.1 | 0.3 | 0.6 |
|---|---|---|---|---|---|
| (mA) | 0.002671 | 0.007226 | 0.005351 | 0.004087 | 0.001591 |
| (mA) | 0.007163 | 0.009023 | 0.007112 | 0.005129 | 0.003440 |
| LI | 0.373 | 0.801 | 0.752 | 0.797 | 0.463 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montoya, M.; Genesca, J.; Montoya, R. The AA7075–CS1018 Galvanic Couple under Evaporating Droplets. Corros. Mater. Degrad. 2024, 5, 92-108. https://doi.org/10.3390/cmd5010005
Montoya M, Genesca J, Montoya R. The AA7075–CS1018 Galvanic Couple under Evaporating Droplets. Corrosion and Materials Degradation. 2024; 5(1):92-108. https://doi.org/10.3390/cmd5010005
Chicago/Turabian StyleMontoya, Marvin, Juan Genesca, and Rodrigo Montoya. 2024. "The AA7075–CS1018 Galvanic Couple under Evaporating Droplets" Corrosion and Materials Degradation 5, no. 1: 92-108. https://doi.org/10.3390/cmd5010005
APA StyleMontoya, M., Genesca, J., & Montoya, R. (2024). The AA7075–CS1018 Galvanic Couple under Evaporating Droplets. Corrosion and Materials Degradation, 5(1), 92-108. https://doi.org/10.3390/cmd5010005