Metallic Iridium Thin-Films as Model Catalysts for the Electrochemical Oxygen Evolution Reaction (OER)—Morphology and Activity

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results and Discussion
3.1. Morphology of Ir Metal Thin-Film Catalysts
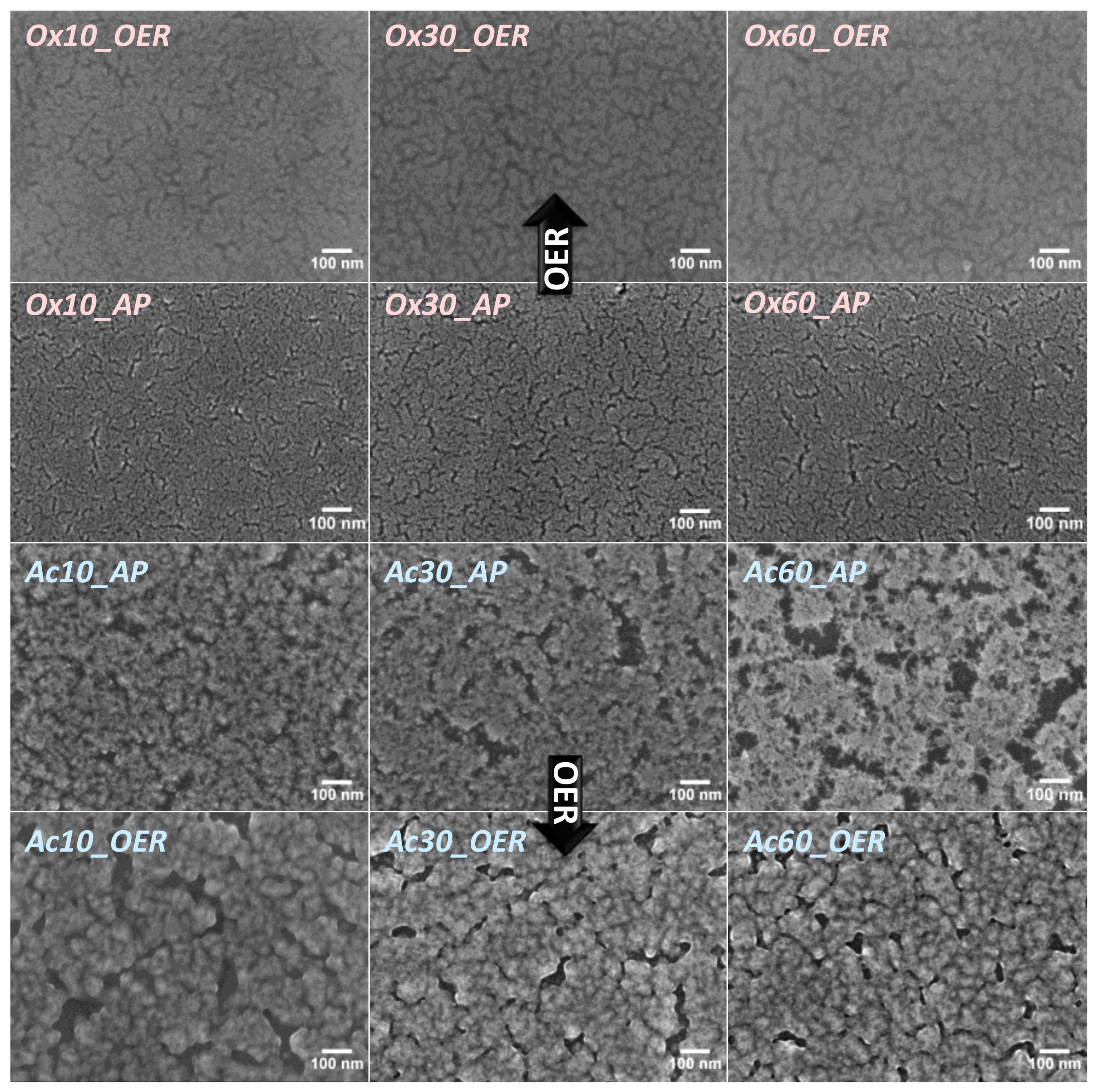
3.1.1. Scanning Electron Microscopy (SEM)
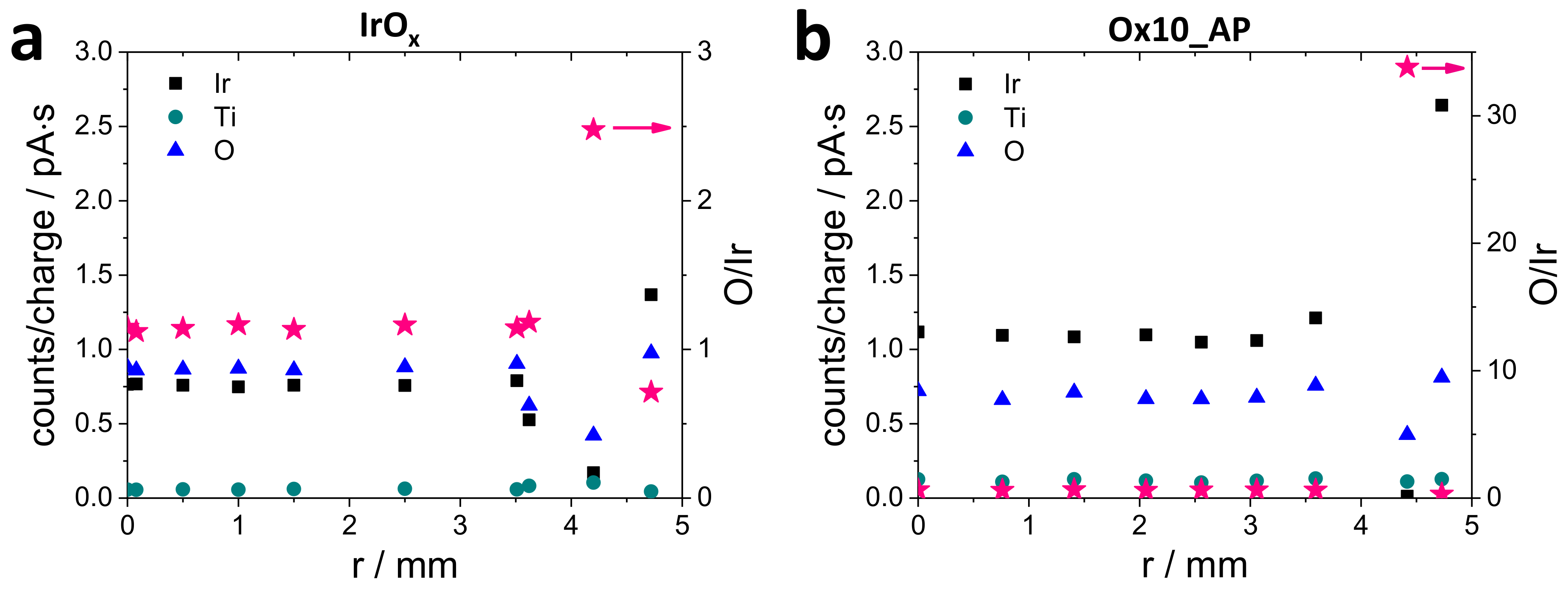
3.1.2. Energy-Dispersive X-ray Spectroscopy Measurements (EDX)
3.2. Structural Characterization of Ir Metal Thin-Film Catalysts
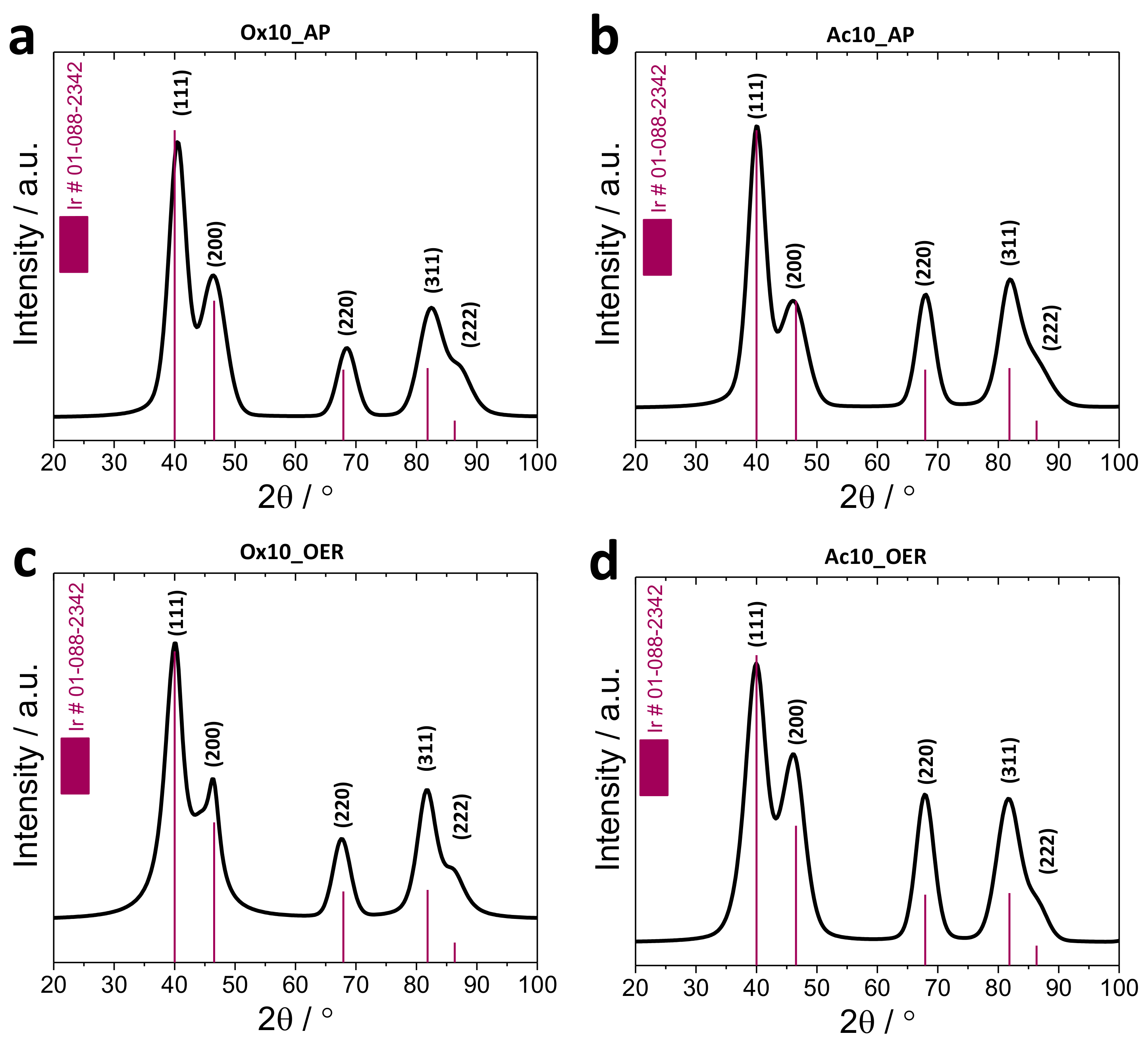
3.2.1. X-Ray Diffraction (XRD)
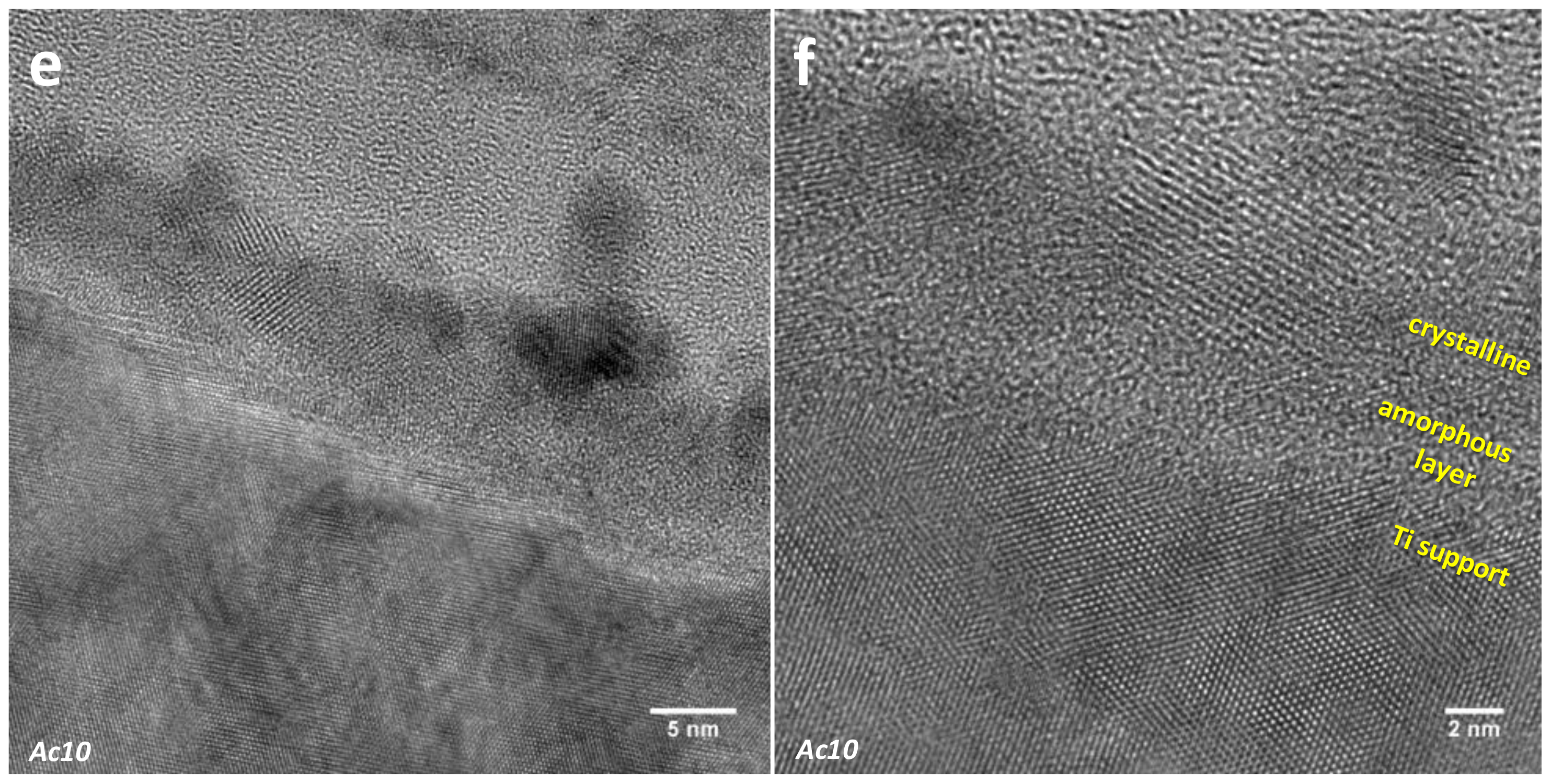
3.2.2. Selected Area Electron Diffraction (SAED)
3.3. Surface Electrochemical Characterization
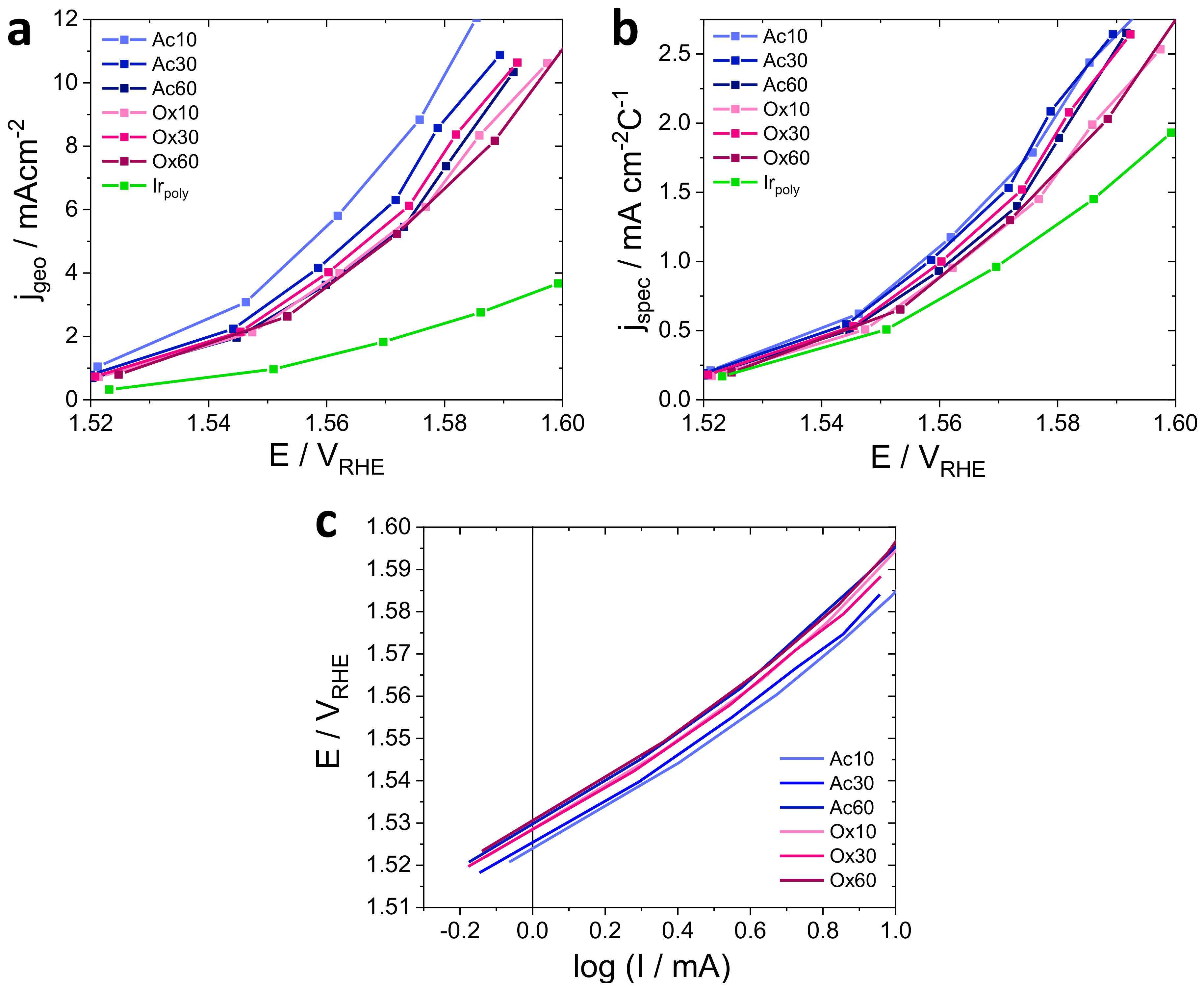
3.3.1. OER Activity
3.3.2. OER Stability
3.3.3. X-Ray Photoelectron Spectroscopy (XPS)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Singh, S.; Jain, S.; Ps, V.; Tiwari, A.K.; Nouni, M.R.; Pandey, J.K.; Goel, S. Hydrogen: A sustainable fuel for future of the transport sector. Renew. Sust. Energ. Rev. 2015, 51, 623–633. [Google Scholar] [CrossRef]
- Carmo, M.; Fritz, D.L.; Mergel, J.; Stolten, D. A comprehensive review on PEM water electrolysis. Int. J. Hydrogen Energ. 2013, 38, 4901–4934. [Google Scholar] [CrossRef]
- McCrory, C.C.L.; Jung, S.; Peters, J.C.; Jaramillo, T.F. Benchmarking heterogeneous electrocatalysts for the oxygen evolution reaction. J. Am. Chem. Soc. 2013, 135, 16977–16987. [Google Scholar] [CrossRef] [PubMed]
- Cherevko, S.; Reier, T.; Zeradjanin, A.R.; Pawolek, Z.; Strasser, P.; Mayrhofer, K.J.J. Stability of nanostructured iridium oxide electrocatalysts during oxygen evolution reaction in acidic environment. Electrochem. Comm. 2014, 48, 81–85. [Google Scholar] [CrossRef]
- Reier, T.; Oezaslan, M.; Strasser, P. Electrocatalytic Oxygen Evolution Reaction (OER) on Ru, Ir, and Pt Catalysts: A Comparative Study of Nanoparticles and Bulk Materials. ACS Catal. 2012, 2, 1765–1772. [Google Scholar] [CrossRef]
- De Oliveira-Sousa, A.; da Silva, M.A.S.; Machado, S.A.S.; Avaca, L.A.; de Lima-Neto, P. Influence of the preparation method on the morphological and electrochemical properties of Ti/IrO2-coated electrodes. Electrochim. Acta 2000, 45, 4467–4473. [Google Scholar] [CrossRef]
- Danilovic, N.; Subbaraman, R.; Chang, K.-C.; Chang, S.H.; Kang, Y.J.; Snyder, J.; Paulikas, A.P.; Strmcnik, D.; Kim, Y.-T.; Myers, D.; et al. Activity-Stability Trends for the Oxygen Evolution Reaction on Monometallic Oxides in Acidic Environments. J. Phys. Chem. Lett. 2014, 5, 2474–2478. [Google Scholar] [CrossRef] [PubMed]
- Cherevko, S.; Geiger, S.; Kasian, O.; Mingers, A.; Mayrhofer, K.J.J. Oxygen evolution activity and stability of iridium in acidic media. Part 2.—Electrochemically grown hydrous iridium oxide. J. Electroanal. Chem. 2016, 774, 102–110. [Google Scholar]
- Bandarenka, A.S.; Koper, M.T.M. Structural and electronic effects in heterogeneous electrocatalysis: Toward a rational design of electrocatalysts. J. Catal. 2013, 308, 11–22. [Google Scholar] [CrossRef]
- Reier, T.; Teschner, D.; Lunkenbein, T.; Bergmann, A.; Selve, S.; Kraehnert, R.; Schlögl, R.; Strasser, P. Electrocatalytic Oxygen Evolution on Iridium Oxide: Uncovering Catalyst-Substrate Interactions and Active Iridium Oxide Species. J. Electrochem. Soc. 2014, 161, F876–F882. [Google Scholar] [CrossRef]
- Özer, E.; Paul, B.; Spöri, C.; Strasser, P. Coupled Inductive Annealing-Electrochemical Setup for Controlled Preparation and Characterization of Alloy Crystal Surface Electrodes. Small Methods 2018, 0, 1800232. [Google Scholar] [CrossRef]
- Gammer, C.; Mangler, C.; Rentenberger, C.; Karnthaler, H.P. Quantitative local profile analysis of nanomaterials by electron diffraction. Scripta Mater. 2010, 63, 312–315. [Google Scholar] [CrossRef]
- PASAD. Available online: http://www.univie.ac.at/pasad/ (accessed on 18 May 2018).
- Pfeifer, V.; Jones, T.E.; Velasco Velez, J.J.; Massue, C.; Greiner, M.T.; Arrigo, R.; Teschner, D.; Girgsdies, F.; Scherzer, M.; Allan, J.; et al. The electronic structure of iridium oxide electrodes active in water splitting. Phys. Chem. Chem. Phys. 2016, 18, 2292–2296. [Google Scholar] [CrossRef] [PubMed]
- Reier, T.; Pawolek, Z.; Cherevko, S.; Bruns, M.; Jones, T.; Teschner, D.; Selve, S.; Bergmann, A.; Nong, H.N.; Schlögl, R.; et al. Molecular Insight in Structure and Activity of Highly Efficient, Low-Ir Ir–Ni Oxide Catalysts for Electrochemical Water Splitting (OER). J. Am. Chem. Soc. 2015, 137, 13031–13040. [Google Scholar] [CrossRef] [PubMed]
- Cherevko, S.; Geiger, S.; Kasian, O.; Mingers, A.; Mayrhofer, K.J.J. Oxygen evolution activity and stability of iridium in acidic media. Part 1.—Metallic iridium. J. Electroanal. Chem. 2016, 773, 69–78. [Google Scholar] [CrossRef]
- Li, T.; Kasian, O.; Cherevko, S.; Zhang, S.; Geiger, S.; Scheu, C.; Felfer, P.; Raabe, D.; Gault, B.; Mayrhofer, K.J.J. Atomic-scale insights into surface species of electrocatalysts in three dimensions. Nat. Catal. 2018, 1, 300–305. [Google Scholar] [CrossRef]
- Sanchez Casalongue, H.G.; Ng, M.L.; Kaya, S.; Friebel, D.; Ogasawara, H.; Nilsson, A. In situ observation of surface species on iridium oxide nanoparticles during the oxygen evolution reaction. Angew. Chem. Int. Ed. 2014, 53, 7169–7172. [Google Scholar] [CrossRef] [PubMed]
- Pfeifer, V.; Jones, T.E.; Velasco Vélez, J.J.; Arrigo, R.; Piccinin, S.; Hävecker, M.; Knop-Gericke, A.; Schlögl, R. In situ observation of reactive oxygen species forming on oxygen-evolving iridium surface. Chem. Sci. 2017, 8, 2143–2249. [Google Scholar] [CrossRef] [PubMed]
- Saveleva, V.A.; Wang, L.; Teschner, D.; Jones, T.; Gago, A.S.; Friedrich, K.A.; Zafeiratos, S.; Schlögl, R.; Savinova, E.R. Operando Evidence for a Universal Oxygen Evolution Mechanism on Thermal and Electrochemical Iridium Oxides. J. Phys. Chem. Lett. 2018, 9, 3154–3160. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Özer, E.; Pawolek, Z.; Kühl, S.; Nong, H.N.; Paul, B.; Selve, S.; Spöri, C.; Bernitzky, C.; Strasser, P. Metallic Iridium Thin-Films as Model Catalysts for the Electrochemical Oxygen Evolution Reaction (OER)—Morphology and Activity. Surfaces 2018, 1, 151-164. https://doi.org/10.3390/surfaces1010012
Özer E, Pawolek Z, Kühl S, Nong HN, Paul B, Selve S, Spöri C, Bernitzky C, Strasser P. Metallic Iridium Thin-Films as Model Catalysts for the Electrochemical Oxygen Evolution Reaction (OER)—Morphology and Activity. Surfaces. 2018; 1(1):151-164. https://doi.org/10.3390/surfaces1010012
Chicago/Turabian StyleÖzer, Ebru, Zarina Pawolek, Stefanie Kühl, Hong Nhan Nong, Benjamin Paul, Sören Selve, Camillo Spöri, Cornelius Bernitzky, and Peter Strasser. 2018. "Metallic Iridium Thin-Films as Model Catalysts for the Electrochemical Oxygen Evolution Reaction (OER)—Morphology and Activity" Surfaces 1, no. 1: 151-164. https://doi.org/10.3390/surfaces1010012
APA StyleÖzer, E., Pawolek, Z., Kühl, S., Nong, H. N., Paul, B., Selve, S., Spöri, C., Bernitzky, C., & Strasser, P. (2018). Metallic Iridium Thin-Films as Model Catalysts for the Electrochemical Oxygen Evolution Reaction (OER)—Morphology and Activity. Surfaces, 1(1), 151-164. https://doi.org/10.3390/surfaces1010012