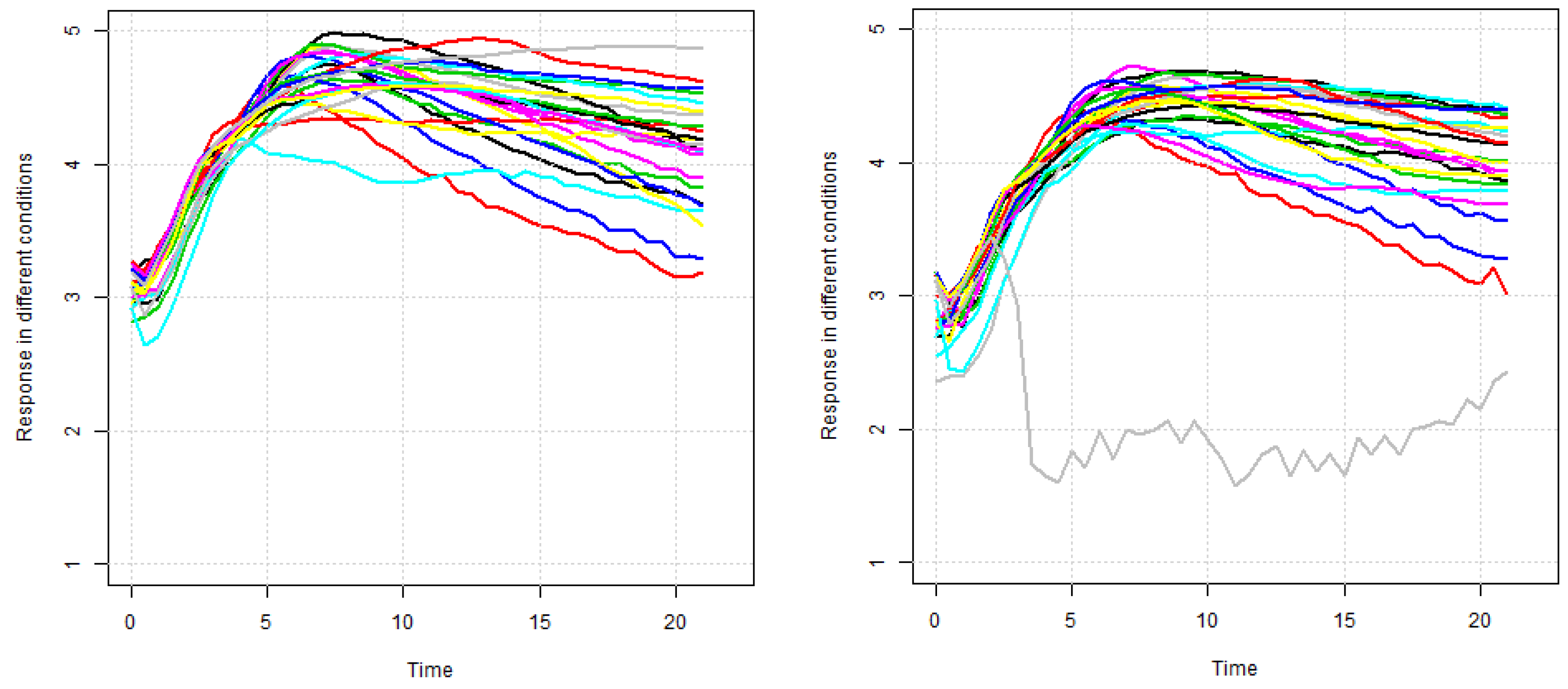
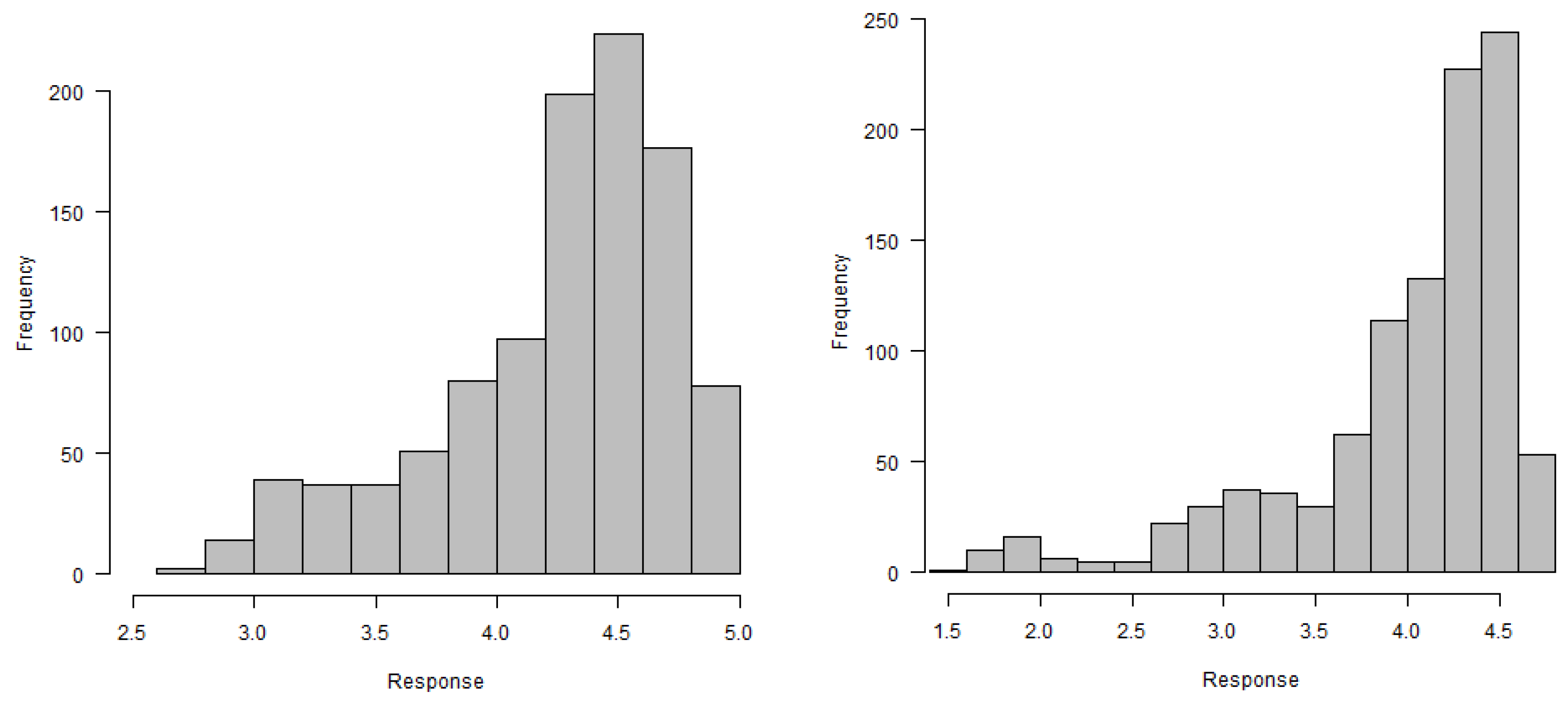
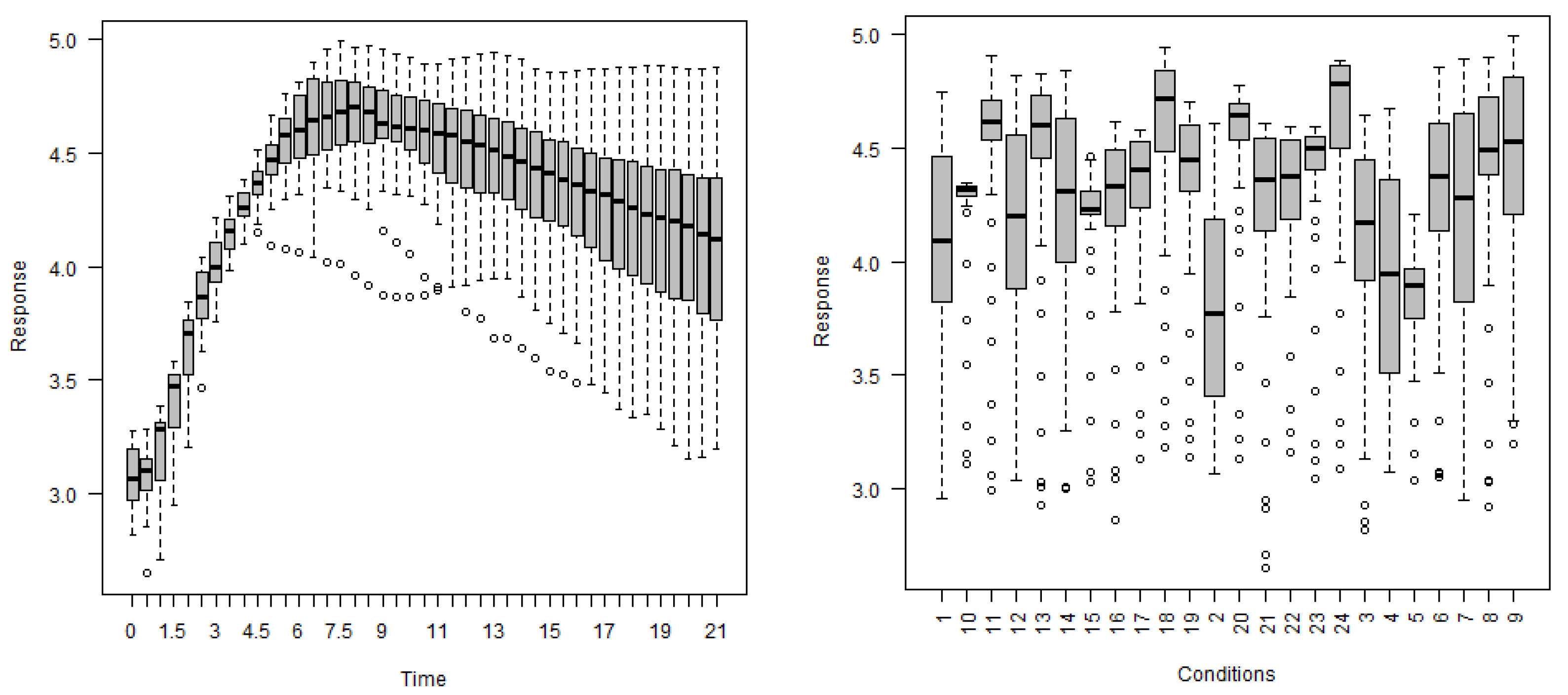
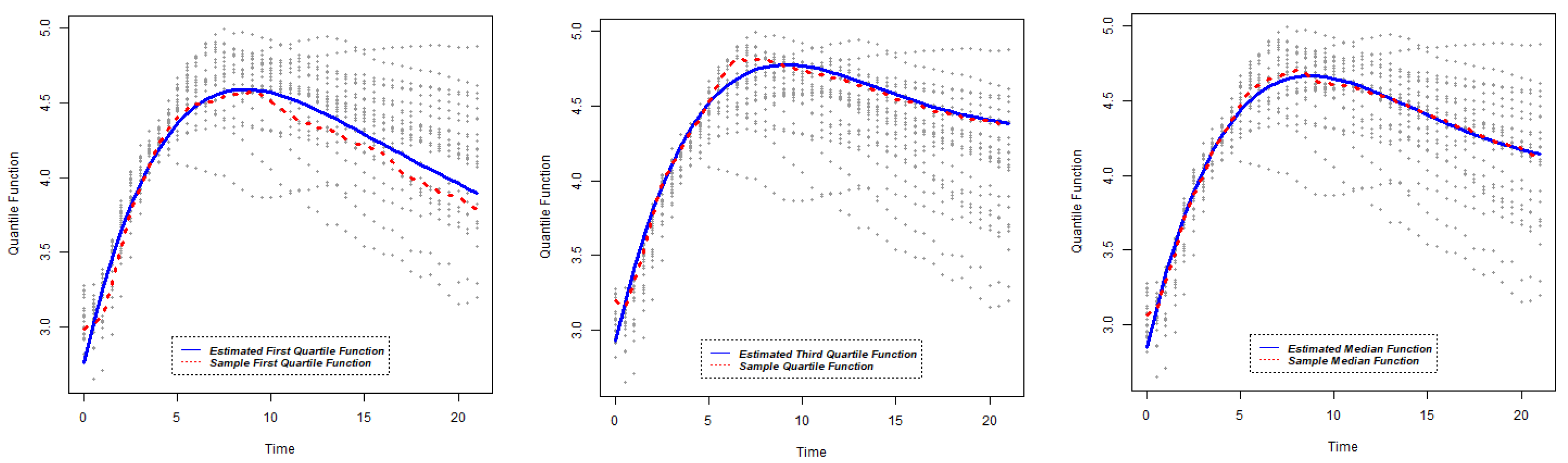
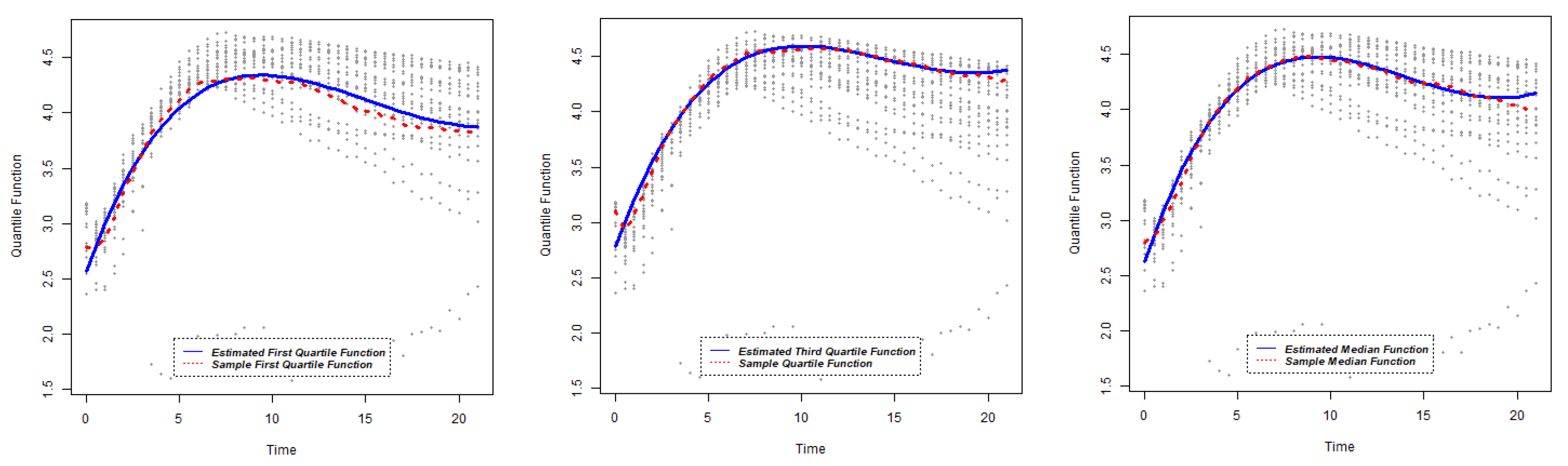
In this section, this research methodology is introduced to analyze the gene expression data. In most cases, for gene expression data, specific characteristics were measured at discrete timepoints and measurements were taken under different biological conditions. As gene expression characteristics were measured over a series of timepoints and repeated under different circumstances, the longitudinal data analysis method can be applied to analyze discrete gene expression data. On the other hand, gene expression measurements show a variety of patterns under different biological conditions over specific times. Therefore, the linear quantile mixed model may be appropriate for the analysis of gene expression data. Let
be the observed measurement of specific gene expression under the i
condition at time
t. To determine the gene trajectory at different quantiles
, the following quantile model for
can be considered.
where,
is the quantile curve at time
t,
is the random noise with zero
th quantile and
N denotes the number of conditions. Note that we are unable to observe
for all timepoints, only at the very specific occasions
at which measurements with errors were taken. Therefore, the observed longitudinal data for a specific gene under i
biological condition consist of the measurements
, where
denotes the number of observed timepoints. Note that, for gene expression data, there is no available covariate. However, there are many methods in the literature that can be used to estimate the quantile function
, including kernel, local polynomial, smoothing splines, regression splines and wavelet-based methods, among others. One simple and straightforward basis is the polynomial basis
, in which the true quantile function of gene trajectory expression is modeled as polynomials of degree
. In this article, the method proposed in Donoho and Johnstone [
15] and further developed in Zhang [
16] is adopted. In this spline method,
is approximated using the linear combination of a set of truncated power basis functions. Given a sequence of
K interior knots
where
T is the end time of observations, the regression spline basis functions of order
p are
. Denoting the vector of
basis functions by
the regression spline smoothing is used to model
using the linear combination of the basis functions
, and the linear quantile mixed model can be written as
where the basis function for the fixed effects parameter is denoted as
and
is considered as basis function for random effects parameter, which could be the
q-dimensional sub-vector of
. In particular, basis functions for the random intercept model and random slope model can be written as follows:
Now, for a longitudinal setup, assuming that
conditioned on random effects,
are independently distributed and this conditional distribution follows the asymmetric Laplace (AL) distribution with location parameter
and scale parameter
, respectively. Therefore, it can be written as
where
denotes the
vector of fixed-effect parameters and
is considered the quantile level. Assume that random effect vectors
are zero
-quantile vectors and independent of the error term
of the model (
). Moreover, assume that random effect vectors
are distributed with the density function
, where
is regarded as the variance–covariance matrix (symmetric positive definite). All parameters depend on the skewness parameter,
. Now, the
th linear quantile mixed model can be written as
where
and i.i.d components of error
follow an asymmetric Laplace distribution. Symbolically,
. Now, in terms of matrix notation,
th linear quantile of response
, denoted as
, can also be expressed as
where,
,
and
.
2.1. Estimation of Parameters
Based on the above discussion, the joint density of
can be written in terms of the
th quantile as follows:
For the random intercept model
, the design matrix for random effects can be written as
. Let,
denote
dimensional Euclidean space. Now, the marginal likelihood can be derived from Equation (5) and written as
Moreover, the marginal log-likelihood function can also be written as
Since the distribution of
is assumed to follow an asymmetric Laplace distribution,
th quantile of
can be estimated using the asymmetric Laplace distribution with location parameter
, common scale
parameter and known skew parameter
. To estimate parameters from joint density
, expressed in Equation (5), it is important to compute the following integral, which is also known as the marginal density of
where,
and
Now, the log-likelihood function for
n conditions can be expressed as
This numerical likelihood can be solved by applying Gaussian quadrature (Gauss–Hermite quadrature or Gauss–Laguerre quadrature) proposed by Geraci and Bottai [
17]. Now, assuming normal random effects
to Equation (9), the Gauss–Hermite quadrature can be applied to approximate the likelihood function with nodes
and weights
, respectively. Integer
M determines the number of points over the real line for each of the
q one-dimensional integrals. The covariance matrix of the random effects is reparameterized by parameters
, i.e.,
, and parameters, characterized by
and
, are denoted by
. Finally, Equation () leads to the following approximate likelihood.
Geraci and Bottai [
17] develop the gradient search (gs) and the derivative free (df) optimization algorithm to maximize likelihood function in Equation (10). This optimization starts with a parameter value and then searches the positive semi-line for a new parameter value where likelihood is larger. This algorithm works until the likelihood change is sufficiently small or less than the pre-specified tolerance (
) level. This algorithm begins estimating by initializing
. The derivative-free optimization algorithm is similar to the gradient search method. This method alternates a loop for
and then updates
.
Now, from the approximate likelihood in Equation (10), the fixed effects parameter
, the random effects parameter
and the error term parameter
can be estimated by using the algorithm given in Geraci and Bottai [
17]. Further, the estimate of quantile function
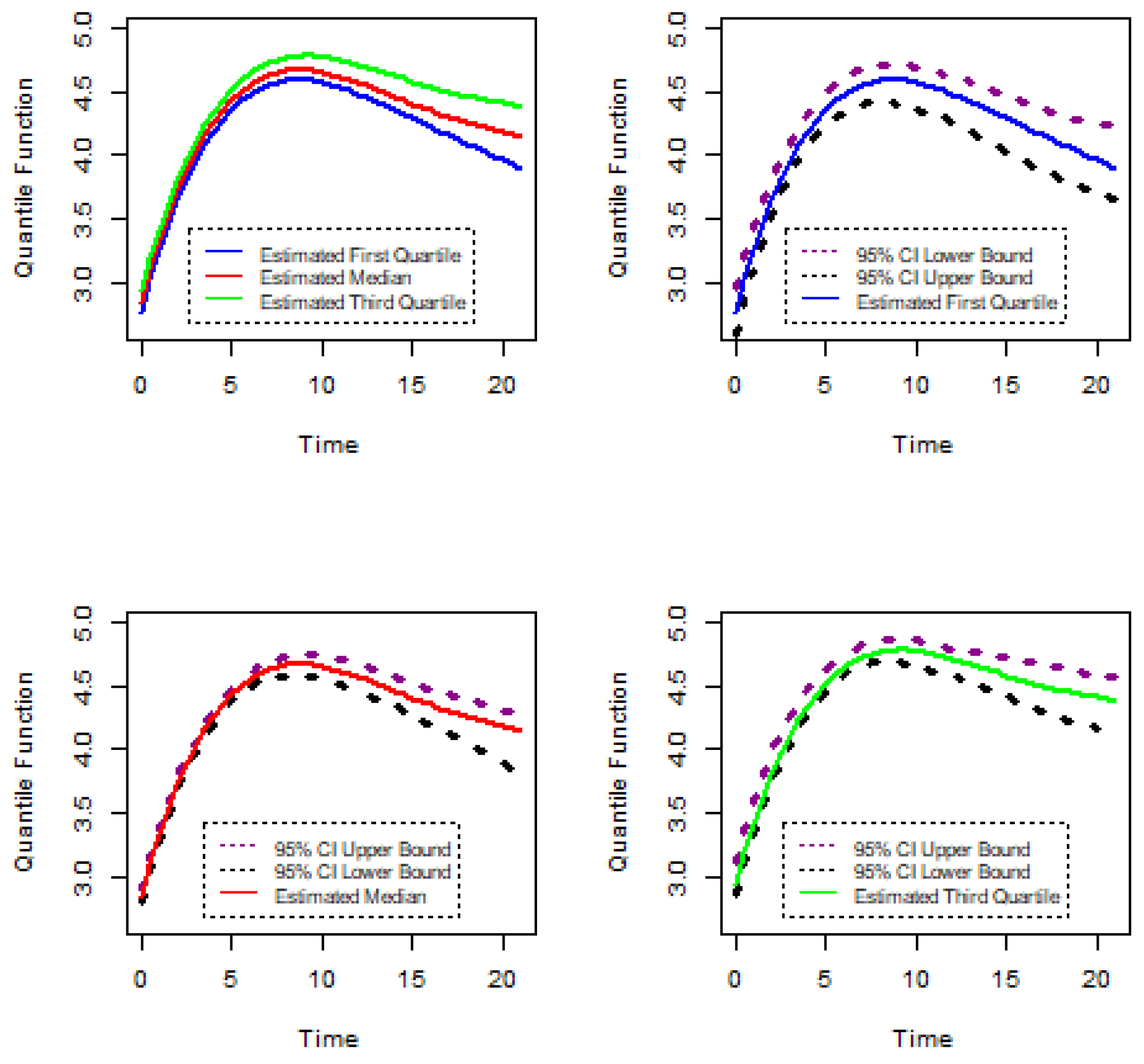
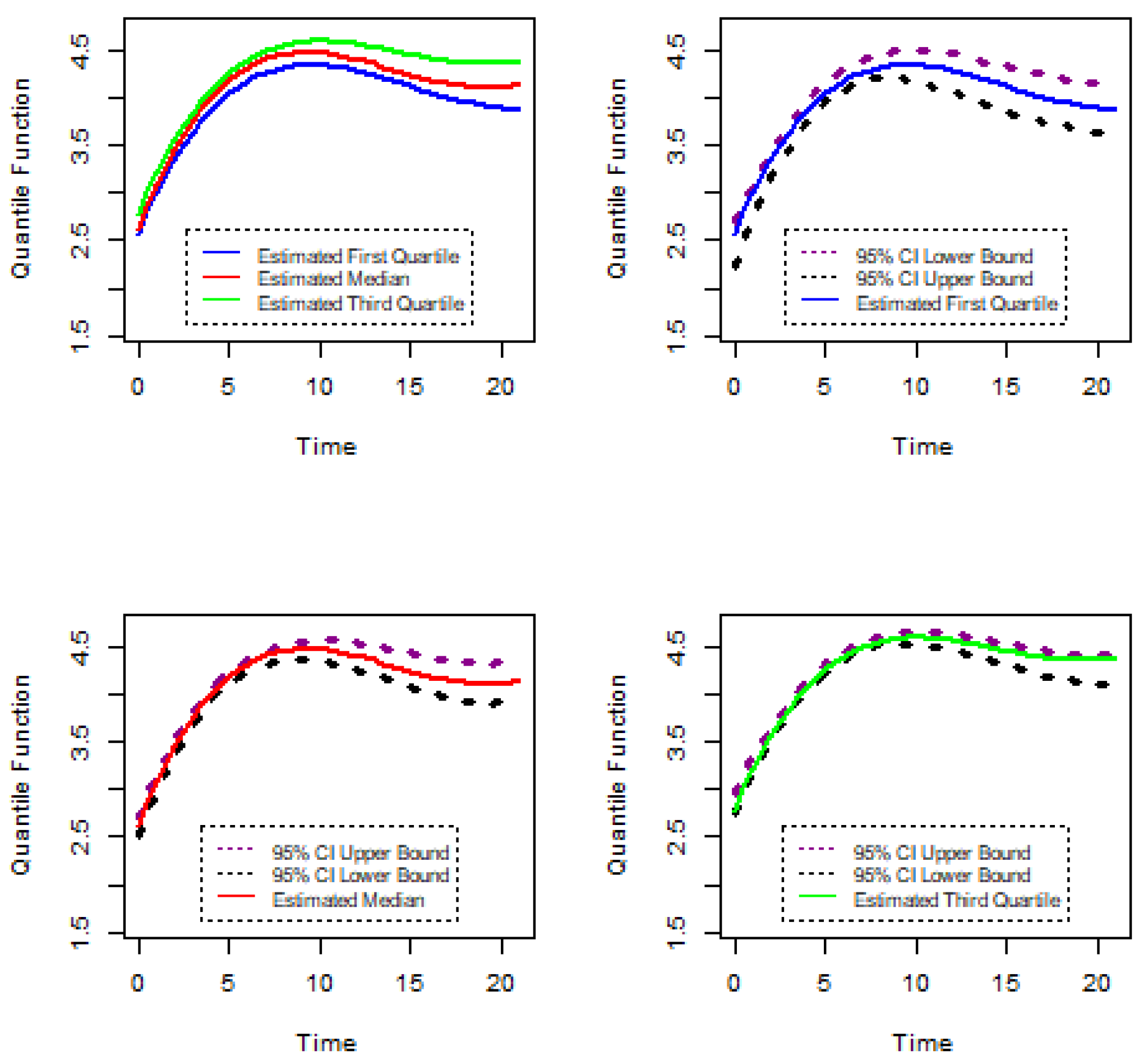
can be written as follows
where
is the estimate of
and
is the estimated best linear predictor of
, which can be expressed as (Geraci and Botai [
17])
where
with
and
with
for
. Furthermore, the asymptotic covariance matrix for
can be derived as
Asymptotically,
with
From the asymptotic normality of
, the approximate
confidence interval for the quantile function
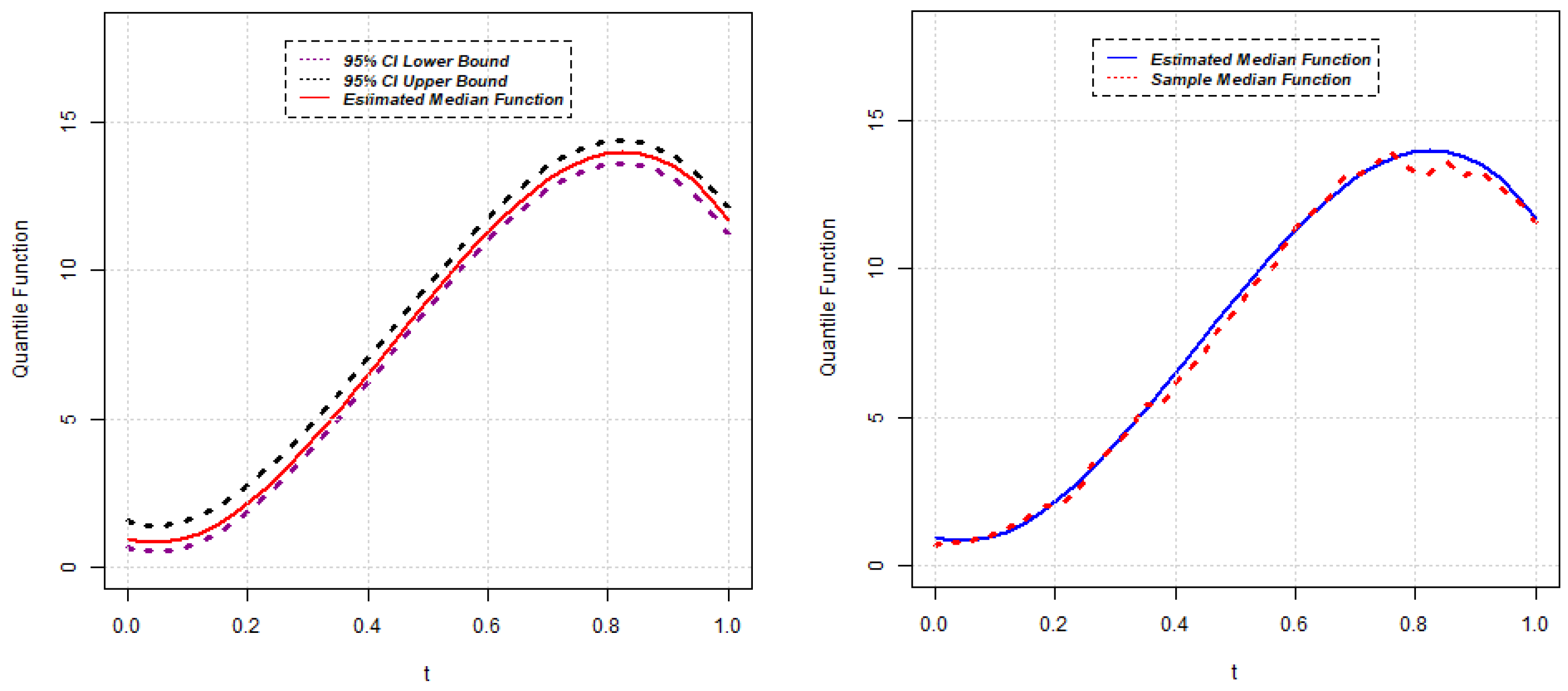
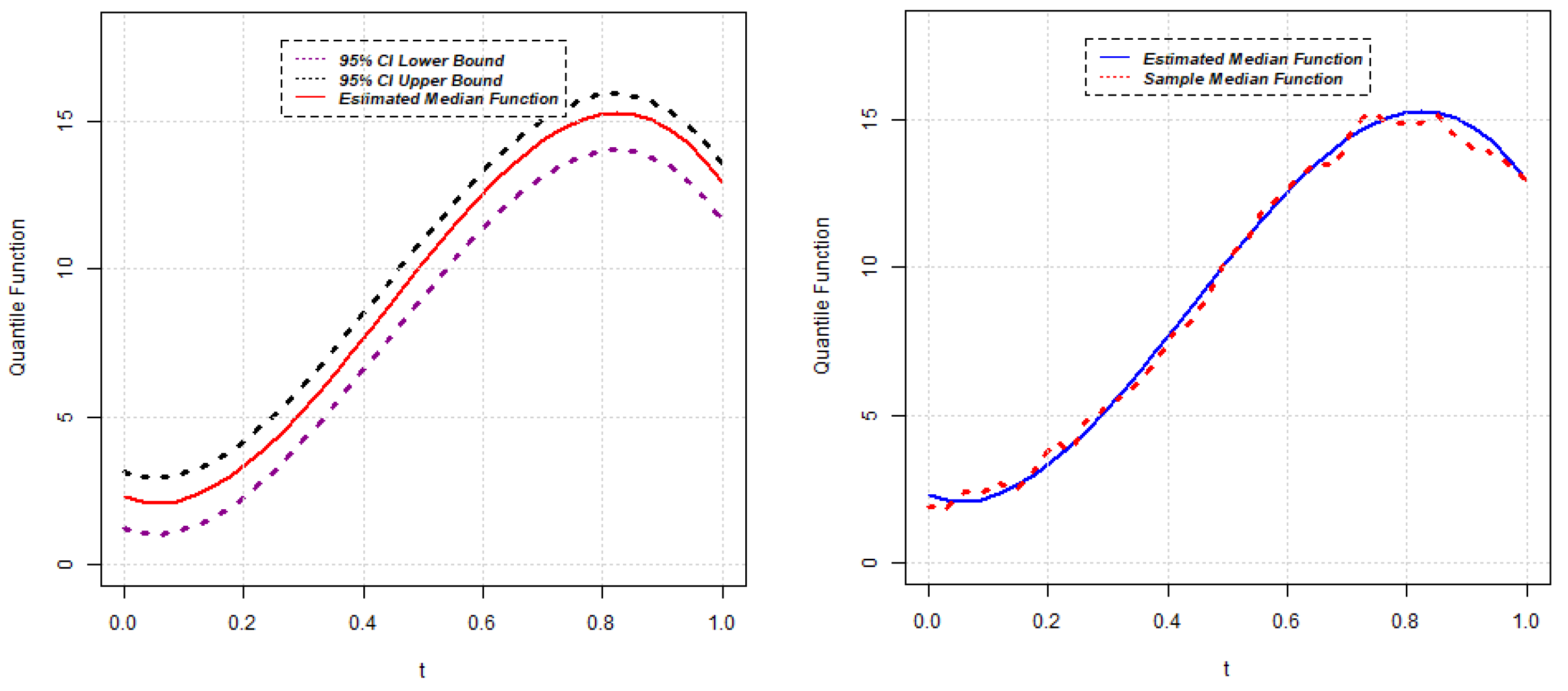
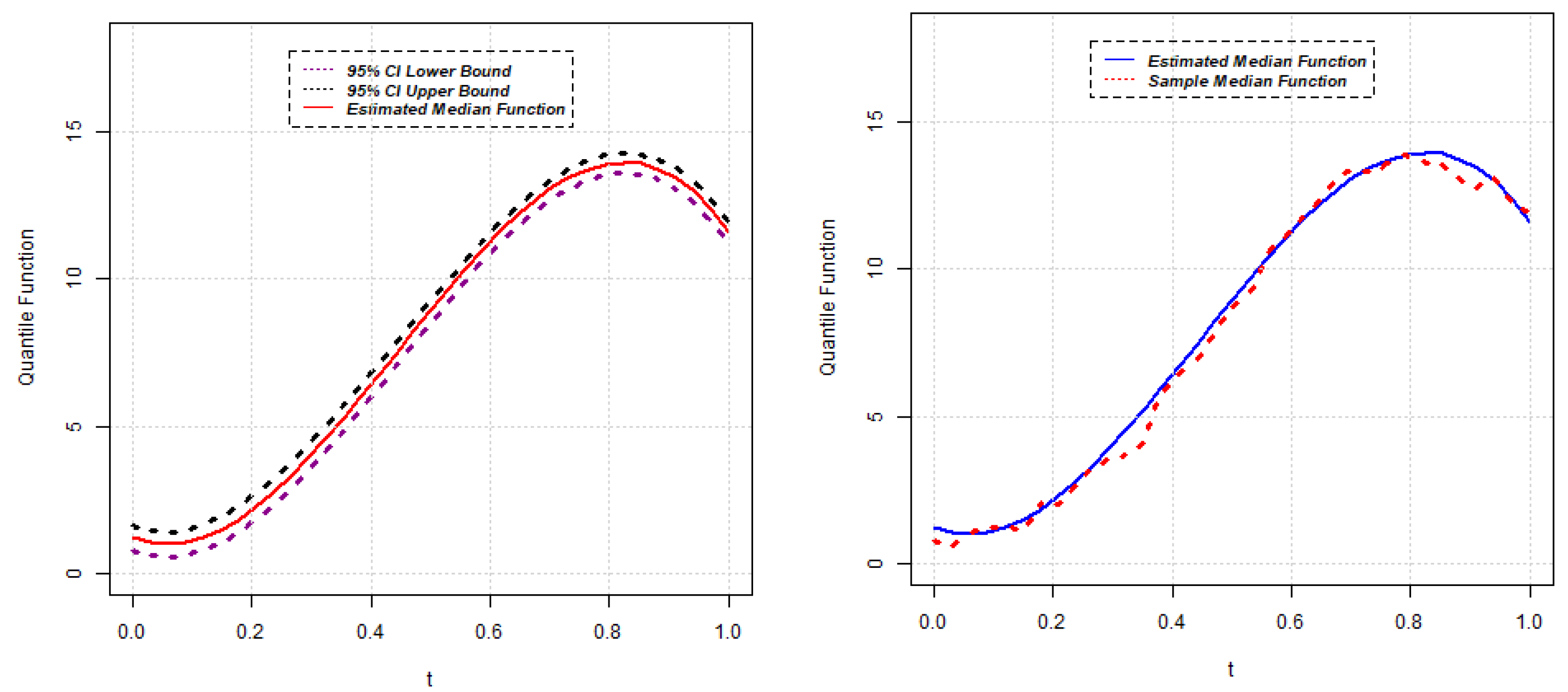
can be constructed as follows:
where
is the upper
percentile of standard normal distribution. Although the asymptotic covariance matrix for
has a closed form (12), there is no expression for the covariance matrix of estimator
, and thus we are unable to find the expression for the estimate of covariance
. However, bootstrap is a very powerful method and can be used in the estimation of covariance matrix for estimators
. Here, we use a block bootstrap approach to find the estimate for
.
The procedures are as follows.
- 1.
Obtain R bootstrap samples from the original data
- 2.
Find the estimated values for the parameters and and then calculate the values of by using formula (11) from each bootstrap sample and denote the obtained values as ; and
- 3.
Set
and calculate the sample means of
R bootstrap estimates for fixed effects parameters and random effects predictors
and
- 4.
Now, the bootstrap estimator for covariance matrix of estimators
can be written as
Furthermore, the estimates of model parameters and covariance matrix of estimators are computed by using the ‘
lqmm’ package developed by Geraci and Bottai [
17] in the
R programming environment. Generally, this ‘
lqmm’ package is used to estimate conditional quantile functions with random effects in linear quantile mixed models.
2.2. Test of the Similarity of Quantile Functions for Two Gene Expressions
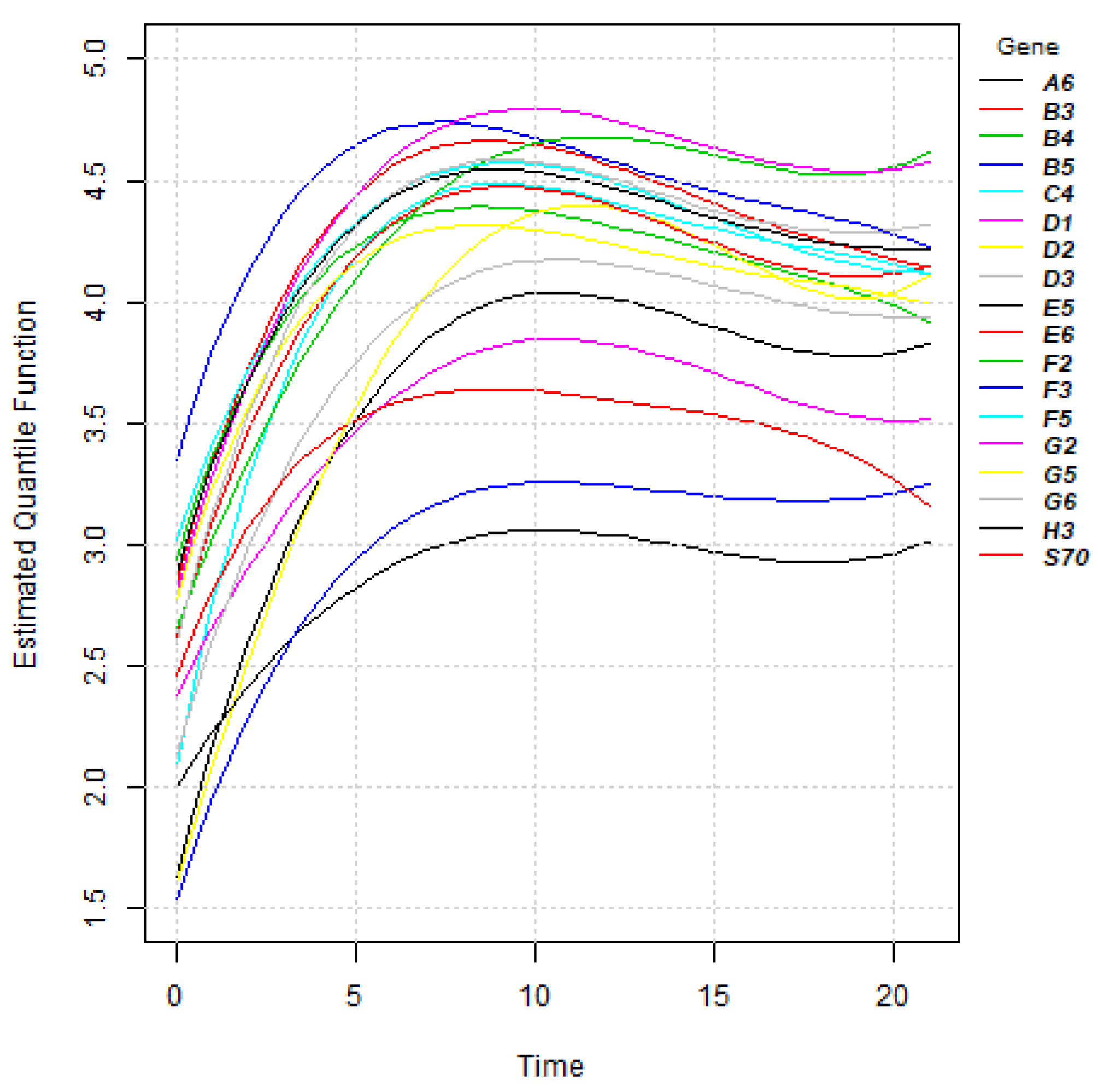
The other purpose of this research is to identify gene similarity based on quantile functions. Using the results obtained in
Section 2.1, the estimates of quantile functions for gene expressions can be obtained. The estimated quantile functions for
g genes can be expressed as
Two genes,
h and
s, are said to be similar if their quantile curve expressions are equal, i.e.,
Therefore, the proposed hypothesis is
Now, suppose that all quantile functions share the same truncated power basis functions
and
. Then, the
–quantile curves of gene expressions depend on the fixed effects parameter
and the random effects components
, and testing the similarity of two gene expression is equivalent to testing the equality of the corresponding fixed effects parameters and random effects components. Further, the predictors of random effects components depend on the estimates of parameters
. Thus, instead of testing the hypothesis (17), one can test the following hypothesis related to the fixed effects parameters and the random effects components.
Based on parameter estimates and its covariance matrix, the following asymptotic statistic for testing the the hypothesis
, is given by
where,
and
and
are the estimated variance–covariance matrices of the estimators
and
, respectively, which can be computed from (16). When
holds,
in Equation (19) has asymptotic chi-squared distribution with
degrees of freedom, where
r is known as the dimension number of
for fixed effects parameters and
q is the dimension number of
for random effects components.
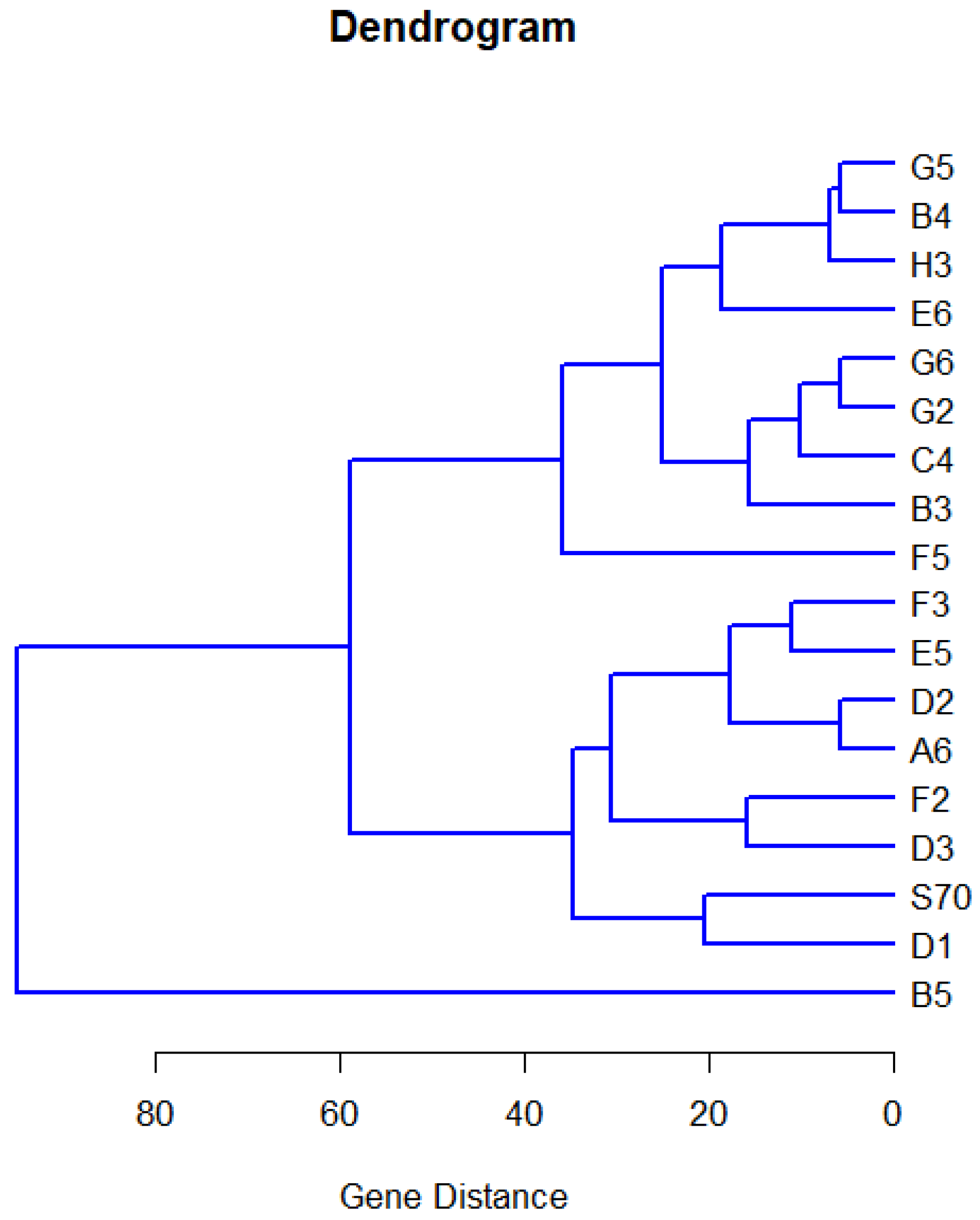
Furthermore, since the quantile function of gene expression is determined by r-dimensional fixed parameters, q-dimensional random effects components, and scale parameter , the pattern of gene expression h can be induced to be a random point in the dimensional Euclidean space, which has the asymptotic multivariate normal distribution with mean and covariance matrix for . From this point of view, the statistic in (19) is also the Mahalanobis distance between random vectors and . In terms of the minimum Mahalanobis distance, we can obtain the clustering tree for g gene expressions.




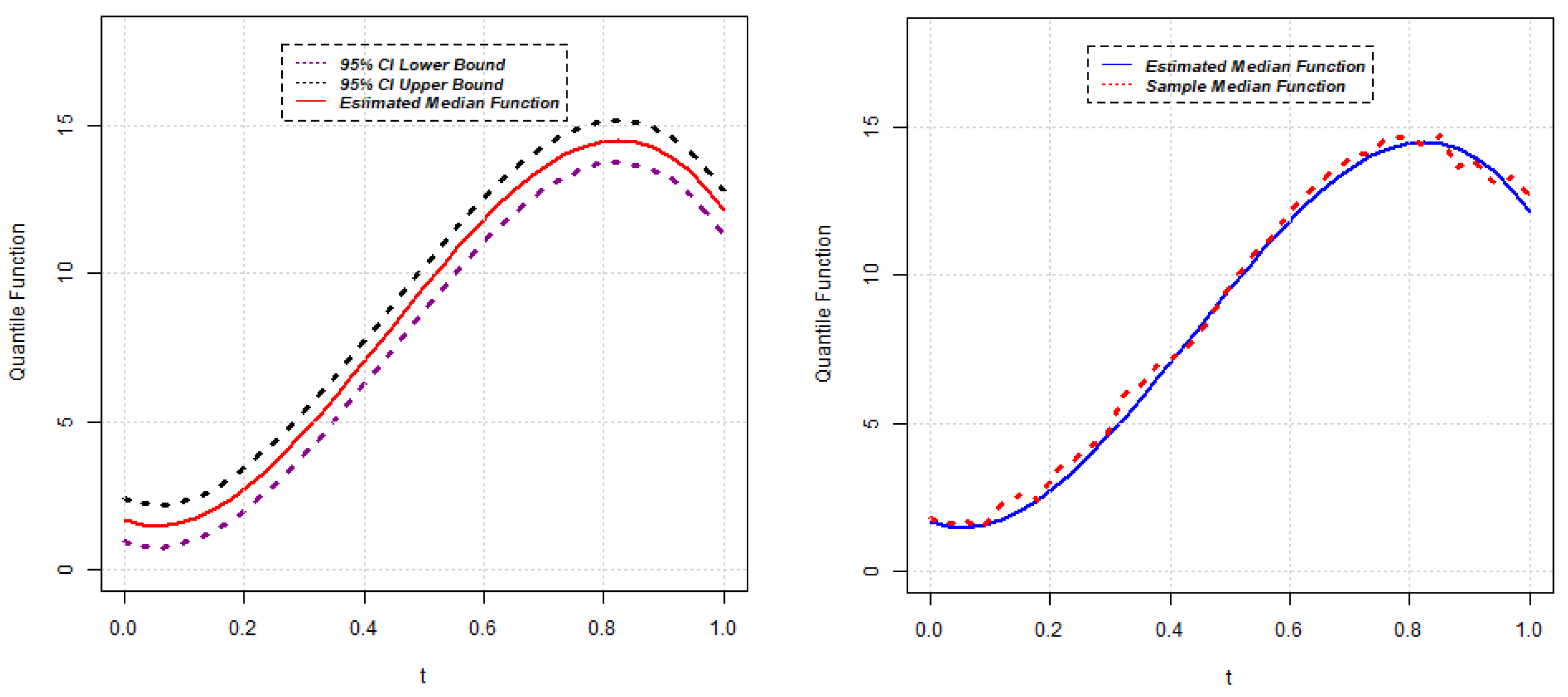

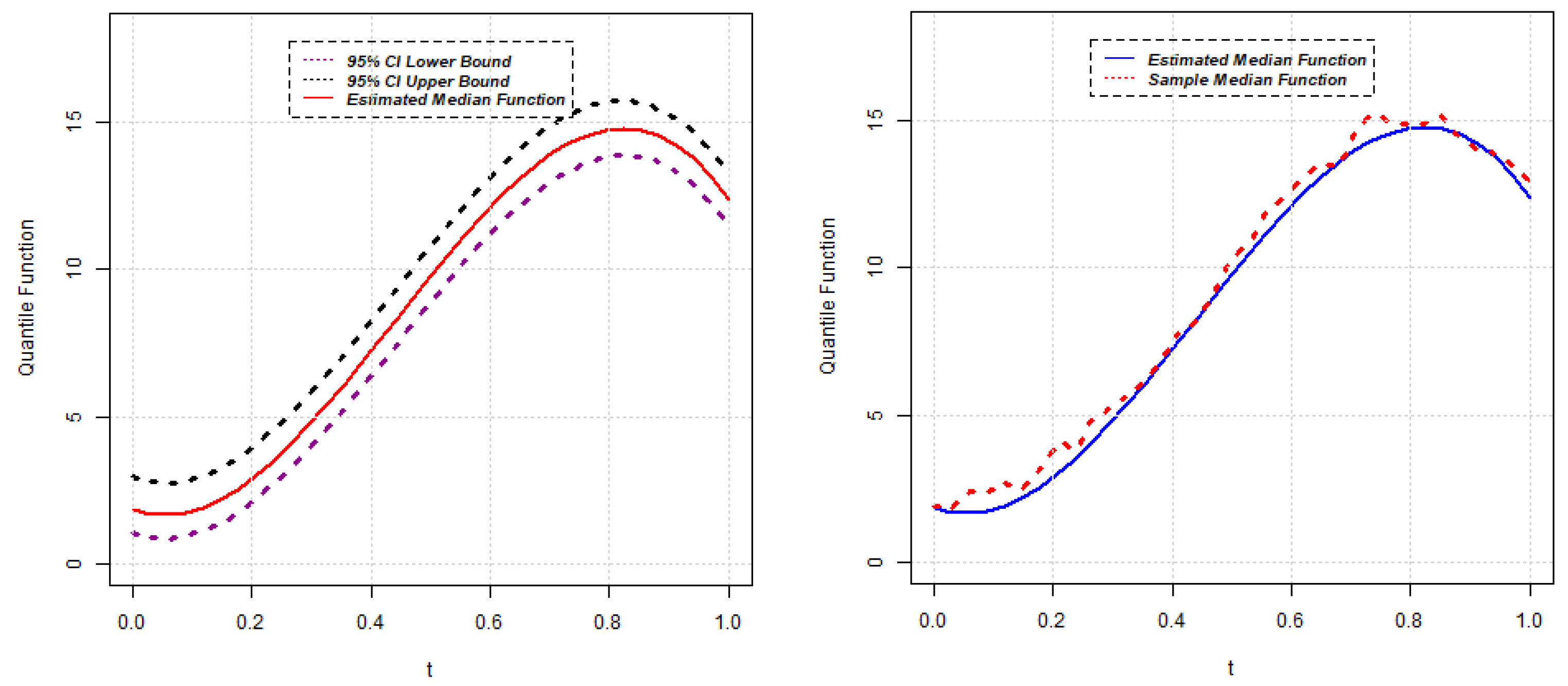
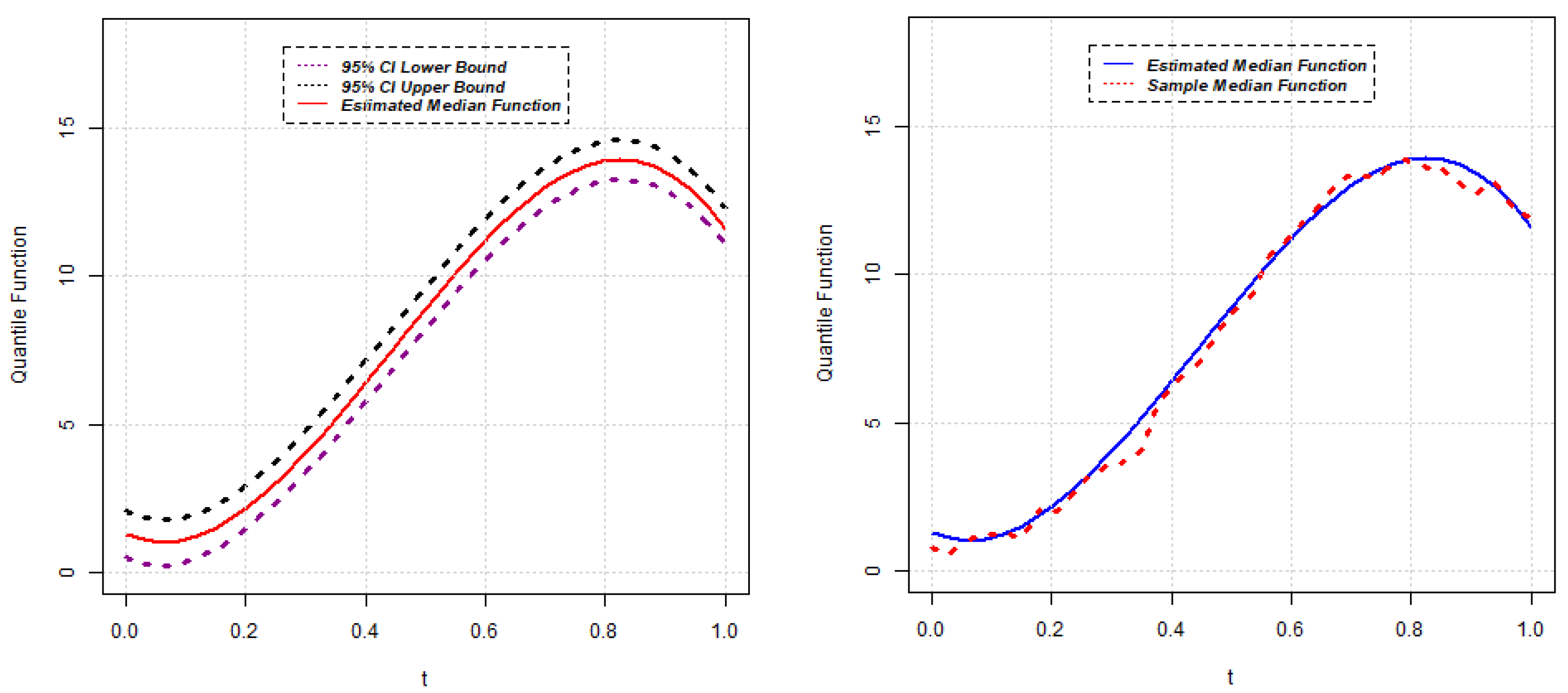
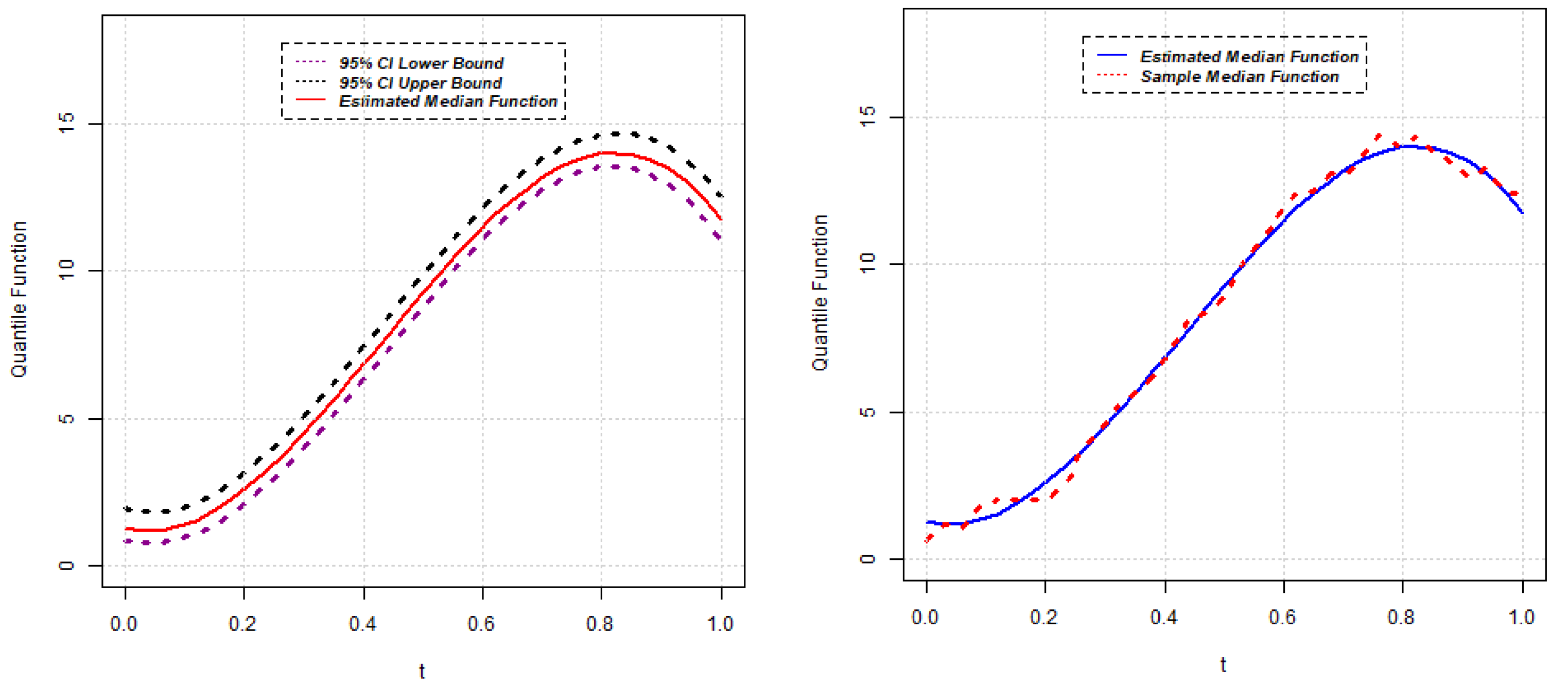



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}