Desorption of Arsenic from Calcareous Mine Affected Soils by Phosphate Fertilizers Application in Relation to Soil Properties and As Partitioning


 ,
,
Abstract
1. Introduction
2. Materials and Methods
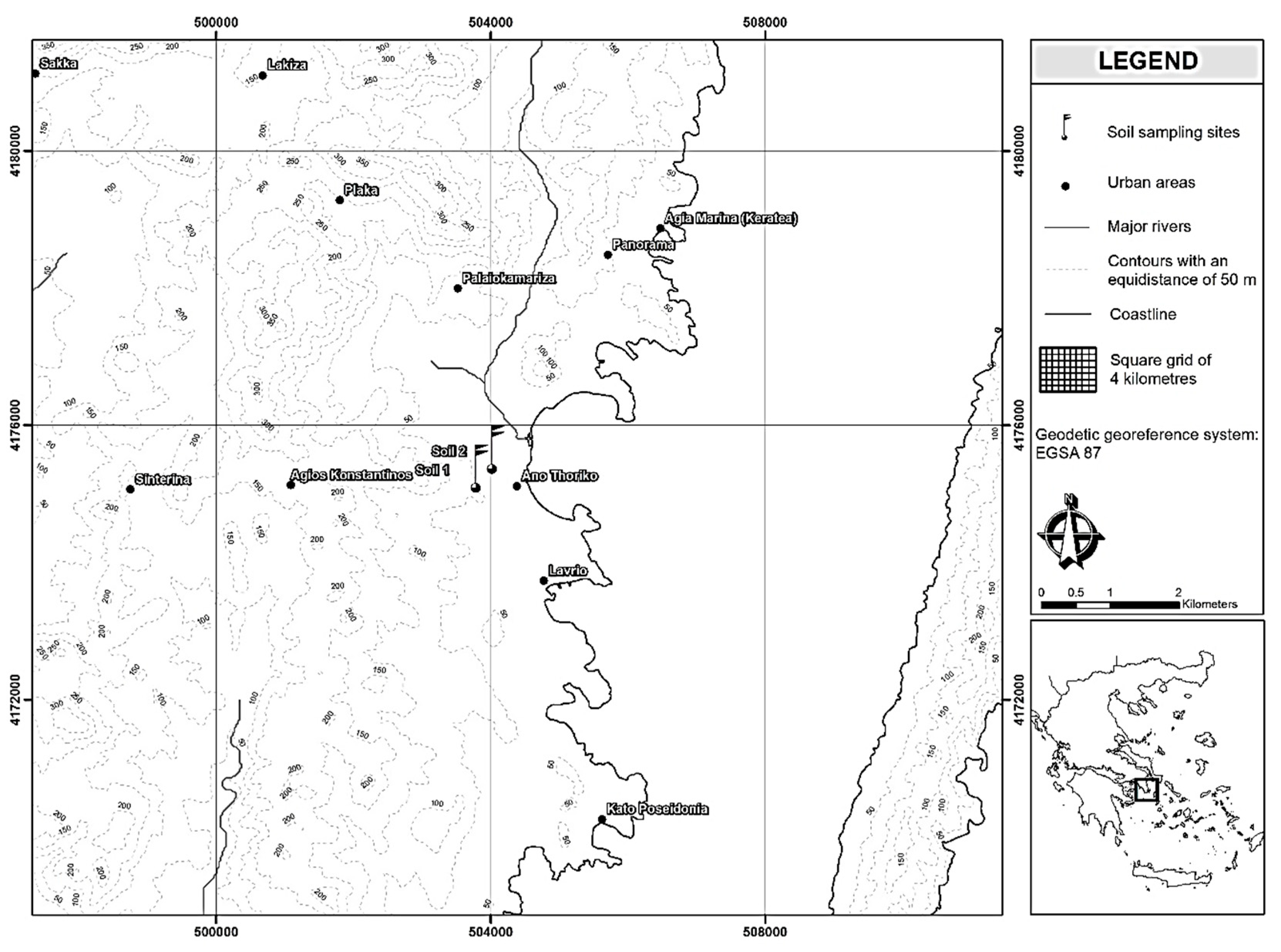
2.1. Soils
2.2. Stock Solutions and Reagents
2.3. Batch Experiments
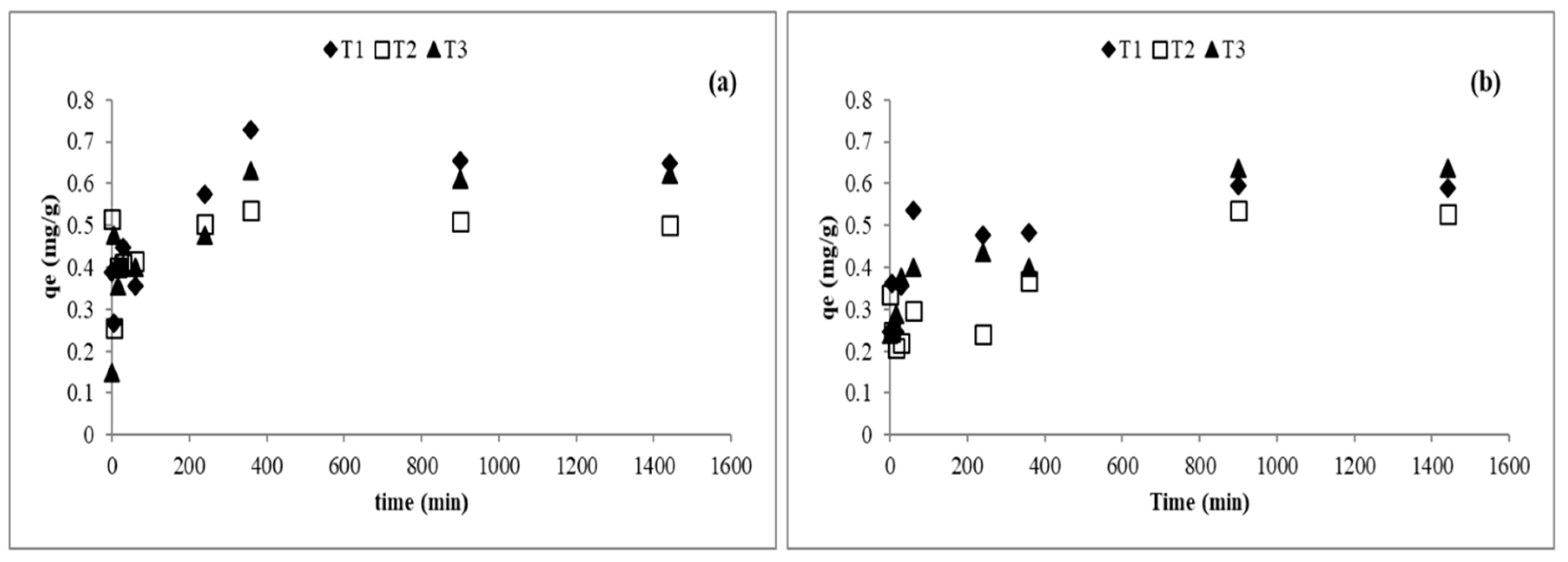
2.3.1. Effect of Contact Time
2.3.2. As Desorption—P Sorption Batch Experiments
2.3.3. Calculation of As Desorption and P Sorption Percentage
2.4. Analytical Determinations
2.5. Statistics
3. Results
3.1. Soil Properties
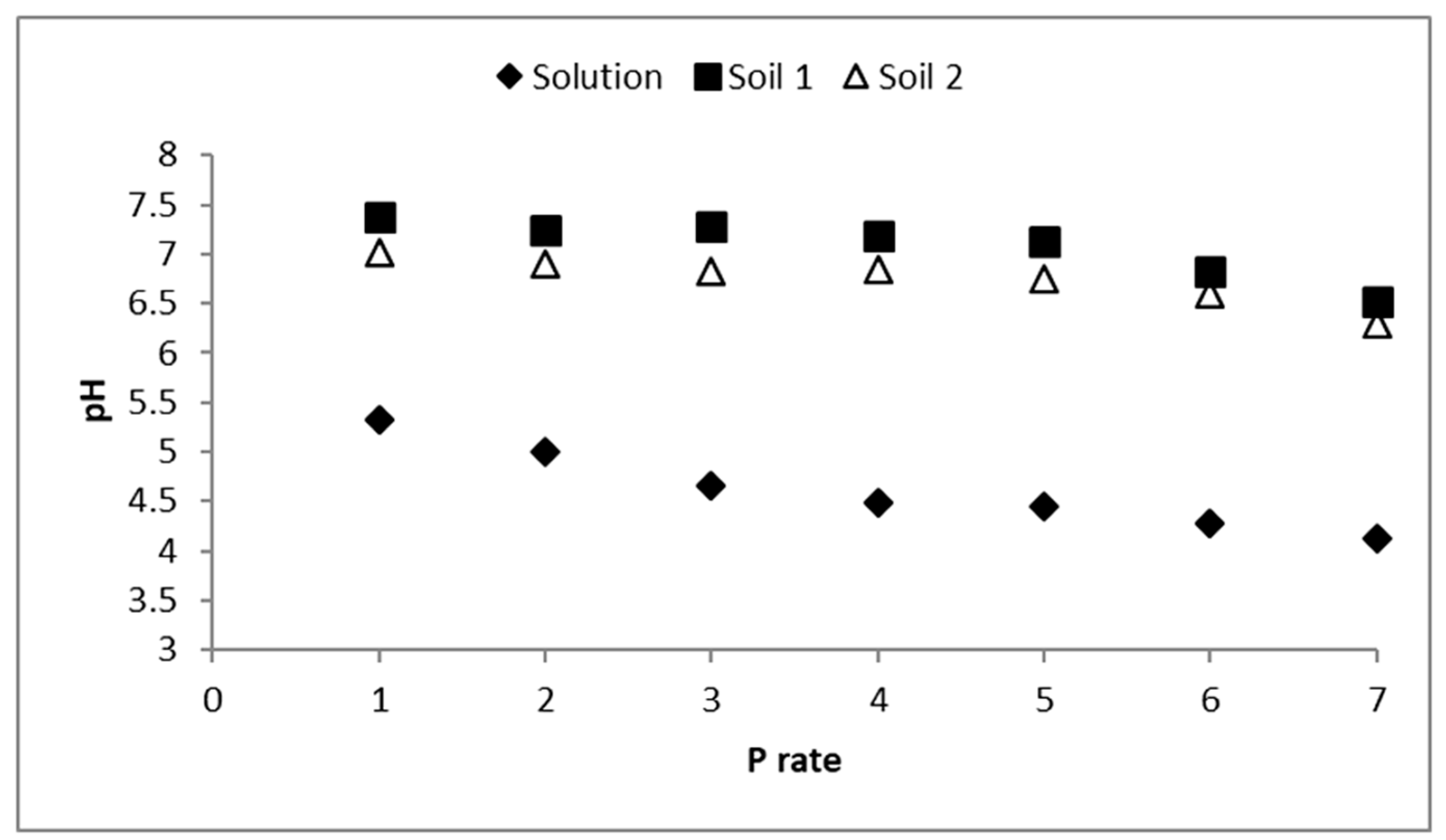
3.2. Solutions pH
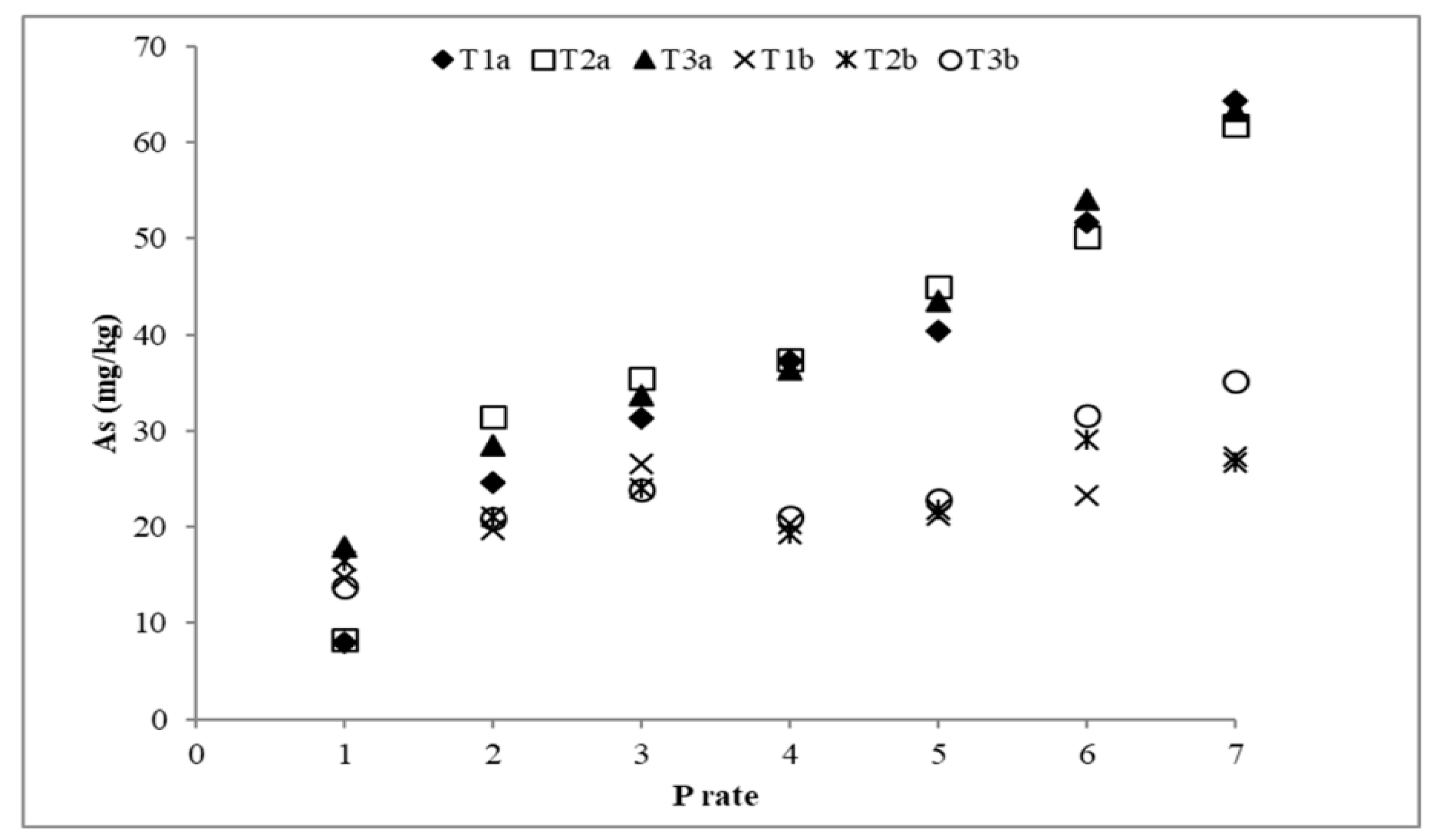
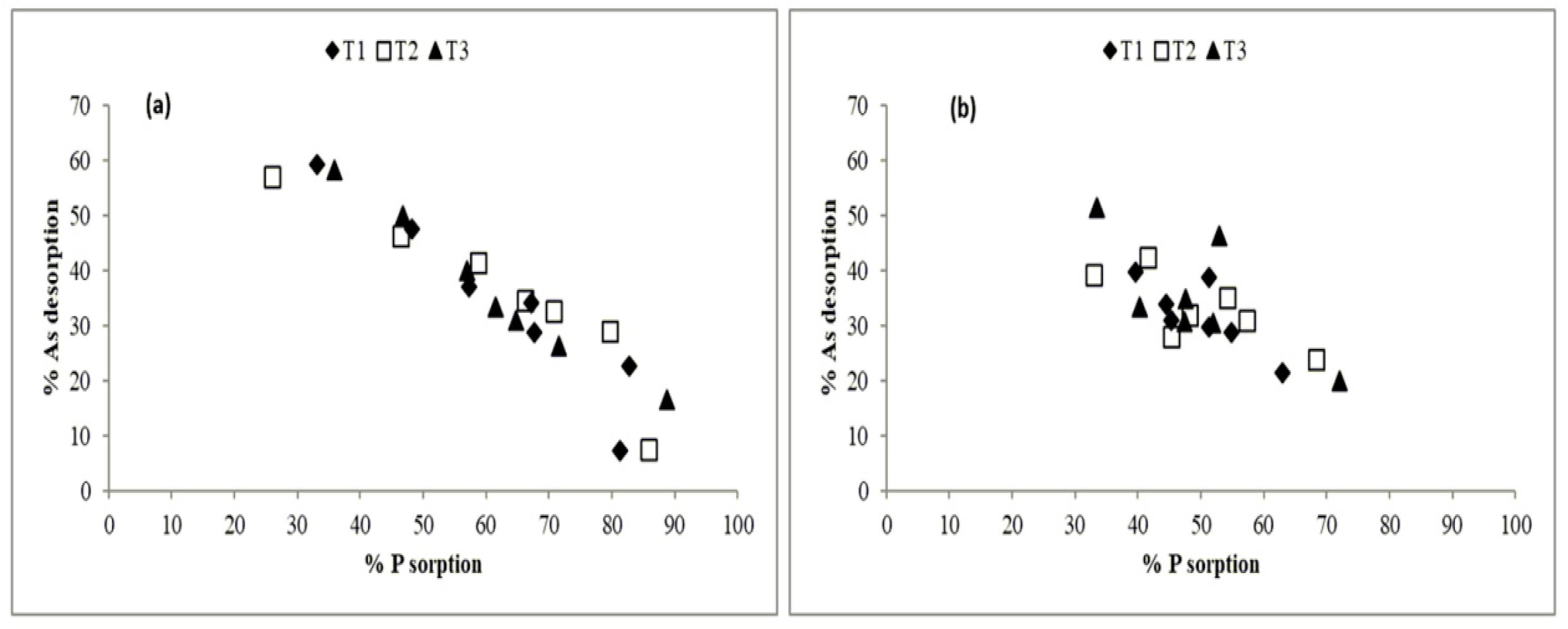
3.3. Arsenic Desorption
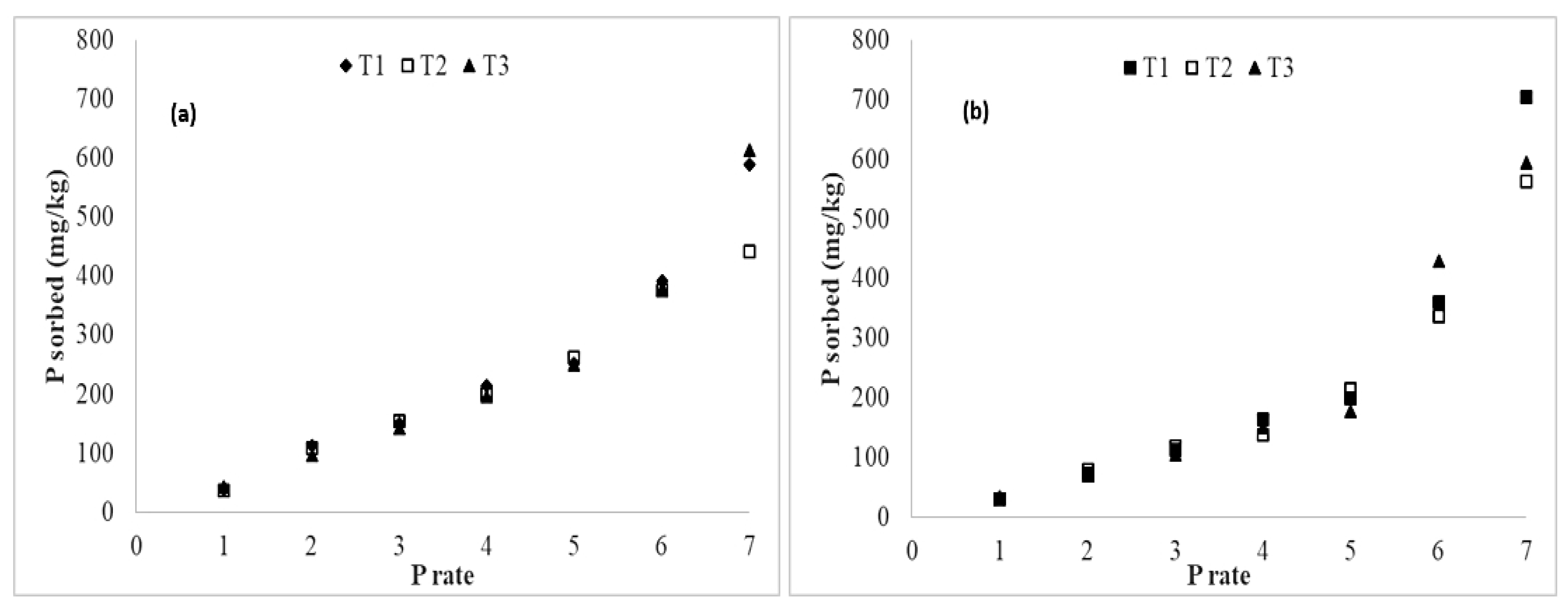
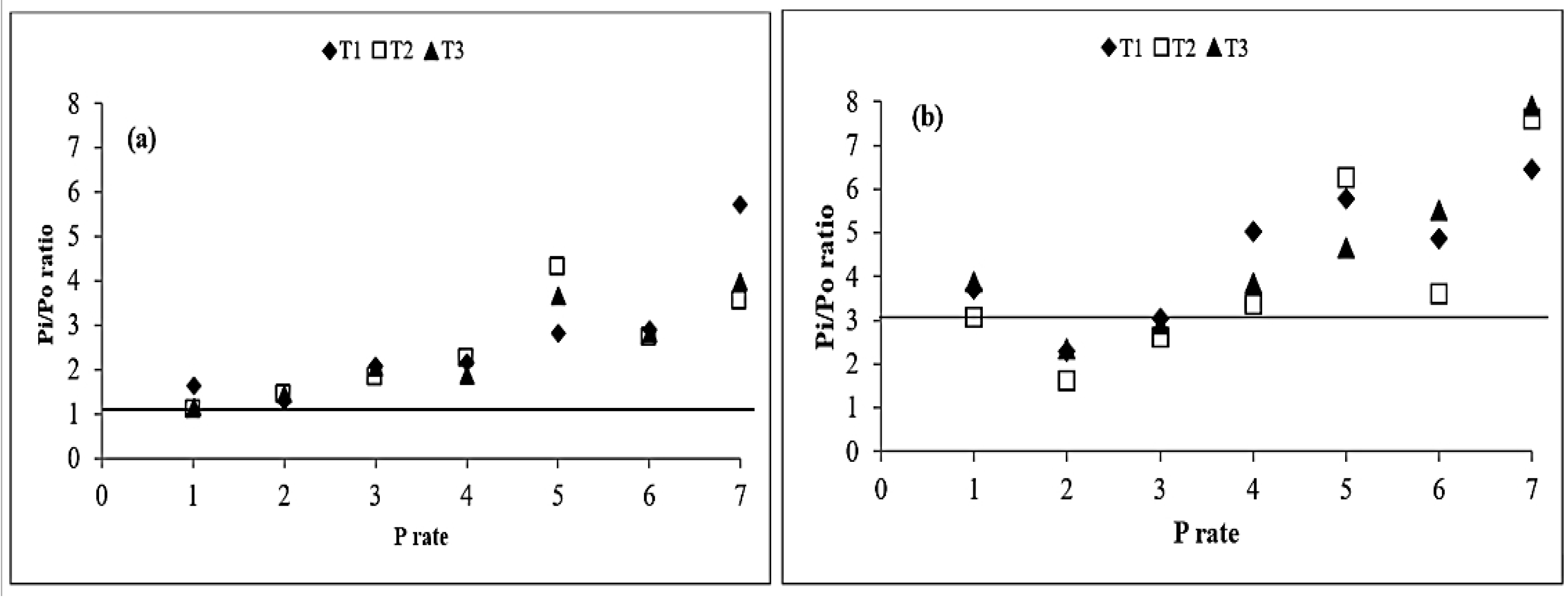
3.4. Added Phosphorus Sorption
3.5. Distribution of Organic and Inorganic Forms of Added P
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Massas, I.; Gasparatos, D.; Ioannou, D.; Kalivas, D. Signs for secondary buildup of heavy metals in soils at the periphery of Athens International Airport, Greece. Environ. Sci. Pollut. Res. 2018, 25, 658–671. [Google Scholar] [CrossRef] [PubMed]
- Gasparatos, D. Sequestration of heavy metals from soil with Fe–Mn concretions and nodules. Environ. Chem. Lett. 2013, 11, 1–9. [Google Scholar] [CrossRef]
- Kalyvas, G.; Tsitselis, G.; Gasparatos, D.; Massas, I. Efficacy of EDTA and olive mill wastewater to enhance As, Pb, and Zn phytoextraction by pteris vittata L. from a soil heavily polluted by mining activities. Sustainability 2018, 10, 1962. [Google Scholar] [CrossRef]
- Anawar, H.M.; Rengel, Z.; Damon, P.; Tibbett, M. Arsenic-phosphorus interactions in the soil-plant-microbe system: Dynamics of uptake, suppression and toxicity to plants. Environ. Pollut. 2018, 233, 1003–1012. [Google Scholar] [CrossRef] [PubMed]
- Tuli, R.; Chakrabarty, D.; Trivedi, P.K.; Tripathi, R.D. Recent advances in arsenic accumulation and metabolism in rice. Mol. Breed. 2010, 26, 307–323. [Google Scholar] [CrossRef]
- Nriagu, J.O.; Pacyna, J.M. Quantitative assessment of worldwide contamination of air, water and soils by trace metals. Nature 1988, 333, 134–139. [Google Scholar] [CrossRef]
- RIVM (National Institute for Public Health and Environmental Protection). Annex A: Target Values, Soil Remediation Intervention Values and Indicative Levels for Serious Contamination, Dutch Target and Intervention Values, (The New Dutch List), Netherlands; RIVM: Bilthoven, The Netherlands, 2000. [Google Scholar]
- Kalyvas, G.; Gasparatos, D.; Massas, I. A critical assessment of arsenic partitioning in mine-affected soils by using two sequential extraction protocols. Arch. Agron. Soil Sci. 2018, 64, 1549–1563. [Google Scholar] [CrossRef]
- Strawn, D.G. Review of interactions between phosphorus and arsenic in soils from four case studies. Geochem. Trans. 2018, 19, 10. [Google Scholar] [CrossRef]
- Lambkin, D.C.; Alloway, B.J. Arsenate-induced phosphate release from soils and its effect on plant phosphorus. Water Air Soil Pollut. 2003, 144, 41–56. [Google Scholar] [CrossRef]
- Bolan, N.; Kunhikrishnan, A.; Thangarajan, R.; Kumpiene, J. Remediation of heavy metal(loid)s contaminated soils—To mobilize or to immobilize? J. Hazard. Mater. 2014, 266, 141–166. [Google Scholar] [CrossRef]
- Feng, Q.; Zhang, Z.; Chen, Y.; Liu, L.; Zhang, Z.; Chen, C. Adsorption and desorption characteristics of arsenic on soils: Kinetics, equilibrium, and effect of Fe(OH)3 colloid, H2SiO3 colloid and phosphate. Procedia Environ. Sci. 2013, 18, 26–36. [Google Scholar] [CrossRef]
- McDowell, R.; Condron, L. Influence of soil constituents on soil phosphorus sorption and desorption. Commun. Soil Sci. Plant Anal. 2001, 32, 2531–2547. [Google Scholar] [CrossRef]
- Gasparatos, D.; Massas, I.; Godelitsas, A. Fe-Mn concretions and nodules formation in redoximorphic soils and their role on soil phosphorus dynamics: Current knowledge and gaps. Catena 2019, 182, 104106. [Google Scholar] [CrossRef]
- Mukhopadhyay, S.; Hashim, M.A.; Allen, M.; Gupta, B.S. Arsenic removal from soil with high iron content using a natural surfactant and phosphate. Int. J. Environ. Sci. Technol. 2015, 12, 617–632. [Google Scholar] [CrossRef]
- Piracha, M.A.; Ashraf, M.; Shahzad, S.M.; Siddiqui, A.R.; Nazeer, S. Arsenic behavior in different textured soils amended with phosphate rock and farm yard manure. J. Environ. Agric. 2016, 1, 55–67. [Google Scholar]
- Violante, A.; Pigna, M. Competitive sorption of As(V) and phosphate on different clay minerals and soils. Soil Sci. Soc. Am. J. 2002, 66, 1788–1796. [Google Scholar] [CrossRef]
- O’Reilly, S.E.; Strawn, D.G.; Sparks, D.L. Residence time effects on arsenate adsorption/desorption mechanisms on goethite. Soil Sci. Soc. Am. J. 2001, 65, 67–77. [Google Scholar] [CrossRef]
- Rhoades, J.D. Cation exchange capacity. In Methods of Soil Analysis, Part 2, Chemical and Microbiological Properties, 2nd ed.; Page, A.L., Miller, R.H., Keeney, D.R., Eds.; ASA-SSSA: Madison, WI, USA, 1982; pp. 149–157. [Google Scholar]
- Nelson, D.W.; Sommers, L.E. Total carbon, organic carbon and organic matter. In Methods of Soil Analysis, Part 2, Chemical and Microbiological Properties; Page, A.L., Miller, R.H., Keeney, D.R., Eds.; ASA-SSSA: Madison, WI, USA, 1982. [Google Scholar]
- Bouyoucos, G.J. A recalibration of the hydrometer method for making mechanical analysis of soils. Agron. J. 1951, 43, 434–438. [Google Scholar] [CrossRef]
- NF ISO 10693. Détermination de la Teneuren Carbonate—Méthode Volumétrique; Qualité des Sols AFNOR: Paris, France, 1995; pp. 177–186. [Google Scholar]
- Loeppert, R.H.; Suarez, D.L. Carbonate and gypsum. In Methods of Soil Analysis, Part 3, Chemical Methods; Bigham, J.M., Bartels, J.M., Eds.; ASA-SSSA: Madison, WI, USA, 1982; pp. 437–474. [Google Scholar]
- Saunders, W.M.H.; Williams, E.G. Observation on the determination of total organic phosphorus in soils. J. Soil Sci. 1955, 6, 254–267. [Google Scholar] [CrossRef]
- Walker, T.; Adams, A. Studies on soil organic matter: I. Influence of phosphorus content of parent materials on accumulations of carbon, nitrogen, sulfur, and organic phosphorus in grassland soils. Soil Sci. 1958, 85, 307–318. [Google Scholar] [CrossRef]
- Olsen, S.R.; Cole, C.V.; Watanabe, F.S.; Dean, L.A. Estimation of available phosphorus in soils by extraction with sodium bicarbonate. US Dept. Agric. Circ. 1954, 939, 1–19. [Google Scholar]
- Mehra, O.P.; Jackson, M.L. Iron oxide removal from soils and clay by a dithionite-citrate system buffered with sodium bicarbonate. Clays Clay Miner. 2013, 7, 317–327. [Google Scholar] [CrossRef]
- Schwertmann, U.; Taylor, R.M. Iron oxides. Minerals in Soil Environments. Dixon, J.B., Weed, S.B., Eds.; 2nd ed. 1989, pp. 379–438. Available online: http://www.scielo.br/scielo.php?script=sci_nlinks&ref=000101&pid=S0103-8478201300060000900024&lng=pt (accessed on 17 August 2019). [CrossRef]
- Gasparatos, D.; Haidouti, C. A comparison of wet oxidation methods for determination of total phosphorus in soils. J. Plant Nutr. Soil Sci. 2001, 164, 435–439. [Google Scholar] [CrossRef]
- Wenzel, W.; Kirchbaumer, N.; Prohaska, T.; Stingeder, G.; Lombi, E.; Adriano, D. Arsenic fractionation in soils using an improved sequential extraction procedure. Anal Chim Acta. 2001, 436, 309–323. [Google Scholar] [CrossRef]
- Shafqat, M.N.; Pierzynski, G.M. The Freudlich adsorption isotherm constants and prediction of phosphorus bioavailability as affected by different phosphorus sources on two Kansas soils. Chemosphere 2014, 99, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Singes-Pastor, A.; Burlo, F.; Mitra, K.; Carbonell-Barrachina, A.A. Arsenic biochemistry as affected by phosphorus fertilizer addition, redox potential and pH in a west Bengal (India) soil. Geoderma 2007, 137, 504–510. [Google Scholar] [CrossRef]
- Smith, E.; Naidu, R. Chemistry of inorganic arsenic in soils: Kinetics of arsenic adsorption–desorption. Environ. Geochem. Health 2009, 31, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.; Naidu, R.; Alston, A.M. Chemistry of inorganic arsenic in soils: II. Effect of phosphorus, sodium and calcium on arsenic sorption. J. Environ. Qual. 2002, 31, 557–563. [Google Scholar] [CrossRef]
- Fayiga, A.O.; Ma, L.Q.; Zhou, O. Effects of plant arsenic uptake and heavy metals on arsenic distribution in an arsenic-contaminated soil. Environ. Pollut. 2007, 147, 737–742. [Google Scholar] [CrossRef]
- Vithanage, M.; Dabrowska, B.B.; Mukherjee, A.B.; Sandhi, A.; Bhattacharya, P. Arsenic uptake by plants and possible phytoremediation applications: A brief overview. Environ. Chem. Lett. 2012, 10, 217–224. [Google Scholar] [CrossRef]
- Fitz, W.J.; Wenzel, W.W. Arsenic transformations in the soil–rhizosphere–plant system: Fundamentals and potential application to phytoremediation. J. Biotechnol. 2002, 99, 259–278. [Google Scholar] [CrossRef]
- Goldberg, S.; Sposito, G. Chemical model of phosphate adsorption by soils—I. Reference oxide minerals. Soil Sci. Soc. Am. J. 1984, 48, 772–778. [Google Scholar] [CrossRef]
- Zhang, W.; Faulkner, J.; Giri, S.; Geohring, L.; Steenhuis, T. Evaluation of two Langmuir models for phosphorus sorption of phosphorus-enriched soils in New York for environmental applications. Soil Sci. 2009, 174, 523–530. [Google Scholar] [CrossRef]
- Kruse, J.; Abraham, M.; Amelung, W.; Baum, C.; Bol, R.; Kühn, O.; Santner, J. Innovative methods in soil phosphorus research: A review. J. Plant Nutr. Soil Sci. 2015, 178, 43–88. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.A.; Golberg, S. Modeling competitive adsorption of arsenate with phosphate and molybdate on oxide minerals. Soil. Sci. Soc. Am. J. 1996, 60, 121–131. [Google Scholar] [CrossRef]
- Hongshao, Z.; Stanforth, R. Competitive adsorption of phosphate and arsenate on goethite. Environ. Sci. Technol. 2001, 35, 4753–4757. [Google Scholar] [CrossRef] [PubMed]
- Riva-Perez, I.M.; Paradelo-Nunez, R.; Novoa-Munoz, J.C.; Arias-Estevez, M.; Fernandez-Sanjurjo, M.; Álvarez-Rodríguez, E.; Núñez-Delgado, A. As(V) and P competitive sorption on soils, by-products and waste materials. Int. J. Environ. Res. Public Health 2015, 12, 15706–15715. [Google Scholar] [CrossRef]
- Ahmad, M.; Ahmad, M.; El-Naggar, A.H.; Usman, A.R.A.; Abduljabbar, A.; Vithanage, M.; Al-Wabel, M.I. Aging effects of organic and inorganic fertilizers on phosphorus fractionation in a calcareous sandy loam soil. Pedosphere 2018, 28, 873–883. [Google Scholar] [CrossRef]
- Yolcubal, I.; Akyol, N.H. Adsorption and transport of arsenate in carbonate-rich soils: Coupled effects of nonlinear and rate-limeted sorption. Chemosphere 2008, 73, 1300–1307. [Google Scholar] [CrossRef]
- Alexendratos, V.G.; Elzinga, E.J.; Reeder, R.J. Arsenate uptake by calcite: Macroscopic and spectroscopic characterization of adsorption and incorporation mechanisms. Geochim. Cosmochim. Acta 2007, 71, 4172–4187. [Google Scholar] [CrossRef]
- Suriyagoda, L.D.B.; Dittert, K.; Lambers, H. Arsenic in rice soils and potential agronomic mitigation strategies to reduce arsenic bioavailability: A review. Pedosphere 2018, 28, 363–382. [Google Scholar] [CrossRef]
- Tu, S.; Ma, L.Q. Effects of arsenate and phosphate on their accumulation by an arsenic-hyperaccumulator Pteris vittata L. Plant Soil 2003, 249, 373–382. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Soil Properties | ||
|---|---|---|
| Soil 1 | Soil 2 | |
| Clay (g kg−1) | 328 | 230 |
| Silt (g kg−1) | 214 | 210 |
| Sand (g kg−1) | 458 | 560 |
| pH (1:1) | 8 | 7.7 |
| CaCO3 (g kg−1) | 115 | 64 |
| Act. CaCO3 (g kg−1) | 27 | 3 |
| O.C. (g kg−1) | 11 | 26 |
| CEC (cmolc kg−1) | 30 | 27 |
| Fed (mg kg−1) | 41.5 | 45.3 |
| Feo (mg kg−1) | 4.8 | 7 |
| Ald (mg kg−1) | 728.3 | 574.8 |
| Alo (mg kg−1) | 2389.5 | 1098 |
| As total (mg kg−1) | 852 | 1745 |
| P total (mg kg−1) | 733 | 1061 |
| P in. (mg kg−1) | 391 | 796 |
| P org. (mg kg−1) | 342 | 265 |
| P avail. (mg kg−1) | 10.7 | 11 |
| As fractions obtained by SEP | ||
| W1: Nonspecifically sorbed As (mg kg−1) | 5 | 4.1 |
| W2: Specifically sorbed As (mg kg−1) | 103.6 | 64.3 |
| W3: As sorbed on amorphous Fe-Al oxides (mg kg−1) | 313.3 | 644.9 |
| W4: As sorbed on crystalline Fe-Al oxides (mg kg−1) | 240.8 | 393.8 |
| W5: Residual As (mg kg−1) | 106 | 302.9 |
| SOIL 1 | ||||
| Factor | SS | DF | MS | p |
| P treatment | 86.17 | 2 | 43.09 | <0.001 |
| P rate | 15116.6 | 6 | 2519.36 | <0.001 |
| P treatment X P rate | 271.81 | 12 | 22.65 | <0.001 |
| SOIL 2 | ||||
| Factor | SS | DF | MS | p |
| P treatment | 58.99 | 2 | 29.50 | <0.05 |
| P rate | 1381.34 | 6 | 230.22 | <0.001 |
| P treatment X P rate | 225.91 | 12 | 18.83 | <0.01 |
| Langmuir Constants | Freundlich Constants | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| qm (mg/g) | bL (L/mg) | R2 | p-Value | KF (mg/g) (L/mg)1/n | 1/n | R2 | p-Value | ||
| Soil 1 | T1 | 0.68 | 0.090 | 0.968 | <0.001 | 12.81 | 0.525 | 0.960 | <0.001 |
| T2 | 0.48 | 0.167 | 0.996 | <0.001 | 12.07 | 0.468 | 0.957 | <0.001 | |
| T3 | 0.73 | 0.070 | 0.932 | <0.01 | 13.06 | 0.511 | 0.995 | <0.001 | |
| Soil 2 | T1 | 1.36 | 0.018 | 0.776 | <0.01 | 31.26 | 0.762 | 0.998 | <0.001 |
| T2 | 0.81 | 0.034 | 0.886 | <0.01 | 26.14 | 0.675 | 0.994 | <0.001 | |
| T3 | 0.99 | 0.024 | 0.648 | <0.05 | 26.58 | 0.676 | 0.946 | <0.01 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zafeiriou, I.; Gasparatos, D.; Kalyvas, G.; Ioannou, D.; Massas, I. Desorption of Arsenic from Calcareous Mine Affected Soils by Phosphate Fertilizers Application in Relation to Soil Properties and As Partitioning. Soil Syst. 2019, 3, 54. https://doi.org/10.3390/soilsystems3030054
Zafeiriou I, Gasparatos D, Kalyvas G, Ioannou D, Massas I. Desorption of Arsenic from Calcareous Mine Affected Soils by Phosphate Fertilizers Application in Relation to Soil Properties and As Partitioning. Soil Systems. 2019; 3(3):54. https://doi.org/10.3390/soilsystems3030054
Chicago/Turabian StyleZafeiriou, Ioannis, Dionisios Gasparatos, Georgios Kalyvas, Dafni Ioannou, and Ioannis Massas. 2019. "Desorption of Arsenic from Calcareous Mine Affected Soils by Phosphate Fertilizers Application in Relation to Soil Properties and As Partitioning" Soil Systems 3, no. 3: 54. https://doi.org/10.3390/soilsystems3030054
APA StyleZafeiriou, I., Gasparatos, D., Kalyvas, G., Ioannou, D., & Massas, I. (2019). Desorption of Arsenic from Calcareous Mine Affected Soils by Phosphate Fertilizers Application in Relation to Soil Properties and As Partitioning. Soil Systems, 3(3), 54. https://doi.org/10.3390/soilsystems3030054