1. Introduction and Clinical Significance
Pulmonary arterial hypertension is a serious disease that can occur in early childhood and can lead to significant morbidity and reduced life expectancy [
1]. Pulmonary arterial hypertension can be associated with congenital heart disease. One of the most common genetic mutations associated with pulmonary arterial hypertension involves mutations in
BMPR2. Rare variants are
SMAD1, SMAD4, SMAD9, CAV1, KCNK3, TBX4, and
ACVRL1 [
2].
An association between Snijders Blok–Campeau syndrome and the development of pulmonary arterial hypertension has not been characterized yet.
Snijders Blok–Campeau syndrome is a rare genetic disease usually caused by pathogenic mutations in the chromodomain helicase DNA binding protein (
CHD3) gene.
CHD3 has a crucial role in chromatin remodeling, thereby modifying gene expressions [
3]. Clinical findings include neurodevelopmental disorders (speech delay, intellectual disability, specific learning difficulties, and behavior disorders like autism spectrum disorder and attention deficit hyperactivity disorder). Facial dysmorphism, with a broad forehead, macrocephaly, hypertelorism, epicanthus, and low-set eyes, has also been previously described [
4].
The Snijders Blok type of X-linked syndromic intellectual disorder is caused by a mutation in the dead-box helicase 3 gene (
DDX3X, OMIM *300160), which encodes a protein acting as an RNA helicase and participates in a variety of cellular processes. It is, among other actors, an important regulator in the Wnt signaling pathway [
5,
6]. Diseases with mutations in the
DDX3X gene can be associated with syndromic disorders with variable clinical presentation, such as varying degrees of intellectual disability, as well as developmental disorders, movement disorders, and behavioral disorders with a gender-specific effect [
5]. In addition, patients show muscular hypotonia, microcephaly with potential cerebral structural abnormalities (corpus callosum hypoplasia, ventricular enlargement, cortical dysplasia), and congenital heart defects (atrial/ventricular septal defect, double orifice mitral valve, mild concentric left hypertrophy and bicuspid aortic valve) [
5,
7,
8].
Here, we describe a female infant with a mutation in the DDX3X gene, pulmonary arterial hypertension, and Snijders Blok–Campeau syndrome.
2. Case Presentation
The parents are healthy and non-consanguineous parents. The two older siblings are healthy, with age-appropriate development. Further family history was unremarkable.
Prenatal ultrasound showed structural abnormalities of the heart, containing findings suggestive of a complex congenital heart disease (functionally univentricular heart with transposition of the great arteries). Consequently, genetic testing was initiated. A chromosome analysis from the amniotic fluid was performed in the 26th week of pregnancy and showed no numerical or structural abnormalities. Due to these findings, further genetic testing was recommended. A whole exome-trio analysis identified a heterozygous mutation (c.1600 > T; p.Arg534Cys; OMIM *300160) within the DDX3X gene. Neither of the parents were carriers, so a de novo mutation in the fetus was assumed.
The index patient was born in gestational week 41 + 2. Birth parameters showed a low birth weight of 2190 g (−8,27z, <1st percentile). Facial abnormalities were described after birth, with retrognathia and auricular dysplasia. Prostaglandin E1 was initiated directly after birth. A complex heart defect was diagnosed by echocardiography, consisting of an univentricular heart with a dominant right ventricle, mitral valve atresia, ventricular septal defect, multifenestrated atrial septal defect, D-transposition of the great arteries, persistent left superior vena cava, patent ductus arteriosus, and coarctation of the aorta and truncus bicaroticus. Six days after birth, the first cardiac surgery with bilateral pulmonary artery banding was performed. One month later, the patient needed a cardiac catheterization with atrial septostomy , and 4 days later, a sinus-Super Flex DS stent (7 × 20 mm optimed, Ettlingen, Germany) was implanted in the patent arterial duct. A cerebral MRI was performed at the age of three months, which showed cerebral abnormalities, such as a hypoplastic cerebellar vermis with enlarged cisterna magna and a cleft palate (
Figure 1).
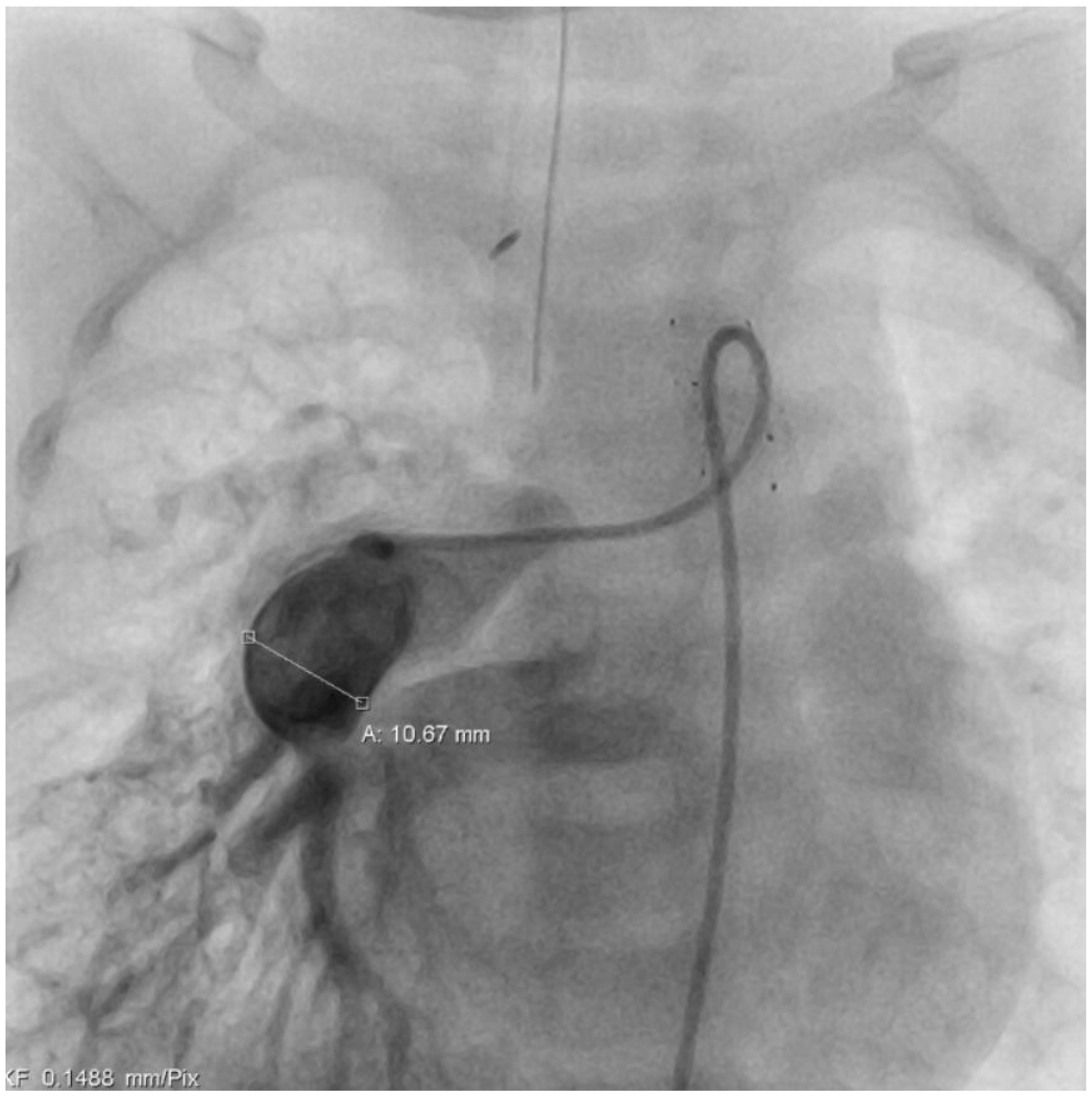
At the age of five months, a cardiac catheter examination was performed, and severe pulmonary arterial hypertension with significantly increased pulmonary resistance was diagnosed (
Table 1) despite the effective and morphologically tight bilateral pulmonary arterial banding (
Figure 2 +
Figure 3). Due to the pulmonary vascular pathology, a cardiosurgical correction operation does not appear to be beneficial. An off-label drug therapy with sildenafil was discussed with the parents. Despite multiple consultations, the parents refused the therapy with sildenafil because of concerns about the off-label use and side effects. Cardiac consultations take place every 3 months including echocardiography, ECG, and clinical examination.
In the long term, the disease will lead to death from right heart failure if left untreated.
3. Discussion
We describe a syndromic infant with a prenatally diagnosed genetic mutation in DDX3X. This gene defect is associated with Snijders Blok–Campeau syndrome. This syndrome is mainly associated with reduced intelligence and developmental delay. In addition to the genetic evidence, the patient has, among other features, a complex cardiac defect. Despite effective pulmonary artery banding, an invasive follow-up revealed severe pulmonary arterial hypertension.
Snijders Blok–Campeau syndrome is presumably a rare neurodevelopmental genetic disease, with only 60 cases described in the literature so far (
https://medlineplus.gov/genetics/condition/snijders-blok-campeau-syndrome/#frequency accessed on 3 April 2024). It is caused by mutations in the chromodomain helicase DNA binding protein 3 on chromosome 17p13, and it is exclusively reported in female patients. All of the previously described mutations were de novo [
10].
The clinical presentation of Snijders Blok–Campeau syndrome is rather broad, with intellectual disability (mild to severe), hypotonia, movement disorders, dyskinesia, spasms, a stiff-legged or wide-base gait, microcephaly, behavioral problems, and epilepsy, as well as additional features like joint hyperlaxity, skin abnormalities, cleft lip, visual impairment, and precocious puberty. MRIs often reveal cerebellar hypoplasia [
5]. The previously described heart defect in our patient does not explain the secondary development of pulmonary arterial hypertension, suggesting a potential primary genetic association.
In our index patient, we found a mutation in the
DDX3X gene, which encodes a certain kind of DEAD-box RNA helicase. Alterations of the Wnt signaling pathway have been described as contributing to pulmonary arterial hypertension [
6,
11].
It is important for transcription, splicing, RNA transport, and translation and has a crucial impact on cellular processes, including cell cycle control, apoptosis, and tumorigenesis. Snijders et al. proved in a knockdown
DDX3X zebrafish a reduced brain size and microcephaly [
5]. Furthermore, Mo et al. described a crucial role for
DDX3X in tumorigenesis to metastasis [
12]. In a study by Chen et al., a correlation between
DDX3X and pulmonary fibrosis was observed. A mouse model of pulmonary fibrosis revealed enhanced survival rates and reduction in lung inflammation after administration of a
DDX3X inhibitor [
13]. You et al. revealed the hypothesis that
DDX3X has the potential to exert an influence on vascular injury in type 2 diabetes mellitus [
14]. A correlation between
DDX3X and the development of pulmonary arterial hypertension has not been described yet.
DDX3X is located in the cytoplasm and nucleus of the cell and has different functions depending on the localization. Rare variants such as
SMAD1,
SMAD4, and
SMAD9 associated with genetics with pulmonary arterial hypertension are also located in the cytoplasm and nucleus. It can therefore be assumed that there is a local relation in the mutation and that cellular processes are influenced accordingly. Presumably, there may be an association of a triggered immune reaction, which can lead to vascular remodeling and thus to pulmonary arterial hypertension [
15,
16].
4. Conclusions
Although the basic biochemical function of DDX3X is well understood, further research on a larger cohort of DDX3X patients is needed to evaluate associations with vasculopathies such as pulmonary arterial hypertension. Another possible way to evaluate DDX3X and PAH would be the development of an animal model (e.g., DDX3X mouse vs. DDX3X knockout mouse).
Author Contributions
Conceptualization, L.P. and V.C.Z.; resources, V.C.Z., M.G. and L.P.; writing—original draft preparation, L.P.; writing—review and editing, V.C.Z. and M.G.; visualization, L.P.; supervision, M.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no specific grant from any funding agency, commercial, or not-for-profit sectors.
Institutional Review Board Statement
This study was conducted in accordance with the Declaration of Helsinki. Ethical approval for the clinical and scientific database for patients with congenital heart disease has been granted by the Ethics Committee of Heidelberg University Hospital for conducting this case report (code: S-547/2016; date: 16 January 2017).
Informed Consent Statement
Written informed consent for publication of this case report was obtained from the patient’s parents.
Data Availability Statement
The data presented in this study are available upon request from the corresponding author due to privacy concerns.
Conflicts of Interest
The authors declare that the research was conducted in absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
| PAPm RPA | mean pulmonary artery pressure measured in the right pulmonary artery |
| PAPm LPA | mean pulmonary artery pressure measured in the left pulmonary artery |
| PCWP | pulmonary artery wedge pressure |
| PVR | pulmonary vascular resistance |
References
- Gorenflo, M.; Ziesenitz, V.C. Treatment of pulmonary arterial hypertension in children. Cardiovasc. Diagn. Ther. 2021, 11, 1144–1159. [Google Scholar] [CrossRef] [PubMed]
- Morrell, N.W.; Aldred, M.A.; Chung, W.K.; Elliott, C.G.; Nichols, W.C.; Soubrier, F.; Trembath, R.C.; Loyd, J.E. Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 2019, 53, 1801899. [Google Scholar] [CrossRef] [PubMed]
- Snijders Blok, L.; Rousseau, J.; Twist, J.; Ehresmann, S.; Takaku, M.; Venselaar, H.; Rodan, L.H.; Nowak, C.B.; Douglas, J.; Swoboda, K.J.; et al. CHD3 helicase domain mutations cause a neurodevelopmental syndrome with macrocephaly and impaired speech and language. Nat. Commun. 2018, 9, 4619. [Google Scholar] [CrossRef] [PubMed]
- Pascual, P.; Tenorio-Castano, J.; Mignot, C.; Afenjar, A.; Arias, P.; Gallego-Zazo, N.; Parra, A.; Miranda, L.; Cazalla, M.; Silván, C.; et al. Snijders Blok–Campeau Syndrome: Description of 20 Additional Individuals with Variants in CHD3 and Literature Review. Genes 2023, 14, 1664. [Google Scholar] [CrossRef] [PubMed]
- Snijders Blok, L.; Madsen, E.; Juusola, J.; Gilissen, C.; Baralle, D.; Reijnders, M.R.F.; Venselaar, H.; Helsmoortel, C.; Cho, M.T.; Hoischen, A.; et al. Mutations in DDX3X Are a Common Cause of Unexplained Intellectual Disability with Gender-Specific Effects on Wnt Signaling. Am. J. Hum. Genet. 2015, 97, 343–352. [Google Scholar] [CrossRef] [PubMed]
- Vuocolo, B.; Holder, J.L. Unwind and Relax: DDX3X RNA Helicase as a Critical Mediator of Cortical Neurogenesis. Neuron 2020, 106, 357–358. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Posey, J.E.; Rosenfeld, J.A.; Bacino, C.A.; Scaglia, F.; Immken, L.; Harris, J.M.; Hickey, S.E.; Mosher, T.M.; Slavotinek, A.; et al. Phenotypic expansion in DDX3X—A common cause of intellectual disability in females. Ann. Clin. Transl. Neurol. 2018, 5, 1277–1285. [Google Scholar] [CrossRef] [PubMed]
- Johnson-Kerner, B.; Snijders Blok, L.; Suit, L.; Thomas, J.; Kleefstra, T.; Sherr, E.H. DDX3X-Related Neurodevelopmental Disorder. In GeneReviews(®); Adam, M.P., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Rosenzweig, E.B.; Abman, S.H.; Adatia, I.; Beghetti, M.; Bonnet, D.; Haworth, S.; Ivy, D.D.; Berger, R.M.F. Paediatric pulmonary arterial hypertension: Updates on definition, classification, diagnostics and management. Eur. Respir. J. 2019, 53, 1801916. [Google Scholar] [CrossRef] [PubMed]
- Drivas, T.G.; Li, D.; Nair, D.; Alaimo, J.T.; Alders, M.; Altmüller, J.; Barakat, T.S.; Bebin, E.M.; Bertsch, N.L.; Blackburn, P.R.; et al. A second cohort of CHD3 patients expands the molecular mechanisms known to cause Snijders Blok-Campeau syndrome. Eur. J. Hum. Genet. 2020, 28, 1422–1431. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Eickhoff, C.; Garcia-Agundez, A.; Bertone, P.; Paudel, S.S.; Tambe, D.T.; Litzky, L.A.; Cox-Flaherty, K.; Klinger, J.R.; Monaghan, S.F.; et al. Transcriptional profiles of pulmonary artery endothelial cells in pulmonary hypertension. Sci. Rep. 2023, 13, 22534. [Google Scholar] [CrossRef] [PubMed]
- Mo, J.; Liang, H.; Su, C.; Li, P.; Chen, J.; Zhang, B. DDX3X: Structure, physiologic functions and cancer. Mol. Cancer 2021, 20, 38. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Pilling, D.; Gomer, R.H. The mRNA-binding protein DDX3 mediates TGF-β1 upregulation of translation and promotes pulmonary fibrosis. JCI Insight 2023, 8, e167566. [Google Scholar] [CrossRef] [PubMed]
- You, S.; Xu, J.; Yin, Z.; Wu, B.; Wang, P.; Hao, M.; Cheng, C.; Liu, M.; Zhao, Y.; Jia, P.; et al. Down-regulation of WWP2 aggravates Type 2 diabetes mellitus-induced vascular endothelial injury through modulating ubiquitination and degradation of DDX3X. Cardiovasc. Diabetol. 2023, 22, 107. [Google Scholar] [CrossRef] [PubMed]
- de Colibus, L.; Stunnenberg, M.; Geijtenbeek, T.B. DDX3X structural analysis: Implications in the pharmacology and innate immunity. Curr. Res. Immunol. 2022, 3, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Li, X.; Cui, X.; Yuan, H.; Wang, H. The roles of immune system and autoimmunity in pulmonary arterial hypertension: A review. Pulm. Pharmacol. Ther. 2022, 72, 102094. [Google Scholar] [CrossRef] [PubMed]
| Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).




{kind=link}
{kind=link}
{kind=link}