New Deletions in the Hermansky-Pudlak Syndrome Type 5 Gene in a Japanese Patient


Abstract
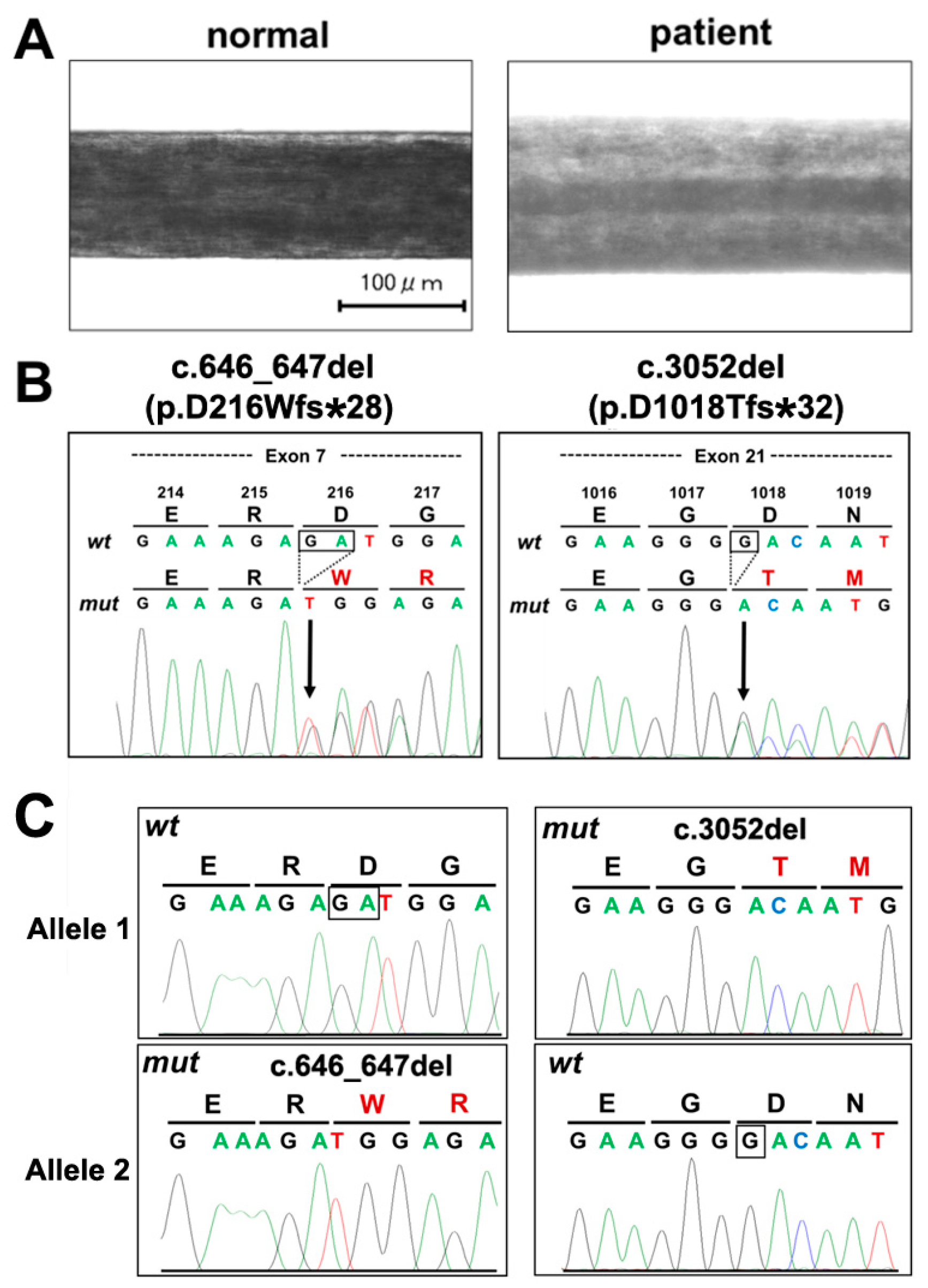
1. Introduction
2. Case Presentation Section
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hermansky, F.; Pudlak, P. Albinism associated with hemorrhagic diathesis and unusual pigmented reticular cells in the bone marrow: Report of two cases with histochemical studies. Blood 1959, 14, 162–169. [Google Scholar] [PubMed]
- Carmona-Rivera, C.; Golas, G.; Hess, R.A.; Cardillo, N.D.; Martin, E.H.; O’Brien, K.; Tsilou, E.; Gochuico, B.R.; White, J.G.; Huizing, M.; et al. Clinical, molecular, and cellular features of non-Puerto Rican Hermansky-Pudlak syndrome patients of Hispanic descent. J. Investig. Dermatol. 2011, 131, 2394–2400. [Google Scholar] [CrossRef] [PubMed]
- Wei, A.H.; Li, W. Hermansky-Pudlak syndrome: Pigmentary and non-pigmentary defects and their pathogenesis. Pigment Cell Melanoma. Res. 2008, 26, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Guiu, I.; Torregrosa, J.M.; Velasco, F.; Antón, A.I.; Lozano, M.L.; Vicente, V.; Rivera, J. Hermansky-Pudlak syndrome. Overview of clinical and molecular features and case report of a new HPS-1 variant. Hamostaseologie. 2014, 34, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, M.; AI-Hashmi, N.; Al-Rashdi, S.; AI-Sukaiti, N.; AI-Adawi, A.; AI-Riyami, M.; AI-Maawali, A. Biallelic mutations in AP3D1 cause Hermansky-Pudlak syndrome type 10 associated with immunodeficiency and seizure disorder. Eur. J. Med. Genet. 2018, 22. [Google Scholar] [CrossRef] [PubMed]
- Wei, M.L. Hermansky-Pudlak syndrome: A disease of protein trafficking and organelle function. Pigment Cell Res. 2006, 19, 19–42. [Google Scholar] [CrossRef]
- Marks, M.S.; Heijnen, H.F.; Raposo, G. Lysosome-related organelles: Unusual compartments become mainstream. Curr. Opin. Cell Biol. 2013, 25, 495–505. [Google Scholar] [CrossRef]
- Adema, G.J.; de Boer, A.J.; Vogel, A.M.; Loenen, W.A.; Figdor, C.G. Molecular characterization of the melanocyte lineage-specific antigen gp100. J. Biol. Chem. 1994, 269, 20126–20133. [Google Scholar]
- Morgan, N.V.; Pasha, S.; Johnson, C.A.; Ainsworth, J.R.; Eady, R.A.; Dawood, B.; McKeown, C.; Trembath, R.C.; Wilde, J.; Watson, S.P.; et al. A germline mutation in BLOC1S3/reduced pigmentation causes a novel variant of Hermansky-Pudlak syndrome (HPS8). Am. J. Hum. Genet. 2006, 78, 160–166. [Google Scholar] [CrossRef]
- Di Pietro, S.M.; Falcon-Perez, J.M.; Dell’Angelica, E.C. Characterization of BLOC-2, a complex containing the Hermansky-Pudlak syndrome proteins HPS3, HPS5 and HPS6. Traffic 2004, 5, 276–283. [Google Scholar] [CrossRef]
- Nazarian, R.; Falcon-Perez, J.M.; Dell’Angelica, E.C. Biogenesis of lysosome-related organelles complex 3 (BLOC-3): A complex containing the Hermansky-Pudlak syndrome (HPS) proteins HPS1 and HPS4. Proc. Natl. Acad. Sci. USA 2003, 100, 8770–8775. [Google Scholar] [CrossRef]
- Korswagen, L.A.; Huizing, M.; Simsek, S.; Janssen, J.J.; Zweegman, S. A novel mutation in a Turkish patient with Hermansky-Pudlak syndrome type 5. Eur. J. Haematol. 2008, 80, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Santiago Borrero, P.J.; Rodríguez-Pérez, Y.; Renta, J.Y.; Izquierdo, N.J.; Del Fierro, L.; Muñoz, D.; Molina, N.L.; Ramírez, S.; Pagán-Mercado, G.; Ortíz, I.; et al. Genetic testing for oculocutaneous albinism type 1 and 2 and Hermansky-Pudlak syndrome type 1 and 3 mutations in Puerto Rico. J. Investig. Dermatol. 2006, 126, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Okamura, K.; Abe, Y.; Araki, Y.; Wakamatsu, K.; Seishima, M.; Umetsu, T.; Kato, A.; Kawaguchi, M.; Hayashi, M.; Hozumi, Y.; et al. Characterization of melanosomes and melanin in Japanese patients with Hermansky-Pudlak syndrome types 1, 4, 6, and 9. Pigment Cell Melanoma. Res. 2018, 31, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Hussain, N.; Quezado, M.; Huizing, M.; Geho, D.; White, J.G.; Gahl, W.; Mannon, P. Intestinal disease in Hermansky-Pudlak syndrome: Occurrence of colitis and relation to genotype. Clin. Gastroenterol. Hepatol. 2006, 4, 73–80. [Google Scholar] [CrossRef]
- Wolkow, N.; Li, Y.; Maminishkis, A.; Song, Y.; Alekseev, O.; Iacovelli, J.; Song, D.; Lee, J.C.; Dunaief, J.L. Iron upregulates melanogenesis in cultured retinal pigment epithelial cells. Exp. Eye Res. 2014, 128, 92–101. [Google Scholar] [CrossRef]
- Itoh, Y.; Nagaoka, Y.; Katakura, Y.; Kawahara, H.; Takemori, H. Simple chronic colitis model using hypopigmented mice with a Hermansky-Pudlak syndrome 5 gene mutation. Pigment Cell Melanoma. Res. 2016, 29, 578–582. [Google Scholar] [CrossRef]
- Felipez, L.M.; Gokhale, R.; Guandalini, S. Hermansky-Pudlak syndrome: Severe colitis and good response to infliximab. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 665–667. [Google Scholar] [CrossRef]
- Huizing, M.; Hess, R.; Dorward, H.; Claassen, D.A.; Helip-Wooley, A.; Kleta, R.; Kaiser-Kupfer, M.I.; White, J.G.; Gahl, W.A. Cellular, molecular and clinical characterization of patients with Hermansky-Pudlak syndrome type 5. Traffic 2004, 5, 711–722. [Google Scholar] [CrossRef]
- Ringeisen, A.L.; Schimmenti, L.A.; White, J.G.; Schoonveld, C.; Summers, C.G. Hermansky-Pudlak syndrome (HPS5) in a nonagenarian. J AAPOS. 2013, 17, 334–336. [Google Scholar] [CrossRef]
- Stephen, J.; Yokoyama, T.; Tolman, N.J.; O’Brien, K.J.; Nicoli, E.R.; Brooks, B.P.; Huryn, L.; Titus, S.A.; Adams, D.R.; Chen, D.; Gahl, W.A.; et al. Cellular and molecular defects in a patient with Hermansky-Pudlak syndrome type 5. PLoS ONE 2017, 12, e0173682. [Google Scholar] [CrossRef] [PubMed]
- Michaud, V.; Lasseaux, E.; Plaisant, C.; Verloes, A.; Perdomo-Trujillo, Y.; Hamel, C.; Elcioglu, N.H.; Leroy, B.; Kaplan, J.; Jouk, P.S.; et al. Clinico-molecular analysis of eleven patients with Hermansky-Pudlak type 5 syndrome, a mild form of HPS. Pigment Cell Melanoma. Res. 2017, 30, 563–570. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Hematology | Coagulation | ||||
|---|---|---|---|---|---|
| WBC | 42 × 102/µL | sedimentation | 19s | ||
| RBC | 409 × 104/µL | PT | 12.7s | ||
| Neutrophil | 47.9% | PT-INR | 1.12 | ||
| Lymphocyte | 34.8% | AP | 79% | ||
| Monocyte | 12.7% | APTT | 28.6 s | ||
| Eosinophil | 2.7% | fibrinogen | 271 mg/dL | ||
| Basophil | 1.9% | FDP | 2.4 µg/dL | ||
| HGB | 6.8 g/dL | L | D dimer | 0.8 µg/dL | |
| HCT | 24.4% | L | BT | 4.5 m | |
| MCV | 59.7 fL | L | |||
| MCH | 16.6% | L | |||
| MCHC | 27.9% | L | |||
| platelet | 21.7 × 104/µL | ||||
| Biochemistry | |||||
| albumin | 4.2 g/dL | Na | 138 mEq/L | ||
| globulin | 3.2 g/dL | K | 4.1 mEq/L | ||
| amylase | 30 U/L | Cl | 103 mEq/L | ||
| AST | 12 U/L | L | Ca | 9.0 mEq/L | |
| ALT | 7 U/L | L | Fe | 10 µg/dL | L |
| LDH | 127 U/L | ferritin | <11.0 ng/ml | ||
| ALP | 117 U/L | TIBC | 393 µg/dL | L | |
| γ-GTP | 10 U/L | UIBC | 383 µg/dL | ||
| TP | 7.4 g/dL | CRP | 0.2 mg/dL | H | |
| ALB | 4.2 g/dL | eGFR | 101 mL/min | ||
| Ch-E | 152 U/L | L | hemolysis | 0 | |
| blood glucose | 87 mg/dL | milky fluid | 0 | ||
| BUN | 10 mg/dL | choloplania | 3 | ||
| CRE | 0.57 mg/dL | ||||
| CK | 23 U/L | L | |||
| UA | 4.1 mg/dL | ||||
| T-CHO | 153 mg/dL | ||||
| TG | 110 mg/dL | ||||
| T-Bil | 0.5 mg/dL | ||||
| DB | 0.1 mg/dL | ||||
| IB | 0.4 mg/dL | ||||
| Exon | Previously Reported Variants (1) | New Variants | ||
|---|---|---|---|---|
| 3 | c.219G>A | |||
| 5 | c.285-10A>G | c.302_305del | c.434G>A | |
| 7 | c.719G>C | c.818_822del | del 1.4 kb | c.646_647del |
| 8 | c.879dup | c.888_889insA | ||
| 12 | c.1417C>T | c.1423del | ||
| 13 | c.1618C>T | c.1634+1G>A | ||
| 16 | c.1871T>G c.1892T>C | c.1900del c.2026_2029del | c.2219T>C | |
| 18 | c.2593C>T | c.2624del | ||
| 19 | c.2750_2751del | |||
| 20 | c.2926_2929dup | |||
| 21 | c.2979_2982del | c.3034A>G | c.3058+3A>G | c.3052del |
| 22 | c.3096_3098del | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kato, S.; Aoe, T.; Hamamoto, A.; Takemori, H.; Nishikubo, T. New Deletions in the Hermansky-Pudlak Syndrome Type 5 Gene in a Japanese Patient. Reports 2019, 2, 15. https://doi.org/10.3390/reports2020015
Kato S, Aoe T, Hamamoto A, Takemori H, Nishikubo T. New Deletions in the Hermansky-Pudlak Syndrome Type 5 Gene in a Japanese Patient. Reports. 2019; 2(2):15. https://doi.org/10.3390/reports2020015
Chicago/Turabian StyleKato, Shinya, Tsugumi Aoe, Akie Hamamoto, Hiroshi Takemori, and Toshiya Nishikubo. 2019. "New Deletions in the Hermansky-Pudlak Syndrome Type 5 Gene in a Japanese Patient" Reports 2, no. 2: 15. https://doi.org/10.3390/reports2020015
APA StyleKato, S., Aoe, T., Hamamoto, A., Takemori, H., & Nishikubo, T. (2019). New Deletions in the Hermansky-Pudlak Syndrome Type 5 Gene in a Japanese Patient. Reports, 2(2), 15. https://doi.org/10.3390/reports2020015