
The Synergistic Roles of Glial Cells and Non-Coding RNAs in the Pathogenesis of Alzheimer’s Disease and Related Dementias (ADRDs)


Abstract
1. Introduction
1.1. Overview of ADRDs
1.2. Glial Cells in ADRDs
1.3. Non-Coding RNAs in ADRDs
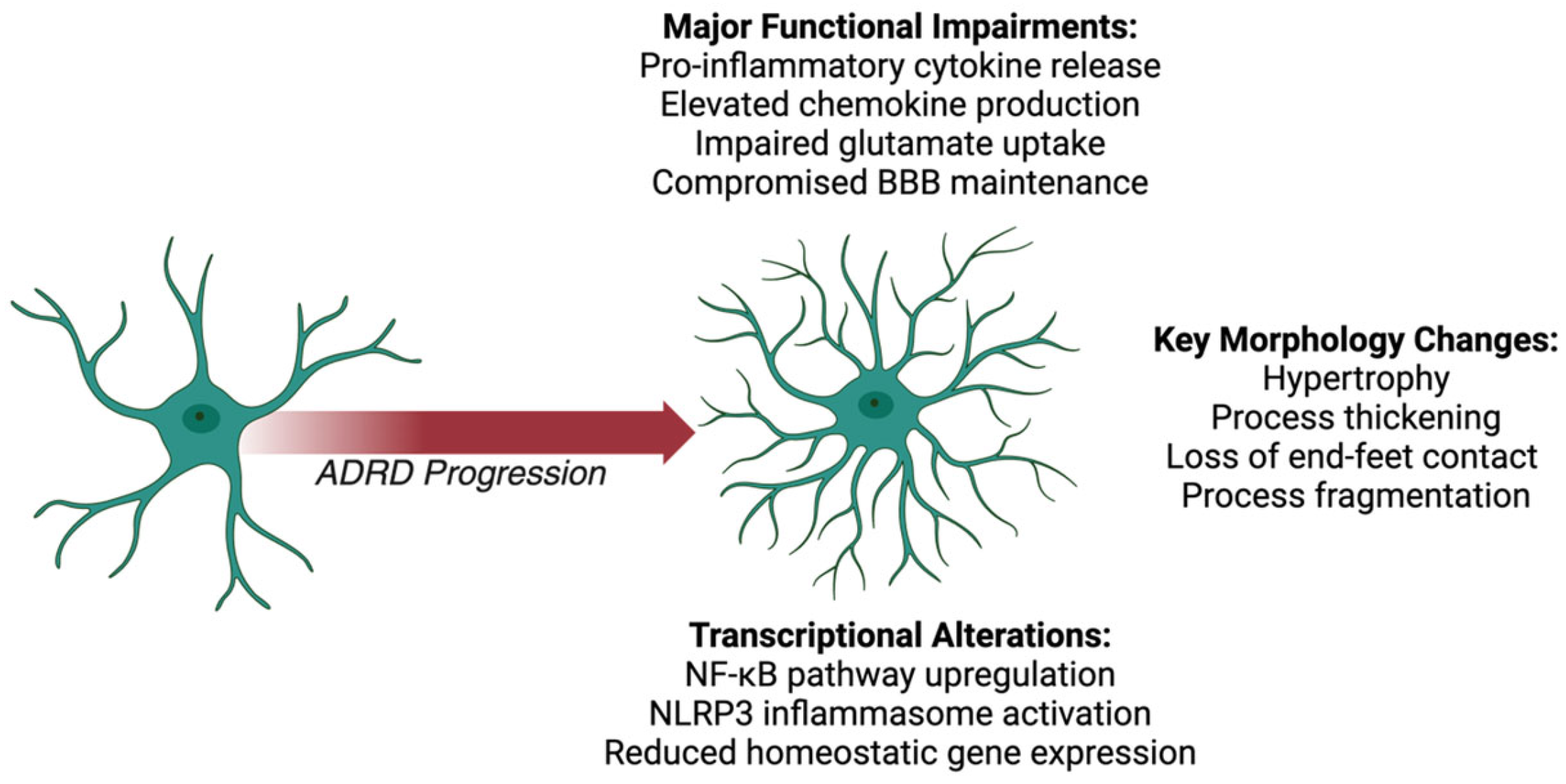
2. Astrocytes
2.1. Astrocyte Morphological Changes
2.2. Astrocyte Functional Impairments
2.3. Astrocyte Transcriptional Alterations

{kind=link}
{kind=link}
{kind=link}
| Morphological Changes | Functional Impairments | Transcriptional Alterations | |
|---|---|---|---|
| All ADRDs |
| ||
| AD | |||
| FTD |
| ||
| LBD | |||
| VCID | |||
| MEDs |
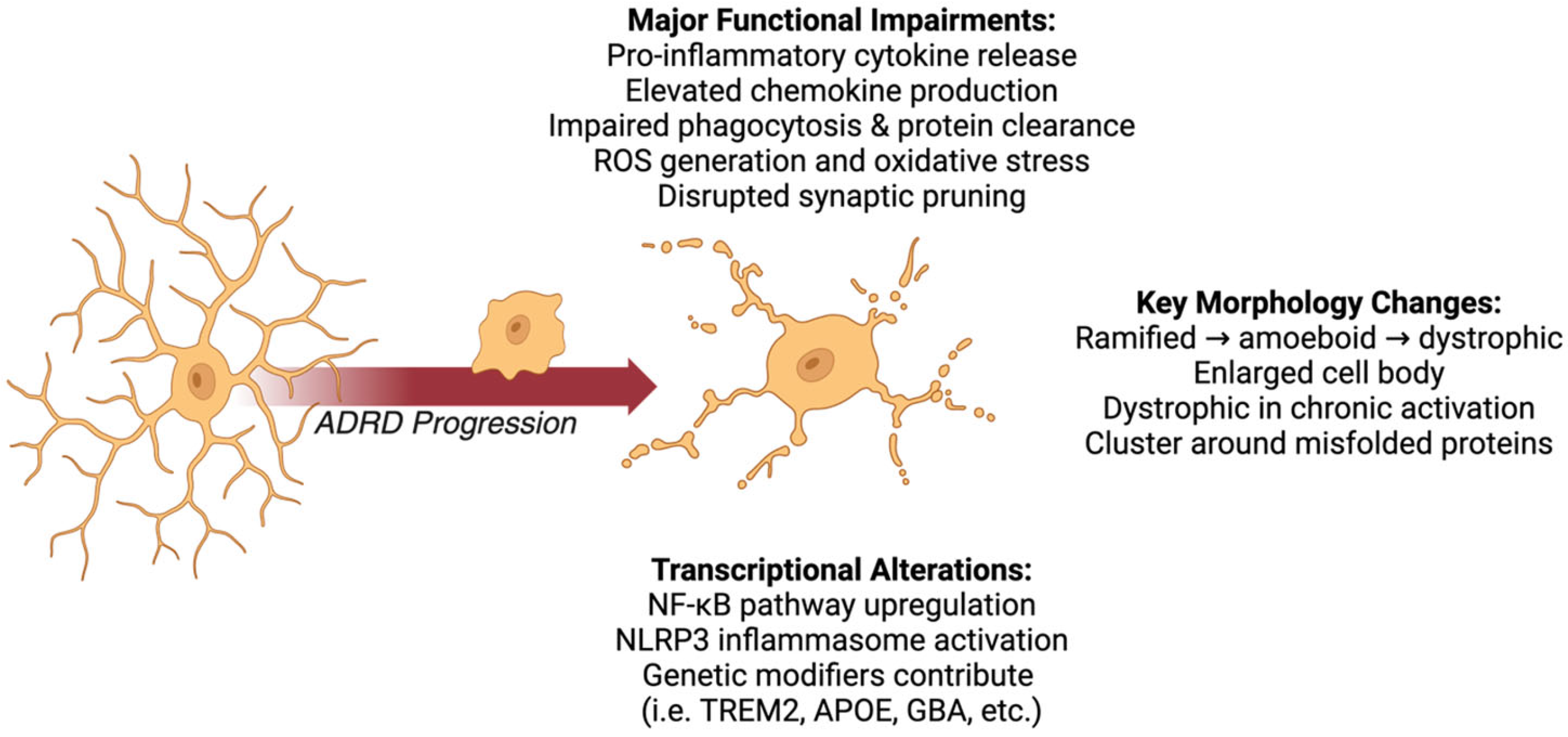
3. Microglia
3.1. Microglial Morphological Changes
3.2. Microglial Functional Impairments
3.3. Microglial Transcriptional Alterations
4. Oligodendrocytes
4.1. Oligodendrocyte Morphological Changes
4.2. Oligodendrocyte Functional Impairments
4.3. Oligodendrocyte Transcriptional Alterations
| Morphological Changes | Functional Impairments | Transcriptional Alterations | |
|---|---|---|---|
| All ADRDs | |||
| AD | |||
| FTD | |||
| LBD | |||
| VCID | |||
| MEDs |
5. Interactions Among Glial Cells
6. Non-Coding RNAs in Glial Cell Ramification, Morphology, and Neuroinflammation
6.1. miRNAs
6.2. lncRNAs
7. Therapeutic Outlooks
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Liang, C.-S.; Li, D.-J.; Yang, F.-C.; Tseng, P.-T.; Carvalho, A.F.; Stubbs, B.; Thompson, T.; Mueller, C.; Shin, J.I.; Radua, J.; et al. Mortality Rates in Alzheimer’s Disease and Non-Alzheimer’s Dementias: A Systematic Review and Meta-Analysis. Lancet Healthy Longev. 2021, 2, e479–e488. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, E.; Lin, P.S.; Kamdar, N.; Gonzales, G.; Norcott, A.; Peterson, M.D. Risk of Early- and Late-onset Alzheimer Disease and Related Dementia in Adults with Cerebral Palsy. Dev. Med. Child Neurol. 2021, 64, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, K.D.; Girling, L.; Berlinger, N. Inclusion of People Living with Alzheimer’s Disease or Related Dementias Who Lack a Study Partner in Social Research: Ethical Considerations from a Qualitative Evidence Synthesis. Dementia 2022, 21, 1200–1218. [Google Scholar] [CrossRef] [PubMed]
- Jicha, G.; Abner, E.L.; Arnold, S.E.; Carrillo, M.C.; Dodge, H.H.; Edland, S.D.; Fargo, K.N.; Feldman, H.; Goldstein, L.B.; Hendrix, J.A.; et al. Committee on High-quality Alzheimer’s Disease Studies (CHADS) Consensus Report. Alzheimer’s Dement. 2021, 18, 1109–1118. [Google Scholar] [CrossRef]
- Jutkowitz, E.; Halladay, C.; Tsai, J.; Hooshyar, D.; Quach, L.; O’Toole, T.; Rudolph, J.L. Prevalence of Alzheimer’s Disease and Related Dementias Among Veterans Experiencing Housing Insecurity. Alzheimer’s Dement. 2021, 18, 1306–1313. [Google Scholar] [CrossRef]
- Keohane, L.M.; Nikpay, S.; Braun, K.; Cheng, A.; Stevenson, D.G.; Buntin, M.B.; Yu, D.; Blot, W.J.; Lipworth, L. Association of Race and Income with Incident Diagnosis of Alzheimer’s Disease and Related Dementias Among Black and White Older Adults. J. Appl. Gerontol. 2022, 42, 898–908. [Google Scholar] [CrossRef]
- Orth, J.; Flowers, D.; Betz, G.; Cagle, J.G. A Systematic Review of Specialty Dementia Care Units in Long-term Care Settings. Int. J. Geriatr. Psychiatry 2023, 38, e5907. [Google Scholar] [CrossRef]
- Trammell, A.R. Characterization of African-American Super-Agers in the National Alzheimer’s Coordinating Center Cohort. J. Am. Geriatr. Soc. 2024, 72, 1995–2005. [Google Scholar] [CrossRef]
- Nguyen, H.Q.; Borson, S.; Khang, P.; Langer-Gould, A.; Wang, S.E.; Carrol, J.A.; Lee, J. Dementia Diagnosis and Utilization Patterns in a Racially Diverse Population Within an Integrated Health Care Delivery System. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2022, 8, e12279. [Google Scholar] [CrossRef]
- Torossian, M. The Dignity of Older Individuals with Alzheimer’s Disease and Related Dementias: A Scoping Review. Dementia 2021, 20, 2891–2915. [Google Scholar] [CrossRef]
- Gianattasio, K.Z.; Moghtaderi, A.; Lupu, D.; Prather, C.; Power, M.C. Evaluation of Federal Policy Changes to the Hospice Benefit and Use of Hospice for Persons With ADRDS. Jama Health Forum 2022, 3, e220900. [Google Scholar] [CrossRef] [PubMed]
- Peacock, S.; Bayly, M.; Duggleby, W.; Ploeg, J.; Pollard, L.; Swindle, J.; Lee, H.J.; Williams, A.; Markle-Reid, M.; McAiney, C. Women’s Caregiving Experience of Older Persons Living with Alzheimer Disease and Related Dementias and Multiple Chronic Conditions: Using Wuest’s Theory. Sage Open Nurs. 2020, 6, 2377960820974816. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.K.; Yelton, B.; Ezeanya, V.; Kannaley, K.; Friedman, D.B. Review of the Content and Quality of Mobile Applications About Alzheimer’s Disease and Related Dementias. J. Appl. Gerontol. 2018, 39, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Pooley, J.; Lee, M.; Shankle, W.R. Understanding Memory Impairment with Memory Models and Hierarchical Bayesian Analysis. J. Math. Psychol. 2011, 55, 47–56. [Google Scholar] [CrossRef]
- Couret, A.; Lapeyre-Mestre, M.; Gombault-Datzenko, E.; Renoux, A.; Villars, H.; Gardette, V. Healthcare Use Patterns Before Alzheimer’s Disease and Related Diseases Identification and Future Healthcare Trajectory. Int. J. Geriatr. Psychiatry 2022, 38, e5849. [Google Scholar] [CrossRef]
- Brown, C.S. Trends in Cause-Specific Mortality Among Persons With Alzheimer’s Disease in South Carolina: 2014 to 2019. Front. Aging Neurosci. 2024, 16, e1387082. [Google Scholar] [CrossRef]
- Langa, K.M.; Foster, N.L.; Larson, E.B. Mixed Dementia: Emerging Concepts and Therapeutic Implications. JAMA 2004, 292, 2901. [Google Scholar] [CrossRef]
- Cross, D.A. Access to Preferred Skilled Nursing Facilities: Transitional Care Pathways for Patients WithAlzheimer’s Disease and Related Dementias. Health Serv. Res. 2023, 59, e14263. [Google Scholar] [CrossRef]
- Shepard, V.; Snih, S.A.; Burke, R.; Downer, B.; Kuo, Y.; Malagaris, I.; Raji, M. Characteristics Associated with Mexican-American Hospice Use: Retrospective Cohort Study Using the Hispanic Established Population for the Epidemiologic Study of the Elderly (H-Epese). Am. J. Hosp. Palliat. Med. 2022, 40, 480–491. [Google Scholar] [CrossRef]
- Lott, S.A. Digital Health Technologies for Alzheimer’s Disease and Related Dementias: Initial Results from a Landscape Analysis and Community Collaborative Effort. J. Prev. Alzheimer’s Dis. 2024, 11, 1480–1489. [Google Scholar] [CrossRef]
- Tosun, D.; Rosen, H.J.; Miller, B.L.; Weiner, M.W.; Schuff, N. MRI Patterns of Atrophy and Hypoperfusion Associations Across Brain Regions in Frontotemporal Dementia. Neuroimage 2012, 59, 2098–2109. [Google Scholar] [CrossRef] [PubMed]
- Mina, C.B.M.; Ana-Marija, V.T.; Olga, Ž.V.; Milana, O.S.; Sanja, P.S.; Sanja, B.S. Kleptomania, a Symptom of Depression or Frontotemporal Dementia? World J. Biol. Pharm. Health Sci. 2021, 6, 29–33. [Google Scholar] [CrossRef]
- Ting, H.S.; Yashodhara, B.M.; Yousuf, U.A.M.; Dar, B.A.; Abas, A.L. A Case of Frontotemporal Dementia. Int. J. Case Rep. Images 2013, 4, 179. [Google Scholar] [CrossRef]
- Bessing, Y.F. Mixed Dementia Alzheimer and Lewy Body with Congenital Diseases: Case Study. World J. Adv. Res. Rev. 2023, 20, 137–140. [Google Scholar] [CrossRef]
- Foster, E.R.; Campbell, M.C.; Burack, M.A.; Hartlein, J.; Flores, H.; Cairns, N.J.; Hershey, T.; Perlmutter, J.S. Amyloid Imaging of Lewy Body-associated Disorders. Mov. Disord. 2010, 25, 2516–2523. [Google Scholar] [CrossRef]
- Al-faham, Z.; Zein, R.; Wong, C.-Y.O. 18f-FDG PET Assessment of Lewy Body Dementia with Cerebellar Diaschisis. J. Nucl. Med. Technol. 2014, 42, 306–307. [Google Scholar] [CrossRef]
- Huang, J.; Li, C.; Shang, H. Astrocytes in Neurodegeneration: Inspiration from Genetics. Front. Neurosci. 2022, 16, e882316. [Google Scholar] [CrossRef]
- Mavroudis, I.; Petridis, F.; Chatzikonstantinou, S.; Mckenn, J.; Karantali, E.; Kazis, D. The Role of Astrocytes in Astrocytes Alzheimer’s Disease. Ann. Acad. Rom. Sci. Ser. Biol. Sci. 2020, 9, 65–79. [Google Scholar] [CrossRef]
- Vicente-Acosta, A.; Giménez-Cassina, A.; Díaz-Nido, J.; Loría, F. The Smoothened Agonist SAG Reduces Mitochondrial Dysfunction and Neurotoxicity of Frataxin-Deficient Astrocytes. J. Neuroinflamm. 2022, 19, 93. [Google Scholar] [CrossRef]
- Peteri, U.-K.; Niukkanen, M.; Castrén, M. Astrocytes in Neuropathologies Affecting the Frontal Cortex. Front. Cell. Neurosci. 2019, 13, 44. [Google Scholar] [CrossRef]
- Early, A.N.; Weekman, E.M.; Lee, T.; Wilcock, D.M. Contribution of Astrocytic MMP9 toward Progression of VCID Pathology. Alzheimer’s Dement. 2022, 18 (Suppl. S3), e065982. [Google Scholar] [CrossRef]
- Sompol, P. Targeting Reactive Astrocytes in Vascular Dementia: Investigation of Neuronal-Astrocyte-Vascular Interactions. J. Exp. Neurosci. 2024, 19, 26331055241255332. [Google Scholar] [CrossRef] [PubMed]
- Altay, M.F.; Liu, A.K.L.; Holton, J.L.; Parkkinen, L.; Lashuel, H.A. Prominent Astrocytic Alpha-Synuclein Pathology with Unique Post-Translational Modification Signatures Unveiled across Lewy Body Disorders. Acta Neuropathol. Commun. 2022, 10, 163. [Google Scholar] [CrossRef] [PubMed]
- Meares, G.P.; Benveniste, E.N. Inflammation and the Pathophysiology of Astrocytes in Neurodegenerative Diseases. In Neuroinflammation and Neurodegeneration; Springer: New York, NY, USA, 2014; pp. 61–80. [Google Scholar] [CrossRef]
- Monterey, M.; Wei, H.; Wu, X.; Wu, J.Q. The Many Faces of Astrocytes in Alzheimer’s Disease. Front. Neurol. 2021, 12, 619626. [Google Scholar] [CrossRef]
- Bi, F.; Huang, C.; Tong, J.; Qiu, G.; Huang, B.; Wu, Q.; Li, F.; Xu, Z.; Bowser, R.; Xia, X.-G.; et al. Reactive astrocytes secrete lcn2 to promote neuron death. Proc. Natl. Acad. Sci. USA 2013, 110, 4069–4074. [Google Scholar] [CrossRef]
- Pérez-Sala, D.; Pajares, M.A. Appraising the Role of Astrocytes as Suppliers of Neuronal Glutathione Precursors. Int. J. Mol. Sci. 2023, 24, 8059. [Google Scholar] [CrossRef]
- Barbierato, M.; Borri, M.; Facci, L.; Zusso, M.; Skaper, S.D.; Giusti, P. Expression and Differential Responsiveness of Central Nervous System Glial Cell Populations to the Acute Phase Protein Serum Amyloid A. Sci. Rep. 2017, 7, 12158. [Google Scholar] [CrossRef]
- Yasuno, F.; Kosaka, J.; Ota, M.; Higuchi, M.; Ito, H.; Fujimura, Y.; Nozaki, S.; Takahashi, S.; Mizukami, K.; Asada, T.; et al. Increased binding of peripheral benzodiazepine receptor in mild cognitive impairment–dementia converters measured by positron emission tomography with [11C]DAA1106. Psychiatry Res. 2012, 203, 67–74. [Google Scholar] [CrossRef]
- Hamelin, L.; Lagarde, J.; Dorothée, G.; Leroy, C.; Labit, M.; Comley, R.A.; de Souza, L.C.; Corne, H.; Dauphinot, L.; Bertoux, M.; et al. Early and protective microglial activation in Alzheimer’s disease: A prospective study using18F-DPA-714 PET imaging. Brain 2016, 139 Pt 4, 1252–1264. [Google Scholar] [CrossRef]
- Carter, S.F.; Herholz, K.; Rosa-Neto, P.; Pellerin, L.; Nordberg, A.; Zimmer, E.R. Astrocyte Biomarkers in Alzheimer’s Disease. Trends Mol. Med. 2019, 25, 77–95. [Google Scholar] [CrossRef]
- Dean, D.C.; Sojkova, J.; Hurley, S.; Kecskemeti, S.; Okonkwo, O.; Bendlin, B.B.; Theisen, F.; Johnson, S.C.; Alexander, A.L.; Gallagher, C.L. Alterations of Myelin Content in Parkinson’s Disease: A Cross-Sectional Neuroimaging Study. PLoS ONE 2016, 11, e0163774. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer Disease—A Brief Review of the Basic Science and Clinical Literature. Cold Spring Harb. Perspect. Med. 2011, 2, a006346. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Kim, W.-K.; Wellman, L.L.; Sanford, L.D.; Guo, M.-L. Short-Term Sleep Fragmentation Dysregulates Autophagy in a Brain Region-Specific Manner. Life 2021, 11, 1098. [Google Scholar] [CrossRef] [PubMed]
- Krupp, S.; Hubbard, I.; Tam, O.; Hammell, G.M.; Dubnau, J. TDP-43 pathology in Drosophila induces glial-cell type specific toxicity that can be ameliorated by knock-down of SF2/SRSF1. PLoS Genet. 2023, 19, e1010973. [Google Scholar] [CrossRef]
- Wahl, D.; Grant, R.A.; LaRocca, T.J. The reverse transcriptase inhibitor 3TC modulates hippocampal transcriptome signatures of inflammation in tauopathy model mice. Exp. Gerontol. 2024, 192, 112458. [Google Scholar] [CrossRef]
- Wahl, D.; Smith, M.E.; McEntee, C.M.; Cavalier, A.N.; Osburn, S.C.; Burke, S.D.; Grant, R.A.; Nerguizian, D.; Lark, D.S.; Link, C.D.; et al. The reverse transcriptase inhibitor 3TC protects against age-related cognitive dysfunction. Aging Cell 2023, 22, e13798. [Google Scholar] [CrossRef]
- Ochoa, E.; Ramirez, P.; Gonzalez, E.; De Mange, J.; Ray, W.J.; Bieniek, K.F.; Frost, B. Pathogenic tau–induced transposable element–derived dsRNA drives neuroinflammation. Sci. Adv. 2023, 9, eabq5423. [Google Scholar] [CrossRef]
- Kempuraj, D.; Khan, M.M.; Thangavel, R.; Xiong, Z.; Yang, E.; Zaheer, A. Glia Maturation Factor Induces Interleukin-33 Release from Astrocytes: Implications for Neurodegenerative Diseases. J. Neuroimmune Pharmacol. 2013, 8, 643–650. [Google Scholar] [CrossRef]
- Thomas, E.O.; Zuniga, G.; Sun, W.; Frost, B. Awakening the dark side: Retrotransposon activation in neurodegenerative disorders. Curr. Opin. Neurobiol. 2020, 61, 65–72. [Google Scholar] [CrossRef]
- Torre, C.D.; Zambello, R.; Cacciavillani, M.; Campagnolo, M.; Berno, T.; Salvalaggio, A.; De March, E.; Barilà, G.; Lico, A.; Lucchetta, M.; et al. Lenalidomide long-term neurotoxicity: Clinical and neurophysiologic prospective study. Neurology 2016, 87, 1161–1166. [Google Scholar] [CrossRef]
- de Medicina, I.P.N.M.E.; Vespero, F.H. Epigenetic Regulation of Gene Expression in the Development of Neurodegenerative Diseases: A Narrative Review. Int. J. Med. Sci. Clin. Res. Stud. 2023, 3, 2385–2393. [Google Scholar] [CrossRef]
- Neal, M.; Richardson, J.R. Epigenetic regulation of astrocyte function in neuroinflammation and neurodegeneration. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2018, 1864, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Reichenbach, N.; Delekate, A.; Plescher, M.; Schmitt, F.; Krauss, S.; Blank, N.; Halle, A.; Petzold, G.C. Inhibition of Stat3-mediated astrogliosis ameliorates pathology in an Alzheimer’s disease model. EMBO Mol. Med. 2019, 11, e9665. [Google Scholar] [CrossRef] [PubMed]
- Cooper, M.L.; Pasini, S.; Lambert, W.S.; D’Alessandro, K.B.; Yao, V.; Risner, M.L.; Calkins, D.J. Redistribution of metabolic resources through astrocyte networks mitigates neurodegenerative stress. Proc. Natl. Acad. Sci. USA 2020, 117, 18810–18821. [Google Scholar] [CrossRef]
- Martorana, F.; Foti, M.; Virtuoso, A.; Gaglio, D.; Aprea, F.; Latronico, T.; Rossano, R.; Riccio, P.; Papa, M.; Alberghina, L.; et al. Differential Modulation of NF-κB in Neurons and Astrocytes Underlies Neuroprotection and Antigliosis Activity of Natural Antioxidant Molecules. Oxidative Med. Cell. Longev. 2019, 2019, 8056904. [Google Scholar] [CrossRef]
- Duran, R.C.-D.; Wang, C.-Y.; Zheng, H.; Deneen, B.; Wu, J.Q. Brain Region-Specific Gene Signatures Revealed by Distinct Astrocyte Subpopulations Unveil Links to Glioma and Neurodegenerative Diseases. ENEURO 2019, 6, ENEURO.0288-18.2019. [Google Scholar] [CrossRef]
- Diaz-Castro, B.; Gangwani, M.R.; Yu, X.; Coppola, G.; Khakh, B.S. Astrocyte molecular signatures in Huntington’s disease. Sci. Transl. Med. 2019, 11, eaaw8546. [Google Scholar] [CrossRef]
- Reid, J.K.; Kuipers, H.F. She Doesn’t Even Go Here: The Role of Inflammatory Astrocytes in CNS Disorders. Front. Cell. Neurosci. 2021, 15, 704884. [Google Scholar] [CrossRef]
- Smith, H.L.; Freeman, O.J.; Butcher, A.J.; Holmqvist, S.; Humoud, I.; Schätzl, T.; Hughes, D.T.; Verity, N.C.; Swinden, D.P.; Hayes, J.; et al. Astrocyte Unfolded Protein Response Induces a Specific Reactivity State that Causes Non-Cell-Autonomous Neuronal Degeneration. Neuron 2020, 105, 855–866.e5. [Google Scholar] [CrossRef]
- Hartmann, K.; Sepulveda-Falla, D.; Rose, I.V.L.; Madore, C.; Muth, C.; Matschke, J.; Butovsky, O.; Liddelow, S.; Glatzel, M.; Krasemann, S. Complement 3+-astrocytes are highly abundant in prion diseases, but their abolishment led to an accelerated disease course and early dysregulation of microglia. Acta Neuropathol. Commun. 2019, 7, 83. [Google Scholar] [CrossRef]
- Vallee, K.-A.J.; Fields, J.A. Caloric Restriction Mimetic 2-Deoxyglucose Reduces Inflammatory Signaling in Human Astrocytes: Implications for Therapeutic Strategies Targeting Neurodegenerative Diseases. Brain Sci. 2022, 12, 308. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.X.; Sibon, O.C.M.; Dijkers, P.F. Inhibition of NF-κB in astrocytes is sufficient to delay neurodegeneration induced by proteotoxicity in neurons. J. Neuroinflamm. 2018, 15, 261. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, R.P.; Koledova, V.V.; Schneider, K.; Sambandan, T.G.; Grayson, A.; Zeidman, G.; Artamonova, A.; Sambanthamurthi, R.; Fairus, S.; Sinskey, A.J.; et al. Palm Fruit Bioactives modulate human astrocyte activity in vitro altering the cytokine secretome reducing levels of TNFα, RANTES and IP-10. Sci. Rep. 2018, 8, 16423. [Google Scholar] [CrossRef] [PubMed]
- Alami, N.O.; Schurr, C.; Heuvel, F.O.; Tang, L.; Li, Q.; Tasdogan, A.; Kimbara, A.; Nettekoven, M.; Ottaviani, G.; Raposo, C.; et al. NF-κB activation in astrocytes drives a stage-specific beneficial neuroimmunological response in ALS. EMBO J. 2018, 37, e98697. [Google Scholar] [CrossRef]
- Oksanen, M.; Lehtonen, S.; Jaronen, M.; Goldsteins, G.; Hämäläinen, R.H.; Koistinaho, J. Astrocyte alterations in neurodegenerative pathologies and their modeling in human induced pluripotent stem cell platforms. Cell. Mol. Life Sci. 2019, 76, 2739–2760. [Google Scholar] [CrossRef]
- Leng, K.; Rooney, B.; McCarthy, F.; Xia, W.; Rose, I.V.L.; Bax, S.; Chin, M.; Fathi, S.; Herrington, K.A.; Leonetti, M.; et al. mTOR activation induces endolysosomal remodeling and nonclassical secretion of IL-32 via exosomes in inflammatory reactive astrocytes. J. Neuroinflamm. 2024, 21, 198. [Google Scholar] [CrossRef]
- Miyazaki, I.; Asanuma, M. Neuron-Astrocyte Interactions in Parkinson’s Disease. Cells 2020, 9, 2623. [Google Scholar] [CrossRef]
- Sofroniew, M.V. Astrocyte Reactivity: Subtypes, States, and Functions in CNS Innate Immunity. Trends Immunol. 2020, 41, 758–770. [Google Scholar] [CrossRef]
- Yin, S.; Ma, X.-Y.; Sun, Y.-F.; Yin, Y.-Q.; Long, Y.; Zhao, C.-L.; Ma, J.-W.; Li, S.; Hu, Y.; Li, M.-T.; et al. RGS5 augments astrocyte activation and facilitates neuroinflammation via TNF signaling. J. Neuroinflamm. 2023, 20, 203. [Google Scholar] [CrossRef]
- Limbad, C.; Oron, T.R.; Alimirah, F.; Davalos, A.R.; Tracy, T.E.; Gan, L.; Desprez, P.-Y.; Campisi, J. Astrocyte senescence promotes glutamate toxicity in cortical neurons. PLoS ONE 2020, 15, e0227887. [Google Scholar] [CrossRef]
- Xingi, E.; Koutsoudaki, P.N.; Thanou, I.; Phan, M.-S.; Margariti, M.; Scheller, A.; Tinevez, J.-Y.; Kirchhoff, F.; Thomaidou, D. LPS-Induced Systemic Inflammation Affects the Dynamic Interactions of Astrocytes and Microglia with the Vasculature of the Mouse Brain Cortex. Cells 2023, 12, 1418. [Google Scholar] [CrossRef] [PubMed]
- Perriot, S.; Mathias, A.; Perriard, G.; Canales, M.; Jonkmans, N.; Merienne, N.; Meunier, C.; El Kassar, L.; Perrier, A.L.; Laplaud, D.-A.; et al. Human Induced Pluripotent Stem Cell-Derived Astrocytes Are Differentially Activated by Multiple Sclerosis-Associated Cytokines. Stem Cell Rep. 2018, 11, 1199–1210. [Google Scholar] [CrossRef] [PubMed]
- Taha, D.M.; Clarke, B.E.; Hall, C.E.; Tyzack, G.E.; Ziff, O.J.; Greensmith, L.; Kalmar, B.; Ahmed, M.; Alam, A.; Thelin, E.P.; et al. Astrocytes display cell autonomous and diverse early reactive states in familial amyotrophic lateral sclerosis. Brain 2022, 145, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B.E.; Taha, D.M.; Tyzack, G.E.; Patani, R. Regionally encoded functional heterogeneity of astrocytes in health and disease: A perspective. Glia 2020, 69, 20–27. [Google Scholar] [CrossRef]
- Michal, I.; Guy, S.S.; Michel, R. Astrocytes in Pathogenesis of Multiple Sclerosis and Potential Translation Into Clinic. Glia Health Dis. 2020, 131. Available online: https://www.intechopen.com/chapters/68613 (accessed on 3 April 2025).
- Tremblay, M.-E.; Cookson, M.R.; Civiero, L. Glial phagocytic clearance in Parkinson’s disease. Mol. Neurodegener. 2019, 14, 16. [Google Scholar] [CrossRef]
- Shigetomi, E.; Saito, K.; Sano, F.; Koizumi, S. Aberrant Calcium Signals in Reactive Astrocytes: A Key Process in Neurological Disorders. Int. J. Mol. Sci. 2019, 20, 996. [Google Scholar] [CrossRef]
- Pathak, D.; Sriram, K. Neuron-astrocyte omnidirectional signaling in neurological health and disease. Front. Mol. Neurosci. 2023, 16, 1169320. [Google Scholar] [CrossRef]
- Huang, X.; Su, Y.; Wang, N.; Li, H.; Li, Z.; Yin, G.; Chen, H.; Niu, J.; Yi, C. Astroglial Connexins in Neurodegenerative Diseases. Front. Mol. Neurosci. 2021, 14, 657514. [Google Scholar] [CrossRef]
- Lee, J.; Kim, S.W.; Kim, K.-T. Region-Specific Characteristics of Astrocytes and Microglia: A Possible Involvement in Aging and Diseases. Cells 2022, 11, 1902. [Google Scholar] [CrossRef]
- Santiago-Balmaseda, A.; Aguirre-Orozco, A.; Valenzuela-Arzeta, I.E.; Villegas-Rojas, M.M.; Pérez-Segura, I.; Jiménez-Barrios, N.; Hurtado-Robles, E.; Rodríguez-Hernández, L.D.; Rivera-German, E.R.; Guerra-Crespo, M.; et al. Neurodegenerative Diseases: Unraveling the Heterogeneity of Astrocytes. Cells 2024, 13, 921. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Ries, M.; Sastre, M. Mechanisms of Aβ Clearance and Degradation by Glial Cells. Front. Aging Neurosci. 2016, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-C.; Hu, J.; Zhao, N.; Wang, J.; Wang, N.; Cirrito, J.R.; Kanekiyo, T.; Holtzman, D.M.; Bu, G. Astrocytic LRP1 Mediates Brain Aβ Clearance and Impacts Amyloid Deposition. J. Neurosci. 2017, 37, 4023–4031. [Google Scholar] [CrossRef]
- Wahrle, S.E.; Jiang, H.; Parsadanian, M.; Legleiter, J.; Han, X.; Fryer, J.D.; Kowalewski, T.; Holtzman, D.M. ABCA1 Is Required for Normal Central Nervous System ApoE Levels and for Lipidation of Astrocyte-secreted apoE. J. Biol. Chem. 2004, 279, 40987–40993. [Google Scholar] [CrossRef]
- Pomilio, C.; Pavia, P.; Gorojod, R.M.; Vinuesa, A.; Alaimo, A.; Galvan, V.; Kotler, M.L.; Beauquis, J.; Saravia, F. Glial alterations from early to late stages in a model of Alzheimer’s disease: Evidence of autophagy involvement in Aβ internalization. Hippocampus 2015, 26, 194–210. [Google Scholar] [CrossRef]
- Prasad, H.; Rao, R. Amyloid clearance defect in ApoE4 astrocytes is reversed by epigenetic correction of endosomal pH. Proc. Natl. Acad. Sci. USA 2018, 115, E6640–E6649. [Google Scholar] [CrossRef]
- Gotoh, M.; Miyamoto, Y.; Ikeshima-Kataoka, H. Astrocytic Neuroimmunological Roles Interacting with Microglial Cells in Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 1599. [Google Scholar] [CrossRef]
- Mulica, P.; Grünewald, A.; Pereira, S.L. Astrocyte-Neuron Metabolic Crosstalk in Neurodegeneration: A Mitochondrial Perspective. Front. Endocrinol. 2021, 12, 668517. [Google Scholar] [CrossRef]
- Suga, M.; Kondo, T.; Inoue, H. Modeling Neurological Disorders with Human Pluripotent Stem Cell-Derived Astrocytes. Int. J. Mol. Sci. 2019, 20, 3862. [Google Scholar] [CrossRef]
- Leventoux, N.; Morimoto, S.; Imaizumi, K.; Sato, Y.; Takahashi, S.; Mashima, K.; Ishikawa, M.; Sonn, I.; Kondo, T.; Watanabe, H.; et al. Human Astrocytes Model Derived from Induced Pluripotent Stem Cells. Cells 2020, 9, 2680. [Google Scholar] [CrossRef] [PubMed]
- Ceyzériat, K.; Ben Haim, L.; Denizot, A.; Pommier, D.; Matos, M.; Guillemaud, O.; Palomares, M.-A.; Abjean, L.; Petit, F.; Gipchtein, P.; et al. Modulation of astrocyte reactivity improves functional deficits in mouse models of Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 104. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kwon, S.-H.; Kam, T.-I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic α-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-S.; Kam, T.-I.; Lee, S.; Park, H.; Oh, Y.; Kwon, S.-H.; Song, J.-J.; Kim, D.; Kim, H.; Jhaldiyal, A.; et al. Blocking microglial activation of reactive astrocytes is neuroprotective in models of Alzheimer’s disease. Acta Neuropathol. Commun. 2021, 9, 78. [Google Scholar] [CrossRef]
- Bai, Y.; Su, X.; Piao, L.; Jin, Z.; Jin, R. Involvement of Astrocytes and microRNA Dysregulation in Neurodegenerative Diseases: From Pathogenesis to Therapeutic Potential. Front. Mol. Neurosci. 2021, 14, 556215. [Google Scholar] [CrossRef]
- Boal, A.M.; Risner, M.L.; Cooper, M.L.; Wareham, L.K.; Calkins, D.J. Astrocyte Networks as Therapeutic Targets in Glaucomatous Neurodegeneration. Cells 2021, 10, 1368. [Google Scholar] [CrossRef]
- Matias, I.; Morgado, J.; Gomes, F.C.A. Astrocyte Heterogeneity: Impact to Brain Aging and Disease. Front. Aging Neurosci. 2019, 11, 59. [Google Scholar] [CrossRef]
- Edison, P. Astroglial activation: Current concepts and future directions. Alzheimer’s Dement. 2024, 20, 3034–3053. [Google Scholar] [CrossRef]
- Jacquet, A.d.R. Preparation and Co-Culture of iPSC-Derived Dopaminergic Neurons and Astrocytes. Curr. Protoc. Cell Biol. 2019, 85, e98. [Google Scholar] [CrossRef]
- Tassoni, A.; Farkhondeh, V.; Itoh, Y.; Itoh, N.; Sofroniew, M.V.; Voskuhl, R.R. The astrocyte transcriptome in EAE optic neuritis shows complement activation and reveals a sex difference in astrocytic C3 expression. Sci. Rep. 2019, 9, 10010. [Google Scholar] [CrossRef]
- Kajiwara, Y.; Wang, E.; Wang, M.; Sin, W.C.; Brennand, K.J.; Schadt, E.; Naus, C.C.; Buxbaum, J.; Zhang, B. GJA1 (connexin43) is a key regulator of Alzheimer’s disease pathogenesis. Acta Neuropathol. Commun. 2018, 6, 144. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, X.; Chen, F.; Song, N.; Xie, J. In vivo Direct Conversion of Astrocytes to Neurons Maybe a Potential Alternative Strategy for Neurodegenerative Diseases. Front. Aging Neurosci. 2021, 13, 689276. [Google Scholar] [CrossRef] [PubMed]
- Leng, K.; Rose, I.V.; Kim, H.; Xia, W.; Romero-Fernández, W.; Rooney, B.; Koontz, M.; Li, E.; Ao, Y.; Wang, S.; et al. CRISPRi Screens in Human Astrocytes Elucidate Regulators of Distinct Inflammatory Reactive States. Nat. Neurosci. 2021, 25, 1528–1542. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Prinz, M.; Priller, J. Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nat. Rev. Neurosci. 2014, 15, 300–312. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Wickstead, E.; Karim, H.A.; Manuel, R.E.; Biggs, C.S.; Getting, S.J.; McArthur, S. Reversal of Β-Amyloid Induced Microglial Toxicityin Vitroby Activation of Fpr2/3. Oxidative Med. Cell. Longev. 2020, 2020, 2139192. [Google Scholar] [CrossRef]
- Xu, J.-J.; Guo, S.; Xue, R.; Xiao, L.; Kou, J.-N.; Liu, Y.-Q.; Han, J.-Y.; Fu, J.-J.; Wei, N. Adalimumab ameliorates memory impairments and neuroinflammation in chronic cerebral hypoperfusion rats. Aging 2021, 13, 14001–14014. [Google Scholar] [CrossRef]
- Chmielarz, M.; Sobieszczańska, B.; Teisseyre, A.; Wawrzyńska, M.; Bożemska, E.; Środa-Pomianek, K. Palmitic Acid Modulates Microglial Cell Response to Metabolic Endotoxemia in an In Vitro Study. Nutrients 2023, 15, 3463. [Google Scholar] [CrossRef]
- Lee, S.-H.; Meilandt, W.J.; Xie, L.; Gandham, V.D.; Ngu, H.; Barck, K.H.; Rezzonico, M.G.; Imperio, J.; Lalehzadeh, G.; Huntley, M.A.; et al. Trem2 restrains the enhancement of tau accumulation and neurodegeneration by β-amyloid pathology. Neuron 2021, 109, 1283–1301.e6. [Google Scholar] [CrossRef]
- Hsieh, C.-F.; Liu, C.-K.; Lee, C.-T.; Yu, L.-E.; Wang, J.-Y. Acute glucose fluctuation impacts microglial activity, leading to inflammatory activation or self-degradation. Sci. Rep. 2019, 9, 840. [Google Scholar] [CrossRef] [PubMed]
- Tsukahara, T.; Hara, H.; Haniu, H.; Matsuda, Y. The Combined Effects of Lysophospholipids against Lipopolysaccharide-induced Inflammation and Oxidative Stress in Microglial Cells. J. Oleo Sci. 2021, 70, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Kloske, C.M.; Dugan, A.J.; Weekman, E.M.; Winder, Z.; Patel, E.; Nelson, P.T.; Fardo, D.W.; Wilcock, D.M. Inflammatory Pathways Are Impaired in Alzheimer Disease and Differentially Associated With Apolipoprotein E Status. J. Neuropathol. Exp. Neurol. 2021, 80, 922–932. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Palavicini, J.P.; Wang, J.; Gonzalez, N.S.; He, S.; Dustin, E.; Zou, C.; Ding, L.; Bhattacharjee, A.; Van Skike, C.E.; et al. Adult-onset CNS myelin sulfatide deficiency is sufficient to cause Alzheimer’s disease-like neuroinflammation and cognitive impairment. Mol. Neurodegener. 2021, 16, 64. [Google Scholar] [CrossRef]
- Mondal, M.; Solomon, S.; Sun, J.; Sampathkumar, N.K.; Carre, I.; Cotel, M.C.; Mehta, P.R.; Rajendran, L.; Vernon, A.C.; Fang, F.; et al. Olanzapine, Risperidone and Clozapine Prescribing Is Associated With Increased Risk for Alzheimer’s Disease Reflecting Antipsychotic-Specific Effects on Microglial Phagocytosis. medRxiv 2023. [Google Scholar] [CrossRef]
- Lokesh, M.; Bandaru, L.J.M.; Rajanna, A.; Dhayal, V.S.; Challa, S. M1 polarization induction by lead and amyloid peptides in microglial cells: Implications for neurodegeneration process. Environ. Toxicol. 2024, 39, 4267–4277. [Google Scholar] [CrossRef]
- Wu, H.; Bao, H.; Liu, C.; Zhang, Q.; Huang, A.; Quan, M.; Li, C.; Xiong, Y.; Chen, G.; Hou, L. Extracellular Nucleosomes Accelerate Microglial Inflammation via C-Type Lectin Receptor 2D and Toll-Like Receptor 9 in mPFC of Mice With Chronic Stress. Front. Immunol. 2022, 13, 854202. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, Y.; Wang, H.; Alam, A.; Kang, S.S.; Ahn, E.H.; Liu, X.; Jia, J.; Ye, K. Gut inflammation triggers C/EBPβ/δ-secretase-dependent gut-to-brain propagation of Aβ and Tau fibrils in Alzheimer’s disease. EMBO J. 2021, 40, e106320. [Google Scholar] [CrossRef]
- Kohli, M.A.; John-Williams, K.; Rajbhandary, R.; Naj, A.; Whitehead, P.; Hamilton, K.; Carney, R.M.; Wright, C.; Crocco, E.; Gwirtzman, H.E.; et al. Repeat expansions in the C9ORF72 gene contribute to Alzheimer’s disease in Caucasians. Neurobiol. Aging 2013, 34, 1519.e5–1519.e12. [Google Scholar] [CrossRef]
- Shi, Y.; Holtzman, D.M. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat. Rev. Immunol. 2018, 18, 759–772. [Google Scholar] [CrossRef]
- Piers, T.M.; East, E.; Villegas-Llerena, C.; Sevastou, I.G.; Matarin, M.; Hardy, J.; Pocock, J.M. Soluble Fibrinogen Triggers Non-cell Autonomous ER Stress-Mediated Microglial-Induced Neurotoxicity. Front. Cell. Neurosci. 2018, 12, 404. [Google Scholar] [CrossRef] [PubMed]
- Essadek, S.; Bouchab, H.; El Kebbaj, R.; Gondcaille, C.; El Kamouni, S.; Savary, S.; Vamecq, J.; Essamadi, A.; Cherkaoui-Malki, M.; Nasser, B.; et al. Effects of a Short-Term Lipopolysaccharides Challenge on Mouse Brain and Liver Peroxisomal Antioxidant and β-oxidative Functions: Protective Action of Argan Oil. Pharmaceuticals 2022, 15, 465. [Google Scholar] [CrossRef] [PubMed]
- Figarella, K.; Uzcategui, N.L.; Mogk, S.; Wild, K.; Fallier-Becker, P.; Neher, J.J.; Duszenko, M. Morphological changes, nitric oxide production, and phagocytosis are triggered in vitro in microglia by bloodstream forms of Trypanosoma brucei. Sci. Rep. 2018, 8, 15002. [Google Scholar] [CrossRef] [PubMed]
- Koyanagi, Y.; Kassai, M.; Yoneyama, H. The Impact of Intestinal Microbiota and Toll-like Receptor 2 Signaling on α-Synuclein Pathology in Nontransgenic Mice Injected with α-Synuclein Preformed Fibrils. Microorganisms 2024, 12, 106. [Google Scholar] [CrossRef]
- Li, Y.; Niu, M.; Zhao, A.; Kang, W.; Chen, Z.; Luo, N.; Zhou, L.; Zhu, X.; Lu, L.; Liu, J. CXCL12 is involved in α-synuclein-triggered neuroinflammation of Parkinson’s disease. J. Neuroinflamm. 2019, 16, 263. [Google Scholar] [CrossRef]
- Raas, Q.; Saih, F.-E.; Gondcaille, C.; Trompier, D.; Hamon, Y.; Leoni, V.; Caccia, C.; Nasser, B.; Jadot, M.; Ménétrier, F.; et al. A microglial cell model for acyl-CoA oxidase 1 deficiency. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2019, 1864, 567–576. [Google Scholar] [CrossRef]
- Abubaker, A.A.; Vara, D.; Visconte, C.; Eggleston, I.; Torti, M.; Canobbio, I.; Pula, G. Amyloid Peptide β1-42 Induces Integrin αIIbβ3 Activation, Platelet Adhesion, and Thrombus Formation in a NADPH Oxidase-Dependent Manner. Oxidative Med. Cell. Longev. 2019, 2019, 1050476. [Google Scholar] [CrossRef]
- Valcarcel-Ares, M.N.; Tucsek, Z.; Kiss, T.; Giles, C.B.; Tarantini, S.; Yabluchanskiy, A.; Balasubramanian, P.; Gautam, T.; Galvan, V.; Ballabh, P.; et al. Obesity in Aging Exacerbates Neuroinflammation, Dysregulating Synaptic Function-Related Genes and Altering Eicosanoid Synthesis in the Mouse Hippocampus: Potential Role in Impaired Synaptic Plasticity and Cognitive Decline. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2018, 74, 290–298. [Google Scholar] [CrossRef]
- Yang, L.; Wang, W.; Chen, J.; Wang, N.; Zheng, G. A comparative study of resveratrol and resveratrol-functional selenium nanoparticles: Inhibiting amyloid β aggregation and reactive oxygen species formation properties. J. Biomed. Mater. Res. A 2018, 106, 3034–3041. [Google Scholar] [CrossRef]
- Kaneshwaran, K.; Olah, M.; Tasaki, S.; Yu, L.; Bradshaw, E.M.; Schneider, J.A.; Buchman, A.S.; Bennett, D.A.; De Jager, P.L.; Lim, A.S.P. Sleep fragmentation, microglial aging, and cognitive impairment in adults with and without Alzheimer’s dementia. Sci. Adv. 2019, 5, eaax7331. [Google Scholar] [CrossRef]
- Li, J.; Xu, X.; Cai, X.; Weng, Y.; Wang, Y.; Shen, Q.; Shi, X. Milk Fat Globule-Epidermal Growth Factor-Factor 8 Reverses Lipopolysaccharide-Induced Microglial Oxidative Stress. Oxidative Med. Cell. Longev. 2019, 2019, 2601394. [Google Scholar] [CrossRef] [PubMed]
- Piers, T.M.; Cosker, K.; Mallach, A.; Johnson, G.T.; Guerreiro, R.; Hardy, J.; Pocock, J.M. A locked immunometabolic switch underlies TREM2 R47H loss of function in human iPSC-derived microglia. FASEB J. 2019, 34, 2436–2450. [Google Scholar] [CrossRef] [PubMed]
- Asthana, S.; Pandey, S.K.; Gautam, A.S.; Singh, R.K. MK2 inhibitor PF-3644022 shows protective effect in mouse microglial N9 cell line induced with cigarette smoke extract. Chem. Biol. Drug Des. 2024, 104, e14592. [Google Scholar] [CrossRef] [PubMed]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Ulland, T.K.; Song, W.M.; Huang, S.C.-C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663.e13. [Google Scholar] [CrossRef]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial Dysfunction and Defective β-Amyloid Clearance Pathways in Aging Alzheimer’s Disease Mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
- Wang, W.-Y.; Tan, M.-S.; Yu, J.-T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.E9. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Enomoto, S.; Ohgidani, M.; Sagata, N.; Inamine, S.; Kato, T.A. Preliminary analysis of hippocampus synaptic apoptosis and microglial phagocytosis induced by severe restraint stress. Neuropsychopharmacol. Rep. 2022, 43, 120–125. [Google Scholar] [CrossRef]
- Howe, A.-M.; Burke, S.; O’reilly, M.E.; McGillicuddy, F.C.; Costello, D.A. Palmitic Acid and Oleic Acid Differently Modulate TLR2-Mediated Inflammatory Responses in Microglia and Macrophages. Mol. Neurobiol. 2022, 59, 2348–2362. [Google Scholar] [CrossRef] [PubMed]
- Irwin, D.J.; Cairns, N.J.; Grossman, M.; McMillan, C.T.; Lee, E.B.; Van Deerlin, V.M.; Lee, V.M.-Y.; Trojanowski, J.Q. Frontotemporal lobar degeneration: Defining phenotypic diversity through personalized medicine. Acta Neuropathol. 2014, 129, 469–491. [Google Scholar] [CrossRef] [PubMed]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef] [PubMed]
- Perea, J.R.; Llorens-Martín, M.; Ávila, J.; Bolós, M. The Role of Microglia in the Spread of Tau: Relevance for Tauopathies. Front. Cell. Neurosci. 2018, 12, 172. [Google Scholar] [CrossRef]
- Striet, W.J.; Xue, Q.-S.; Tischer, J.; Bechmann, I. Microglial pathology. Acta Neuropathol. Commun. 2014, 2, 142. [Google Scholar] [CrossRef]
- Kurioka, T.; Mogi, S.; Yamashita, T. Decreasing auditory input induces neurogenesis impairment in the hippocampus. Sci. Rep. 2021, 11, 423. [Google Scholar] [CrossRef]
- Malko, P.; Mortadza, S.A.S.; McWilliam, J.; Jiang, L.-H. TRPM2 Channel in Microglia as a New Player in Neuroinflammation Associated With a Spectrum of Central Nervous System Pathologies. Front. Pharmacol. 2019, 10, 239. [Google Scholar] [CrossRef]
- Minami, Y.; Sonoda, N.; Hayashida, E.; Makimura, H.; Ide, M.; Ikeda, N.; Ohgidani, M.; Kato, T.A.; Seki, Y.; Maeda, Y.; et al. p66Shc Signaling Mediates Diabetes-Related Cognitive Decline. Sci. Rep. 2018, 8, 3213. [Google Scholar] [CrossRef]
- Ide, M.; Sonoda, N.; Inoue, T.; Kimura, S.; Minami, Y.; Makimura, H.; Hayashida, E.; Hyodo, F.; Yamato, M.; Takayanagi, R.; et al. The dipeptidyl peptidase-4 inhibitor, linagliptin, improves cognitive impairment in streptozotocin-induced diabetic mice by inhibiting oxidative stress and microglial activation. PLoS ONE 2020, 15, e0228750. [Google Scholar] [CrossRef]
- Trares, K.; Gào, X.; Perna, L.; Rujescu, D.; Stocker, H.; Möllers, T.; Beyreuther, K.; Brenner, H.; Schöttker, B. Associations of urinary 8-iso-prostaglandin F2α levels with all-cause dementia, Alzheimer’s disease, and vascular dementia incidence: Results from a prospective cohort study. Alzheimer’s Dement. 2020, 16, 804–813. [Google Scholar] [CrossRef]
- Hu, B.; Duan, S.; Wang, Z.; Li, X.; Zhou, Y.; Zhang, X.; Zhang, Y.-W.; Xu, H.; Zheng, H. Insights Into the Role of CSF1R in the Central Nervous System and Neurological Disorders. Front. Aging Neurosci. 2021, 13, 789834. [Google Scholar] [CrossRef]
- Afridi, R.; Lee, W.-H.; Suk, K. Microglia Gone Awry: Linking Immunometabolism to Neurodegeneration. Front. Cell. Neurosci. 2020, 14, 246. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Rosenzweig, N.; Kleemann, K.L.; Zhang, X.; Brandão, W.; Margeta, M.A.; Schroeder, C.; Sivanathan, K.N.; Silveira, S.; Gauthier, C.; et al. APOE4 impairs the microglial response in Alzheimer’s disease by inducing TGFβ-mediated checkpoints. Nat. Immunol. 2023, 24, 1839–1853. [Google Scholar] [CrossRef] [PubMed]
- Malpetti, M.; Jones, P.S.; Tsvetanov, K.A.; Rittman, T.; van Swieten, J.C.; Borroni, B.; Sanchez-Valle, R.; Moreno, F.; Laforce, R.; Graff, C.; et al. Apathy in presymptomatic genetic frontotemporal dementia predicts cognitive decline and is driven by structural brain changes. Alzheimer’s Dement. 2020, 17, 969–983. [Google Scholar] [CrossRef]
- Ferrari-Souza, J.P.; Lussier, F.Z.; Leffa, D.T.; Therriault, J.; Tissot, C.; Bellaver, B.; Ferreira, P.C.L.; Malpetti, M.; Wang, Y.-T.; Povala, G.; et al. APOE ε4 associates with microglial activation independently of Aβ plaques and tau tangles. Sci. Adv. 2023, 9, eade1474. [Google Scholar] [CrossRef]
- Weigand, A.J.; Thomas, K.R.; Bangen, K.J.; Eglit, G.M.; Delano-Wood, L.; Gilbert, P.E.; Brickman, A.M.; Bondi, M.W.; Alzheimer’s Disease Neuroimaging Initiative. APOE interacts with tau PET to influence memory independently of amyloid PET in older adults without dementia. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2021, 17, 61–69. [Google Scholar] [CrossRef]
- Giacomucci, G.; Mazzeo, S.; Bagnoli, S.; Casini, M.; Padiglioni, S.; Polito, C.; Berti, V.; Balestrini, J.; Ferrari, C.; Lombardi, G.; et al. Matching Clinical Diagnosis and Amyloid Biomarkers in Alzheimer’s Disease and Frontotemporal Dementia. J. Pers. Med. 2021, 11, 47. [Google Scholar] [CrossRef]
- Xia, Y.; Zhang, G.; Kou, L.; Yin, S.; Han, C.; Hu, J.; Wan, F.; Sun, Y.; Wu, J.; Li, Y.; et al. Reactive microglia enhance the transmission of exosomal α-synuclein via toll-like receptor 2. Brain 2021, 144, 2024–2037. [Google Scholar] [CrossRef]
- Iannucci, J.; Sen, A.; Grammas, P. Isoform-Specific Effects of Apolipoprotein E on Markers of Inflammation and Toxicity in Brain Glia and Neuronal Cells In Vitro. Curr. Issues Mol. Biol. 2021, 43, 215–225. [Google Scholar] [CrossRef]
- Panitch, R.; Hu, J.; Chung, J.; Zhu, C.; Meng, G.; Xia, W.; Bennett, D.A.; Lunetta, K.L.; Ikezu, T.; Au, R.; et al. Integrative brain transcriptome analysis links complement component 4 and HSPA2 to the APOE ε2 protective effect in Alzheimer disease. Mol. Psychiatry 2021, 26, 6054–6064. [Google Scholar] [CrossRef] [PubMed]
- Saab, A.S.; Nave, K.-A. Myelin dynamics: Protecting and shaping neuronal functions. Curr. Opin. Neurobiol. 2017, 47, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Ettle, B.; Schlachetzki, J.C.M.; Winkler, J. Oligodendroglia and Myelin in Neurodegenerative Diseases: More Than Just Bystanders? Mol. Neurobiol. 2016, 53, 3046–3062. [Google Scholar] [CrossRef] [PubMed]
- Rauchmann, B.S.; Brendel, M.; Franzmeier, N.; Trappmann, L.; Zaganjori, M.; Morenas-Rodriguez, E.; Guersel, S.; Burow, L.; Kurz, C.; Haeckert, J.; et al. Microglial Activation and Connectivity in Alzheimer Disease and Aging. Ann. Neurol. 2022, 92, 768–781. [Google Scholar] [CrossRef]
- Taha, M.; Eldemerdash, O.M.; Elshaffei, I.M.; Yousef, E.M.; Soliman, A.S.; Senousy, M.A. Apigenin Attenuates Hippocampal Microglial Activation and Restores Cognitive Function in Methotrexate-Treated Rats: Targeting the miR-15a/ROCK-1/ERK1/2 Pathway. Mol. Neurobiol. 2023, 60, 3770–3787. [Google Scholar] [CrossRef]
- Raas, Q.; Gondcaille, C.; Hamon, Y.; Leoni, V.; Caccia, C.; Ménétrier, F.; Lizard, G.; Trompier, D.; Savary, S. CRISPR/Cas9-mediated knockout of Abcd1 and Abcd2 genes in BV-2 cells: Novel microglial models for X-linked Adrenoleukodystrophy. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2019, 1864, 704–714. [Google Scholar] [CrossRef]
- Essadek, S.; Gondcaille, C.; Savary, S.; Samadi, M.; Vamecq, J.; Lizard, G.; El Kebbaj, R.; Latruffe, N.; Benani, A.; Nasser, B.; et al. Two Argan Oil Phytosterols, Schottenol and Spinasterol, Attenuate Oxidative Stress and Restore LPS-Dysregulated Peroxisomal Functions in Acox1−/− and Wild-Type BV-2 Microglial Cells. Antioxidants 2023, 12, 168. [Google Scholar] [CrossRef]
- Wan, Y.; Gao, W.; Zhou, K.; Liu, X.; Jiang, W.; Xue, R.; Wu, W. Role of IGF-1 in neuroinflammation and cognition deficits induced by sleep deprivation. Neurosci. Lett. 2022, 776, 136575. [Google Scholar] [CrossRef]
- Xin, W.; Mironova, Y.A.; Shen, H.; Marino, R.A.; Waisman, A.; Lamers, W.H.; Bergles, D.E.; Bonci, A. Oligodendrocytes Support Neuronal Glutamatergic Transmission via Expression of Glutamine Synthetase. Cell Rep. 2019, 27, 2262–2271.e5. [Google Scholar] [CrossRef]
- Meyer, N.; Richter, N.; Fan, Z.; Siemonsmeier, G.; Pivneva, T.; Jordan, P.; Steinhäuser, C.; Semtner, M.; Nolte, C.; Kettenmann, H. Oligodendrocytes in the Mouse Corpus Callosum Maintain Axonal Function by Delivery of Glucose. Cell Rep. 2018, 22, 2383–2394. [Google Scholar] [CrossRef]
- Saito, E.; Miller, J.; Harari, O.; Cruchaga, C.; Mihindukulasuriya, K.; Kauwe, J.; Bikman, B. Alzheimer’s Disease Alters Oligodendrocytic Glycolytic and Ketolytic Gene Expression. FASEB J. 2021, 35, 1474–1486. [Google Scholar] [CrossRef]
- Hughes, A.N.; Appel, B. Oligodendrocytes express synaptic proteins that modulate myelin sheath formation. Nature communications 2019, 10, 4125. [Google Scholar] [CrossRef] [PubMed]
- Panlilio, J.M.; Hammar, K.M.; Aluru, N.; Hahn, M.E. Developmental exposure to domoic acid targets reticulospinal neurons and leads to aberrant myelination in the spinal cord. Sci. Rep. 2023, 13, 2587. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, J.; Macrì, R.; Bava, I.; Gliozzi, M.; Musolino, V.; Nucera, S.; Carresi, C.; Scicchitano, M.; Bosco, F.; Scarano, F.; et al. Myelin Disturbances Produced by Sub-Toxic Concentration of Heavy Metals: The Role of Oligodendrocyte Dysfunction. Int. J. Mol. Sci. 2019, 20, 4554. [Google Scholar] [CrossRef]
- Moore, S.; Meschkat, M.; Ruhwedel, T.; Trevisiol, A.; Tzvetanova, I.D.; Battefeld, A.; Kusch, K.; Kole, M.H.P.; Strenzke, N.; Möbius, W.; et al. A role of oligodendrocytes in information processing. Nat. Commun. 2020, 11, 5497. [Google Scholar] [CrossRef]
- Depp, C.M.; Nave, K.; Lab, K.N. Ageing-Associated Myelin Dysfunction Drives Amyloid Deposition in Mouse Models of Alzheimer’s Disease. Alzheimer’s Dement. 2022, 18, e061183. [Google Scholar] [CrossRef]
- Green, L.A.; Gallant, R.M.; Brandt, J.P.; Nichols, E.L.; Smith, C.J. A Subset of Oligodendrocyte Lineage Cells Interact With the Developing Dorsal Root Entry Zone During Its Genesis. Front. Cell. Neurosci. 2022, 16, 893629. [Google Scholar] [CrossRef]
- Miedema, S.S.M.; Mol, M.O.; Koopmans, F.T.W.; Hondius, D.C.; van Nierop, P.; Menden, K.; Mestdagh, C.F.d.V.; van Rooij, J.; Ganz, A.B.; Paliukhovich, I.; et al. Distinct cell type-specific protein signatures in GRN and MAPT genetic subtypes of frontotemporal dementia. Acta Neuropathol. Commun. 2022, 10, 100. [Google Scholar] [CrossRef]
- Yoon, Y.-S.; Ahn, W.J.; Ricarte, D.; Ortiz, D.; Shin, C.Y.; Lee, S.-J.; Lee, H.-J. Alpha-Synuclein Inclusion Formation in Human Oligodendrocytes. Biomol. Ther. 2021, 29, 83–89. [Google Scholar] [CrossRef]
- Xin, W.; Kaneko, M.; Roth, R.H.; Zhang, A.; Nocera, S.; Ding, J.B.; Stryker, M.P.; Chan, J.R. Oligodendrocytes and myelin limit neuronal plasticity in visual cortex. Nature 2024, 633, 856–863. [Google Scholar] [CrossRef]
- Zhao, C.; Dong, C.; Frah, M.; Deng, Y.; Marie, C.; Zhang, F.; Xu, L.; Ma, Z.; Dong, X.; Lin, Y.; et al. Dual Requirement of CHD8 for Chromatin Landscape Establishment and Histone Methyltransferase Recruitment to Promote CNS Myelination and Repair. Dev. Cell 2018, 45, 753–768.e8. [Google Scholar] [CrossRef] [PubMed]
- Frühbeis, C.; Kuo-Elsner, W.P.; Müller, C.; Barth, K.; Peris, L.; Tenzer, S.; Möbius, W.; Werner, H.B.; Nave, K.-A.; Fröhlich, D.; et al. Oligodendrocytes support axonal transport and maintenance via exosome secretion. PLoS Biol. 2020, 18, e3000621. [Google Scholar] [CrossRef] [PubMed]
- Mitew, S.; Gobius, I.; Fenlon, L.R.; McDougall, S.J.; Hawkes, D.; Xing, Y.L.; Bujalka, H.; Gundlach, A.L.; Richards, L.J.; Kilpatrick, T.J.; et al. Pharmacogenetic stimulation of neuronal activity increases myelination in an axon-specific manner. Nat. Commun. 2018, 9, 306. [Google Scholar] [CrossRef] [PubMed]
- Hinman, J.D.; Ngo, K.J.; Kim, D.; Chen, C.; Abraham, C.R.; Ghanbari, M.; Ikram, M.A.; Kushner, S.A.; Kawaguchi, R.; Coppola, G.; et al. miR-142-3p regulates cortical oligodendrocyte gene co-expression networks associated with tauopathy. Hum. Mol. Genet. 2021, 30, 103–118. [Google Scholar] [CrossRef]
- Müller, K.; Schnatz, A.; Schillner, M.; Woertge, S.; Müller, C.; von Graevenitz, I.; Waisman, A.; van Minnen, J.; Vogelaar, C.F. A predominantly glial origin of axonal ribosomes after nerve injury. Glia 2018, 66, 1591–1610. [Google Scholar] [CrossRef]
- Chen, K.; Cambi, F.; Kozai, T.D. Pro-myelinating clemastine administration improves recording performance of chronically implanted microelectrodes and nearby neuronal health. Biomaterials 2023, 301, 122210. [Google Scholar] [CrossRef]
- Eykens, C.; Rossaert, E.; Duqué, S.; Rué, L.; Bento-Abreu, A.; Hersmus, N.; Lenaerts, A.; Kerstens, A.; Corthout, N.; Munck, S.; et al. AAV9-mediated gene delivery of MCT1 to oligodendrocytes does not provide a therapeutic benefit in a mouse model of ALS. Mol. Ther.-Methods Clin. Dev. 2021, 20, 508–519. [Google Scholar] [CrossRef]
- Banerjee, S.; Ghoshal, S.; Girardet, C.; DeMars, K.M.; Yang, C.; Niehoff, M.L.; Nguyen, A.D.; Jayanth, P.; Mersman, B.A.; Xu, F.; et al. Adropin Expression Correlates with Age-Related Neuropathologies in the Human Brain and Improves Neuroinflammation and Cognitive Function in Aging Mice. 2021. Available online: https://pdfs.semanticscholar.org/5a6a/99a4e3bba9a697186be8582c7eeed7fc9dfa.pdf (accessed on 20 December 2024). [CrossRef]
- Forbes, T.A.; Goldstein, E.Z.; Dupree, J.L.; Jablonska, B.; Scafidi, J.; Adams, K.L.; Imamura, Y.; Hashimoto-Torii, K.; Gallo, V. Environmental enrichment ameliorates perinatal brain injury and promotes functional white matter recovery. Nat. Commun. 2020, 11, 964. [Google Scholar] [CrossRef]
- Mukherjee, C.; Kling, T.; Russo, B.; Miebach, K.; Kess, E.; Schifferer, M.; Pedro, L.D.; Weikert, U.; Fard, M.K.; Kannaiyan, N.; et al. Oligodendrocytes Provide Antioxidant Defense Function for Neurons by Secreting Ferritin Heavy Chain. Cell Metab. 2020, 32, 259–272.e10. [Google Scholar] [CrossRef]
- Marton, R.M.; Miura, Y.; Sloan, S.A.; Li, Q.; Revah, O.; Levy, R.J.; Huguenard, J.R.; Pașca, S.P. Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat. Neurosci. 2019, 22, 484–491. [Google Scholar] [CrossRef]
- Butt, T.H.; Tobiume, M.; Re, D.B.; Kariya, S. Physical Exercise Counteracts Aging-Associated White Matter Demyelination Causing Cognitive Decline. Aging Dis. 2024, 15, 2136–2148. [Google Scholar] [CrossRef] [PubMed]
- Tiane, A.; Schepers, M.; Rombaut, B.; Hupperts, R.; Prickaerts, J.; Hellings, N.; van den Hove, D.; Vanmierlo, T. From OPC to Oligodendrocyte: An Epigenetic Journey. Cells 2019, 8, 1236. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.; Dobrowolski, M.; Sockanathan, S. GDE2 expression in oligodendroglia regulates the pace of oligodendrocyte maturation. Dev. Dyn. 2020, 250, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Moloney, R.A.; Pavy, C.L.; Kahl, R.G.S.; Palliser, H.K.; Hirst, J.J.; Shaw, J.C. Dual isolation of primary neurons and oligodendrocytes from guinea pig frontal cortex. Front. Cell. Neurosci. 2024, 17, 1298685. [Google Scholar] [CrossRef]
- Psenicka, M.W.; Smith, B.C.; Tinkey, R.A.; Williams, J.L. Connecting Neuroinflammation and Neurodegeneration in Multiple Sclerosis: Are Oligodendrocyte Precursor Cells a Nexus of Disease? Front. Cell. Neurosci. 2021, 15, 654284. [Google Scholar] [CrossRef]
- Chia, R.; Sabir, M.S.; Bandres-Ciga, S.; Saez-Atienzar, S.; Reynolds, R.H.; Gustavsson, E.; Walton, R.L.; Ahmed, S.; Viollet, C.; Ding, J.; et al. Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat. Genet. 2021, 53, 294–303. [Google Scholar] [CrossRef]
- Jiang, C.; Hopfner, F.; Katsikoudi, A.; Hein, R.; Catli, C.; Evetts, S.; Huang, Y.; Wang, H.; Ryder, J.W.; Kuhlenbaeumer, G.; et al. Serum neuronal exosomes predict and differentiate Parkinson’s disease from atypical parkinsonism. J. Neurol. Neurosurg. Psychiatry 2020, 91, 720–729. [Google Scholar] [CrossRef]
- Do, J.; McKinney, C.; Sharma, P.; Sidransky, E. Glucocerebrosidase and its relevance to Parkinson disease. Mol. Neurodegener. 2019, 14, 36. [Google Scholar] [CrossRef]
- Kuo, S.-H.; Tasset, I.; Cheng, M.M.; Diaz, A.; Pan, M.-K.; Lieberman, O.J.; Hutten, S.J.; Alcalay, R.N.; Kim, S.; Ximénez-Embún, P.; et al. Mutant glucocerebrosidase impairs α-synuclein degradation by blockade of chaperone-mediated autophagy. Sci. Adv. 2022, 8, eabm6393. [Google Scholar] [CrossRef]
- Johnson, M.E.; Stecher, B.; Labrie, V.; Brundin, L.; Brundin, P. Triggers, Facilitators, and Aggravators: Redefining Parkinson’s Disease Pathogenesis. Trends Neurosci. 2019, 42, 4–13. [Google Scholar] [CrossRef]
- Madureira, M.; Connor-Robson, N.; Wade-Martins, R. “LRRK2: Autophagy and Lysosomal Activity”. Front. Neurosci. 2020, 14, 498. [Google Scholar] [CrossRef] [PubMed]
- Farfel-Becker, T.; Roney, J.C.; Cheng, X.-T.; Li, S.; Cuddy, S.R.; Sheng, Z.-H. Neuronal Soma-Derived Degradative Lysosomes Are Continuously Delivered to Distal Axons to Maintain Local Degradation Capacity. Cell Rep. 2019, 28, 51–64.e4. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Romero, A.; Montpeyó, M.; Martinez-Vicente, M. The Emerging Role of the Lysosome in Parkinson’s Disease. Cells 2020, 9, 2399. [Google Scholar] [CrossRef] [PubMed]
- Matejuk, A.; Ransohoff, R.M. Crosstalk Between Astrocytes and Microglia: An Overview. Front. Immunol. 2020, 11, 1416. [Google Scholar] [CrossRef]
- Zhang, Y.; Chao, F.-L.; Zhou, C.-N.; Jiang, L.; Zhang, L.; Liang, X.; Tang, J.; Tang, Y. Atrophy of lacunosum moleculare layer is important for learning and memory in APP/PS1 transgenic mice. NeuroReport 2021, 32, 596–602. [Google Scholar] [CrossRef]
- Chen, S.; Chang, Y.; Li, L.; Acosta, D.; Li, Y.; Guo, Q.; Wang, C.; Turkes, E.; Morrison, C.; Julian, D.; et al. Spatially resolved transcriptomics reveals genes associated with the vulnerability of middle temporal gyrus in Alzheimer’s disease. Acta Neuropathol Commun. 2022, 10, 188. [Google Scholar] [CrossRef]
- Muraoka, S.; Jedrychowski, M.P.; Iwahara, N.; Abdullah, M.; Onos, K.D.; Keezer, K.J.; Hu, J.; Ikezu, S.; Howell, G.R.; Gygi, S.P.; et al. Enrichment of Neurodegenerative Microglia Signature in Brain-Derived Extracellular Vesicles Isolated from Alzheimer’s Disease Mouse Models. J. Proteome Res. 2021, 20, 1733–1743. [Google Scholar] [CrossRef]
- Gushchina, S.; Pryce, G.; Yip, P.K.; Wu, D.; Pallier, P.; Giovannoni, G.; Baker, D.; Bo, X. Increased expression of colony-stimulating factor-1 in mouse spinal cord with experimental autoimmune encephalomyelitis correlates with microglial activation and neuronal loss. Glia 2018, 66, 2108–2125. [Google Scholar] [CrossRef]
- Wang, G.; Jin, S.; Liu, J.; Li, X.; Dai, P.; Wang, Y.; Hou, S.X. A neuron-immune circuit regulates neurodegeneration in the hindbrain and spinal cord of Arf1-ablated mice. Natl. Sci. Rev. 2023, 12, 222. [Google Scholar] [CrossRef]
- Liu, Y.; Given, K.S.; Harlow, D.E.; Matschulat, A.M.; Macklin, W.B.; Bennett, J.L.; Owens, G.P. Myelin-specific multiple sclerosis antibodies cause complement-dependent oligodendrocyte loss and demyelination. Acta Neuropathol. Commun. 2017, 5, 25. [Google Scholar] [CrossRef]
- Linden, J.R.; Ma, Y.; Zhao, B.; Harris, J.M.; Rumah, K.R.; Schaeren-Wiemers, N.; Vartanian, T. Clostridium perfringens Epsilon Toxin Causes Selective Death of Mature Oligodendrocytes and Central Nervous System Demyelination. mBio 2015, 6, e02513-14. [Google Scholar] [CrossRef] [PubMed]
- Gingele, S.; Merkel, L.; Prajeeth, C.K.; Kronenberg, J.; von Hoevel, F.F.; Skripuletz, T.; Gudi, V.; Stangel, M. Polarized microglia do not influence oligodendrocyte lineage cells via astrocytes. Int. J. Dev. Neurosci. 2019, 77, 39–47. [Google Scholar] [CrossRef] [PubMed]
- Amaral, A.I.; Meisingset, T.W.; Kotter, M.R.; Sonnewald, U. Metabolic Aspects of Neuron-Oligodendrocyte-Astrocyte Interactions. Front. Endocrinol. 2013, 4, 54. [Google Scholar] [CrossRef] [PubMed]
- Verrier, J.D.; Jackson, T.C.; Gillespie, D.G.; Janesko-Feldman, K.; Bansal, R.; Goebbels, S.; Nave, K.-A.; Kochanek, P.M.; Jackson, E.K. Role of CNPase in the oligodendrocytic extracellular 2′,3′-cAMP-adenosine pathway. Glia 2013, 61, 1595–1606. [Google Scholar] [CrossRef]
- Jansen, A.H.; van Hal, M.; Kelder, I.C.O.D.; Meier, R.T.; de Ruiter, A.; Schut, M.H.; Smith, D.L.; Grit, C.; Brouwer, N.; Kamphuis, W.; et al. Frequency of nuclear mutant huntingtin inclusion formation in neurons and glia is cell-type-specific. Glia 2016, 65, 50–61. [Google Scholar] [CrossRef]
- Stevenson, T.J.; Murray, H.C.; Turner, C.; Faull, R.L.M.; Dieriks, B.V.; Curtis, M.A. α-synuclein inclusions are abundant in non-neuronal cells in the anterior olfactory nucleus of the Parkinson’s disease olfactory bulb. Sci. Rep. 2020, 10, 6682. [Google Scholar] [CrossRef]
- Zhan, Z.; Wu, Y.; Liu, Z.; Quan, Y.; Li, D.; Huang, Y.; Yang, S.; Wu, K.; Huang, L.; Yu, M. Reduced Dendritic Spines in the Visual Cortex Contralateral to the Optic Nerve Crush Eye in Adult Mice. Investig. Opthalmol. Vis. Sci. 2020, 61, 55. [Google Scholar] [CrossRef]
- Joshi, A.U.; Minhas, P.S.; Liddelow, S.A.; Haileselassie, B.; Andreasson, K.I.; Dorn, G.W., II; Mochly-Rosen, D. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci. 2019, 22, 1635–1648. [Google Scholar] [CrossRef]
- Alshaebi, F.; Sciortino, A.; Kayed, R. The Role of Glial Cell Senescence in Alzheimer’s Disease. J. Neurochem. 2025, 169, e70051. [Google Scholar] [CrossRef]
- Mattick, J.S.; Amaral, P.P.; Carninci, P.; Carpenter, S.; Chang, H.Y.; Chen, L.-L.; Chen, R.; Dean, C.; Dinger, M.E.; Fitzgerald, K.A. Long non-coding RNAs: Definitions, functions, challenges and recommendations. Nat. Rev. Mol. Cell biology 2023, 24, 430–447. [Google Scholar] [CrossRef]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers in sight? Nat. Rev. Genet. 2008, 9, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Si, L.; Bai, J.; Fu, H.; Qiu, H.; Guo, R. The functions and potential roles of extracellular vesicle noncoding RNAs in gynecological malignancies. Cell Death Discov. 2021, 7, 258. [Google Scholar] [CrossRef] [PubMed]
- Loganathan, T.; Doss C, G.P. Non-coding RNAs in human health and disease: Potential function as biomarkers and therapeutic targets. Funct. Integr. Genom. 2023, 23, 33. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V.; Bartel, B.; Bartel, D.P.; Burge, C.B.; Carrington, J.C.; Chen, X.; Dreyfuss, G.; Eddy, S.R.; Griffiths-Jones, S.; Marshall, M.; et al. A uniform system for microRNA annotation. RNA 2003, 9, 277–279. [Google Scholar] [CrossRef]
- Shang, R.; Lee, S.; Senavirathne, G.; Lai, E.C. microRNAs in action: Biogenesis, function and regulation. Nat. Rev. Genet. 2023, 24, 816–833. [Google Scholar] [CrossRef]
- Gil-Jaramillo, N.; Aristizábal-Pachón, A.F.; Aleman, M.A.L.; Gómez, V.G.; Hurtado, H.D.E.; Pinto, L.C.G.; Camacho, J.S.J.; Rojas-Cruz, A.F.; González-Giraldo, Y.; Pinzón, A.; et al. Competing endogenous RNAs in human astrocytes: Crosstalk and interacting networks in response to lipotoxicity. Front. Neurosci. 2023, 17, 1195840. [Google Scholar] [CrossRef]
- Prada, I.; Gabrielli, M.; Turola, E.; Iorio, A.; D’arrigo, G.; Parolisi, R.; De Luca, M.; Pacifici, M.; Bastoni, M.; Lombardi, M.; et al. Glia-to-neuron transfer of miRNAs via extracellular vesicles: A new mechanism underlying inflammation-induced synaptic alterations. Acta Neuropathol. 2018, 135, 529–550. [Google Scholar] [CrossRef]
- Phatnani, H.; Maniatis, T. Astrocytes in neurodegenerative disease. Cold Spring Harb. Perspect. Biol. 2015, 7, a020628. [Google Scholar] [CrossRef]
- Meraz, R. Medication Nonadherence or Self-care? Understanding the Medication Decision-Making Process and Experiences of Older Adults With Heart Failure. J. Cardiovasc. Nurs. 2019, 35, 26–34. [Google Scholar] [CrossRef]
- Wang, Y.; Han, Z.; Fan, Y.; Zhang, J.; Chen, K.; Gao, L.; Zeng, H.; Cao, J.; Wang, C. MicroRNA-9 Inhibits NLRP3 Inflammasome Activation in Human Atherosclerosis Inflammation Cell Models through the JAK1/STAT Signaling Pathway. Cell. Physiol. Biochem. 2017, 41, 1555–1571. [Google Scholar] [CrossRef]
- Wongrakpanich, A.; Mudunkotuwa, I.A.; Geary, S.M.; Morris, A.S.; Mapuskar, K.A.; Spitz, D.R.; Grassian, V.H.; Salem, A.K. Size-dependent cytotoxicity of copper oxide nanoparticles in lung epithelial cells. Environ. Sci. Nano 2016, 3, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Ding, J.; Yang, J.; Guo, X.; Zheng, Y. MicroRNA Roles in the Nuclear Factor Kappa B Signaling Pathway in Cancer. Front. Immunol. 2018, 9, 546. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Reed, K.M.; Carter, A.; Cheng, Y.; Roodsari, S.K.; Pineda, D.M.; Wellman, L.L.; Sanford, L.D.; Guo, M.-L. Sleep-Disturbance-Induced Microglial Activation Involves CRH-Mediated Galectin 3 and Autophagy Dysregulation. Cells 2022, 12, 160. [Google Scholar] [CrossRef]
- Fan, X.-M.; Luo, Y.; Cao, Y.-M.; Xiong, T.-W.; Song, S.; Liu, J.; Fan, Q.-Y. Chronic Manganese Administration with Longer Intervals Between Injections Produced Neurotoxicity and Hepatotoxicity in Rats. Neurochem. Res. 2020, 45, 1941–1952. [Google Scholar] [CrossRef]
- Conrad, A.T.; Dittel, B.N. Taming of macrophage and microglial cell activation by microRNA-124. Cell Res. 2011, 21, 213–216. [Google Scholar] [CrossRef]
- Coolen, M.; Katz, S.; Bally-Cuif, L. miR-9: A versatile regulator of neurogenesis. Front. Cell. Neurosci. 2013, 7, 220. [Google Scholar] [CrossRef]
- Delgado-García, L.M.; Ojalvo-Sanz, A.C.; Nakamura, T.K.E.; Martín-López, E.; Porcionatto, M.; Lopez-Mascaraque, L. Dissecting reactive astrocyte responses: Lineage tracing and morphology-based clustering. Biol. Res. 2024, 57, 54. [Google Scholar] [CrossRef]
- Fonken, L.K.; Frank, M.G.; Gaudet, A.D.; Maier, S.F. Stress and aging act through common mechanisms to elicit neuroinflammatory priming. Brain, Behav. Immun. 2018, 73, 133–148. [Google Scholar] [CrossRef]
- Gaudet, A.D.; Fonken, L.K.; Watkins, L.R.; Nelson, R.J.; Popovich, P.G. MicroRNAs: Roles in Regulating Neuroinflammation. Neurosci. 2017, 24, 221–245. [Google Scholar] [CrossRef]
- Pogue, A.; Cui, J.; Li, Y.; Zhao, Y.; Culicchia, F.; Lukiw, W. Micro RNA-125b (miRNA-125b) function in astrogliosis and glial cell proliferation. Neurosci. Lett. 2010, 476, 18–22. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Song, S.; Pan, X. Knockdown of miR-155 protects microglia against LPS-induced inflammatory injury via targeting RACK1: A novel research for intracranial infection. J. Inflamm. 2017, 14, 17. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Sun, J.; Dai, T.; Xu, Q.; Ding, Y. miR-29c-5p knockdown reduces inflammation and blood–brain barrier disruption by upregulating LRP6. Open Med. 2022, 17, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.; Zurolo, E.; Prabowo, A.; Fluiter, K.; Spliet, W.G.M.; van Rijen, P.C.; Gorter, J.A.; Aronica, E. MicroRNA-146a: A Key Regulator of Astrocyte-Mediated Inflammatory Response. PLoS ONE 2012, 7, e44789. [Google Scholar] [CrossRef]
- Chipman, L.B.; Pasquinelli, A.E. miRNA Targeting: Growing beyond the Seed. Trends Genet. 2019, 35, 215–222. [Google Scholar] [CrossRef]
- Iyer, M.; Kantarci, H.; Cooper, M.H.; Ambiel, N.; Novak, S.W.; Andrade, L.R.; Lam, M.; Jones, G.; Münch, A.E.; Yu, X.; et al. Oligodendrocyte calcium signaling promotes actin-dependent myelin sheath extension. Nat. Commun. 2024, 15, 265. [Google Scholar] [CrossRef]
- Kieran, N.W.; Suresh, R.; Dorion, M.-F.; MacDonald, A.; Blain, M.; Wen, D.; Fuh, S.-C.; Ryan, F.; Diaz, R.J.; Stratton, J.A.; et al. MicroRNA-210 regulates the metabolic and inflammatory status of primary human astrocytes. J. Neuroinflamm. 2022, 19, 10. [Google Scholar] [CrossRef]
- Chen, Y.; Schlotterer, A.; Kurowski, L.; Li, L.; Dannehl, M.; Hammes, H.-P.; Lin, J. miRNA-124 Prevents Rat Diabetic Retinopathy by Inhibiting the Microglial Inflammatory Response. Int. J. Mol. Sci. 2023, 24, 2291. [Google Scholar] [CrossRef]
- Yang, R.; Yang, B.; Liu, W.; Tan, C.; Chen, H.; Wang, X. Emerging role of non-coding RNAs in neuroinflammation mediated by microglia and astrocytes. J. Neuroinflamm. 2023, 20, 173. [Google Scholar] [CrossRef]
- Zeng, M.; Zhang, T.; Lin, Y.; Lin, Y.; Wu, Z. The Common LncRNAs of Neuroinflammation-Related Diseases. Mol. Pharmacol. 2022, 103, 113–131. [Google Scholar] [CrossRef]
- Sharma, S.; Houfani, A.A.; Foster, L.J. Pivotal functions and impact of long con-coding RNAs on cellular processes and genome integrity. J. Biomed. Sci. 2024, 31, 52. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Chen, B.; Yao, X.; Lei, Y.; Ou, F.; Huang, F. Upregulation of the lncRNA MEG3 improves cognitive impairment, alleviates neuronal damage, and inhibits activation of astrocytes in hippocampus tissues in Alzheimer’s disease through inactivating the PI3K/Akt signaling pathway. J. Cell. Biochem. 2019, 120, 18053–18065. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Yu, Z.; Fang, X.; Liu, M.; Pu, Y.; Shao, Q.; Wang, D.; Zhao, X.; Huang, A.; Xiang, Z.; et al. Lnc RNA GAS 5 inhibits microglial M2 polarization and exacerbates demyelination. Embo Rep. 2017, 18, 1801–1816. [Google Scholar] [CrossRef] [PubMed]
- Takei, N.; Inamura, N.; Kawamura, M.; Namba, H.; Hara, K.; Yonezawa, K.; Nawa, H. Brain-Derived Neurotrophic Factor Induces Mammalian Target of Rapamycin-Dependent Local Activation of Translation Machinery and Protein Synthesis in Neuronal Dendrites. J. Neurosci. 2004, 24, 9760–9769. [Google Scholar] [CrossRef]
- Ni, X.; Su, Q.; Xia, W.; Zhang, Y.; Jia, K.; Su, Z.; Li, G. Knockdown lncRNA NEAT1 regulates the activation of microglia and reduces AKT signaling and neuronal apoptosis after cerebral ischemic reperfusion. Sci. Rep. 2020, 10, 19658. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, T.; Zhao, Z.; Wei, W.; Yang, X.; Wang, X.; Xin, W. Novel Insights into the Emerging Role of Neat1 and Its Effects Downstream in the Regulation of Inflammation. J. Inflamm. Res. 2022, 15, 557–571. [Google Scholar] [CrossRef]
- Zeng, C.; Hu, J.; Chen, F.; Huang, T.; Zhang, L. The Coordination of mTOR Signaling and Non-Coding RNA in Regulating Epileptic Neuroinflammation. Front. Immunol. 2022, 13, 924642. [Google Scholar] [CrossRef]
- Han, C.-L.; Ge, M.; Liu, Y.-P.; Zhao, X.-M.; Wang, K.-L.; Chen, N.; Meng, W.-J.; Hu, W.; Zhang, J.-G.; Li, L.; et al. LncRNA H19 contributes to hippocampal glial cell activation via JAK/STAT signaling in a rat model of temporal lobe epilepsy. J. Neuroinflamm. 2018, 15, 103. [Google Scholar] [CrossRef]
- Passamonti, L.; Tsvetanov, K.A.; Jones, P.S.; Bevan-Jones, W.R.; Arnold, R.; Borchert, R.J.; Mak, E.; Su, L.; O’brien, J.T.; Rowe, J.B. Neuroinflammation and Functional Connectivity in Alzheimer’s Disease: Interactive Influences on Cognitive Performance. J. Neurosci. 2019, 39, 7218–7226. [Google Scholar] [CrossRef]
- Griciuc, A.; Tanzi, R.E. The role of innate immune genes in Alzheimer’s disease. Curr. Opin. Neurol. 2021, 34, 228–236. [Google Scholar] [CrossRef]
- Tejera, D.; Mercan, D.; Sanchez-Caro, J.M.; Hanan, M.; Greenberg, D.; Soreq, H.; Latz, E.; Golenbock, D.; Heneka, M.T. Systemic inflammation impairs microglial Aβ clearance through NLRP 3 inflammasome. EMBO J. 2019, 38, e101064. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Gong, J.; Li, S.; Wang, P.; Han, X.; Xu, C.; Luan, H.; Li, R.; Wen, B.; Wei, C. Bibliometric analysis of neuroinflammation and Alzheimer’s disease. Front. Aging Neurosci. 2024, 16, 1423139. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.E.; Jung, I.; Na, J.Y.; Lee, Y.; Lee, J.; Lee, J.S.; Lee, J.S. Pseudane-VII Regulates LPS-Induced Neuroinflammation in Brain Microglia Cells through the Inhibition of iNOS Expression. Molecules 2018, 23, 3196. [Google Scholar] [CrossRef] [PubMed]
- Bellver-Landete, V.; Bretheau, F.; Mailhot, B.; Vallières, N.; Lessard, M.; Janelle, M.-E.; Vernoux, N.; Tremblay, M.; Fuehrmann, T.; Shoichet, M.S.; et al. Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat. Commun. 2019, 10, 518. [Google Scholar] [CrossRef]
- Tarczyluk-Wells, M.A.; Salzlechner, C.; Najafi, A.R.; Lim, M.J.; Smith, D.; Platt, F.M.; Williams, B.P.; Cooper, J.D. Combined Anti-inflammatory and Neuroprotective Treatments Have the Potential to Impact Disease Phenotypes in Cln3−/− Mice. Front. Neurol. 2019, 10, 963. [Google Scholar] [CrossRef]
- Frigerio, C.S.; Wolfs, L.; Fattorelli, N.; Thrupp, N.; Voytyuk, I.; Schmidt, I.; Mancuso, R.; Chen, W.-T.; Woodbury, M.E.; Srivastava, G.; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep. 2019, 27, 1293–1306.e6. [Google Scholar] [CrossRef]
- Barroeta-Espar, I.; Weinstock, L.D.; Perez-Nievas, B.G.; Meltzer, A.C.; Chong, M.S.T.; Amaral, A.C.; Murray, M.E.; Moulder, K.L.; Morris, J.C.; Cairns, N.J.; et al. Distinct cytokine profiles in human brains resilient to Alzheimer’s pathology. Neurobiol. Dis. 2019, 121, 327–337. [Google Scholar] [CrossRef]
- Bradburn, S.; Murgatroyd, C.; Ray, N. Neuroinflammation in mild cognitive impairment and Alzheimer’s disease: A meta-analysis. Ageing Res. Rev. 2019, 50, 1–8. [Google Scholar] [CrossRef]
- Cheng, Y.; Saville, L.; Gollen, B.; Veronesi, A.A.; Mohajerani, M.; Joseph, J.T.; Zovoilis, A. Increased Alu RNA processing in Alzheimer brains is linked to gene expression changes. Embo Rep. 2021, 22, e52255. [Google Scholar] [CrossRef]
- Muthukumarasamy, I.; Buel, S.M.; Hurley, J.M.; Dordick, J.S. NOX2 inhibition enables retention of the circadian clock in BV2 microglia and primary macrophages. Front. Immunol. 2023, 14, 1106515. [Google Scholar] [CrossRef]
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood–brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Jiao, F.; Gong, Z. The Beneficial Roles of SIRT1 in Neuroinflammation-Related Diseases. Oxidative Med. Cell. Longev. 2020, 2020, 6782872. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.; Wang, Y.; Zhang, X.; Sun, X. Recent Studies on Protective Effects of Walnuts against Neuroinflammation. Nutrients 2022, 14, 4360. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.O.; Lee, S.J.; Pyo, J.-S. Effect of acetylcholinesterase inhibitors on post-stroke cognitive impairment and vascular dementia: A meta-analysis. PLoS ONE 2020, 15, e0227820. [Google Scholar] [CrossRef]
- Tan, Q.; Zhang, C.; Rao, X.; Wan, W.; Lin, W.; Huang, S.; Ying, J.; Lin, Y.; Hua, F. The interaction of lipocalin-2 and astrocytes in neuroinflammation: Mechanisms and therapeutic application. Front. Immunol. 2024, 15, 1358719. [Google Scholar] [CrossRef]
- Angelbello, A.J.; Chen, J.L.; Disney, M.D. Small molecule targeting of RNA structures in neurological disorders. Ann. N. Y. Acad. Sci. 2019, 1471, 57–71. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Spangenberg, E.; Severson, P.L.; Hohsfield, L.A.; Crapser, J.; Zhang, J.; Burton, E.A.; Zhang, Y.; Spevak, W.; Lin, J.; Phan, N.Y.; et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat. Commun. 2019, 10, 3758. [Google Scholar] [CrossRef]
- Nasrabady, S.E.; Rizvi, B.; Goldman, J.E.; Brickman, A.M. White matter changes in Alzheimer’s disease: A focus on myelin and oligodendrocytes. Acta Neuropathol. Commun. 2018, 6, 22. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Wang, J.; Tian, T.; Li, X.; Zhang, Y. Noncoding RNAs Emerging as Drugs or Drug Targets: Their Chemical Modification, Bio-Conjugation and Intracellular Regulation. Molecules 2022, 27, 6717. [Google Scholar] [CrossRef] [PubMed]
- El Idrissi, F.; Gressier, B.; Devos, D.; Belarbi, K. A Computational Exploration of the Molecular Network Associated to Neuroinflammation in Alzheimer’s Disease. Front. Pharmacol. 2021, 12, 630003. [Google Scholar] [CrossRef] [PubMed]
- Bruffaerts, R.; Gors, D.; Gallardo, A.B.; Vandenbulcke, M.; Van Damme, P.; Suetens, P.; van Swieten, J.C.; Borroni, B.; Sanchez-Valle, R.; Moreno, F.; et al. Hierarchical spectral clustering reveals brain size and shape changes in asymptomatic carriers of C9orf72. Brain Commun. 2022, 4, fcac182. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Deng, Y.; Duan, D.; Sun, S.; Ge, L.; Zhuo, Y.; Yuan, T.; Wu, P.; Wang, H.; Lu, M.; et al. Hyperthermia influences fate determination of neural stem cells with lncRNAs alterations in the early differentiation. PLoS ONE 2017, 12, e0171359. [Google Scholar] [CrossRef]
- Policarpo, R.; Sierksma, A.; De Strooper, B.; D’ydewalle, C. From Junk to Function: LncRNAs in CNS Health and Disease. Front. Mol. Neurosci. 2021, 14, 714768. [Google Scholar] [CrossRef]
- Ye, Z.; Yuan, T. Role and Therapeutic Potential of Non-Coding RNAs in Astrocytes During Neonatal Brain Injury. 2023. Available online: https://www.preprints.org/manuscript/202312.0927/download/final_file (accessed on 20 December 2024). [CrossRef]
- Qureshi, I.A.; Mehler, M.F. Emerging roles of non-coding RNAs in brain evolution, development, plasticity and disease. Nat. Rev. Neurosci. 2012, 13, 528–541. [Google Scholar] [CrossRef]
- Schirmer, L.; Velmeshev, D.; Holmqvist, S.; Kaufmann, M.; Werneburg, S.; Jung, D.; Vistnes, S.; Stockley, J.H.; Young, A.; Steindel, M.; et al. Neuronal vulnerability and multilineage diversity in multiple sclerosis. Nature 2019, 573, 75–82. [Google Scholar] [CrossRef]
- Taylor, D.H.; Chu, E.T.; Spektor, R.; Soloway, P.D. Long non-coding RNA regulation of reproduction and development. Mol. Reprod. Dev. 2015, 82, 932–956. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Shi, Q.; Colodner, K.J.; Matousek, S.B.; Merry, K.; Hong, S.; Kenison, J.E.; Frost, J.L.; Le, K.X.; Li, S.; Dodart, J.-C.; et al. Complement C3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. J. Neurosci. 2015, 35, 13029–13042. [Google Scholar] [CrossRef]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Lansita, J.A.; Mease, K.M.; Qiu, H.; Yednock, T.; Sankaranarayanan, S.; Kramer, S. Nonclinical Development of ANX005: A Humanized Anti-C1q Antibody for Treatment of Autoimmune and Neurodegenerative Diseases. Int. J. Toxicol. 2017, 36, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Costales, M.G.; Aikawa, H.; Li, Y.; Childs-Disney, J.L.; Abegg, D.; Hoch, D.G.; Velagapudi, S.P.; Nakai, Y.; Khan, T.; Wang, K.W.; et al. Small-molecule targeted recruitment of a nuclease to cleave an oncogenic RNA in a mouse model of metastatic cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 2406–2411. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Martin, P.; Fogarty, B.; Brown, A.; Schurman, K.; Phipps, R.; Yin, V.P.; Lockman, P.; Bai, S. Exosome Delivered Anticancer Drugs Across the Blood-Brain Barrier for Brain Cancer Therapy in Danio Rerio. Pharm. Res. 2015, 32, 2003–2014. [Google Scholar] [CrossRef]
- Zheng, X.; Song, L.; Cao, C.; Sun, S. Multiple roles of circular RNAs in prostate cancer: From the biological basis to potential clinical applications. Eur. J. Med. Res. 2025, 30, 140. [Google Scholar] [CrossRef]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef]
- (Stămat), L.-R.B.; Dinescu, S.; Costache, M. Regulation of Inflammasome by microRNAs in Triple-Negative Breast Cancer: New Opportunities for Therapy. Int. J. Mol. Sci. 2023, 24, 3245. [Google Scholar] [CrossRef]
- Antoniou, D.; Stergiopoulos, A.; Politis, P.K. Recent advances in the involvement of long non-coding RNAs in neural stem cell biology and brain pathophysiology. Front. Physiol. 2014, 5, 155. [Google Scholar] [CrossRef]

| Effect on Glial Cells | |
|---|---|
| miR-124 | Regulates glial cell morphology, influencing astrocyte branching. Affects neuronal health and differentiation by regulating the mTOR pathway. Downregulated during the microglial transition to a reactive state, contributing to neuroinflammation. |
| miR-9 | Modulates neuroinflammation by targeting genes in the NF-κB pathway. Affects neurogenesis, neuronal differentiation, and astrocyte morphology by regulating actin- and tubulin-related genes. |
| miR-146 | Linked to reactive astrocytes, promoting neuroinflammation, impaired repair mechanisms, and neuronal damage. |
| miR-223 | Associated with changes in astrocyte morphology and reactive astrogliosis, contributing to neurotoxicity and neuroinflammation. |
| miR-155 | Implicated in neuroinflammation and changes in astrocyte morphology, contributing to reactive gliosis and neurotoxic responses. |
| miR-146a | Induces reactive microglia and promotes neuroinflammation, synaptic dysfunction, and neuronal damage. |
| Bdnf-AS | Modulates astrocyte morphology and branching by interacting with the mTOR signaling pathway. |
| NEAT1 | Regulates glial activation and inflammation and interacts with the NF-κB pathway. Modulates glial response, including microglia transition from the M2 state to the M1 state. Decreasing NEAT1 is neuroprotective. |
| H19 | Induces activation of astrocytes and microglia, promoting the release of pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α). |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Risen, S.J.; Wahl, D.; LaRocca, T.J.; Moreno, J.A. The Synergistic Roles of Glial Cells and Non-Coding RNAs in the Pathogenesis of Alzheimer’s Disease and Related Dementias (ADRDs). Neuroglia 2025, 6, 22. https://doi.org/10.3390/neuroglia6020022
Risen SJ, Wahl D, LaRocca TJ, Moreno JA. The Synergistic Roles of Glial Cells and Non-Coding RNAs in the Pathogenesis of Alzheimer’s Disease and Related Dementias (ADRDs). Neuroglia. 2025; 6(2):22. https://doi.org/10.3390/neuroglia6020022
Chicago/Turabian StyleRisen, Sydney J., Devin Wahl, Thomas J. LaRocca, and Julie A. Moreno. 2025. "The Synergistic Roles of Glial Cells and Non-Coding RNAs in the Pathogenesis of Alzheimer’s Disease and Related Dementias (ADRDs)" Neuroglia 6, no. 2: 22. https://doi.org/10.3390/neuroglia6020022
APA StyleRisen, S. J., Wahl, D., LaRocca, T. J., & Moreno, J. A. (2025). The Synergistic Roles of Glial Cells and Non-Coding RNAs in the Pathogenesis of Alzheimer’s Disease and Related Dementias (ADRDs). Neuroglia, 6(2), 22. https://doi.org/10.3390/neuroglia6020022