Abstract
Background: Chronic neuroinflammation mediated by persistent microglial activation is strongly linked to neurodegeneration. Therefore, targeting microglial activation could be beneficial in treating neurodegenerative disorders. Angiotensin receptor blockers (ARBs), commonly prescribed for high blood pressure, exhibit prominent anti-inflammatory effects in the brain and are considered potential therapies for neurodegenerative diseases and neurotrauma. Although all ARBs are angiotensin II receptor type I antagonists, some ARBs act through other signaling pathways, allowing for multiple mechanisms of action. The anti-inflammatory mechanisms of ARBs are not well understood. Methods: In this study, we compared eight different FDA-approved ARBs for their ability to reduce the LPS stimulation of primary microglia or BV2 cells through analyses of nitric oxide production, reactive oxygen species generation, and the mRNA of proinflammatory cytokines. Finding specific and unique effects of telmisartan, we interrogated signaling pathways and other downstream effectors of telmisartan activity on microglia. Results: Our findings indicate that telmisartan showed the greatest efficacy in reducing the LPS induction of reactive oxygen species (ROS) and nitric oxide production in microglia. Uniquely amongst ARBs, telmisartan activated AMPK phosphorylation and inhibited mTOR phosphorylation. Telmisartan’s anti-inflammatory activity was partially inhibited by the AMPK inhibitor compound C. Furthermore, telmisartan uniquely induced markers of autophagy in microglia through an AMPK–mTOR–autophagy pathway. Telmisartan also reduced microglial viability. Telmisartan’s cytotoxicity was partially ameliorated by an autophagy inhibitor and a pan-caspase inhibitor, indicating a link between microglial autophagy and apoptosis. Conclusions: We conclude that telmisartan has unique properties relative to other ARBs, including potent anti-inflammatory actions and an induction of microglial autophagy, which may enable specific therapeutic uses.
1. Introduction
Inflammation within the central nervous system (CNS) is an integral feature of neurodegenerative diseases as well as neurotrauma [1,2,3,4]. Although this inflammation may initially be beneficial, chronic neuroinflammation can escalate or worsen neuropathology and cause disease progression. Indeed, chronic neuroinflammation has been strongly correlated with neurodegeneration [1,5,6,7]. Drugs ameliorating neuroinflammation are a reasonable therapeutic option for neurodegeneration and neurotrauma [3,6,8].
Microglia are an important cellular contributor to the development and modulation of inflammation in the CNS [8,9,10]. Exposure of microglia to signals from pathogens or damaged cellular components activates microglia to a proinflammatory phenotype, where they increase their production of proinflammatory cytokines such as IL-1β and TNFα and upregulate the production of nitric oxide (NO) and reactive oxygen species (ROS) [11,12]. Although this activated phenotype can be useful for host defense and phagocytosis, the products from these activated microglia are harmful to neurons, and prolonged activation can lead to a damaging hyperinflammatory state [13,14]. Microglial activation following some CNS insults may persist in these activated proinflammatory states, leading to neuronal injury and neurodegeneration [7,11,15]. However, anti-inflammatory signals can alter the microglial phenotype to an alternate state that either enhances phagocytic activity or promotes tissue repair and remodeling [13]. Although this polar definition of microglial phenotypes is increasingly disputed as many microglia express markers of both states [16], the retention of a more proinflammatory phenotype is prevalent in microglia after a traumatic brain injury (TBI) or in neurodegenerative diseases [17,18]. Reducing this proinflammatory state, and/or shifting microglial phenotypes to a less inflammatory state, is an important therapeutic target for preventing and mitigating neurodegeneration.
Angiotensin receptor blockers (ARBs) are FDA-approved drugs used extensively in the treatment of cardiovascular disorders, including hypertension, heart failure, and diabetic nephropathy [19,20,21]. ARBs act as antagonists at the angiotensin II (Ang II) type I receptor (AT1R) to block Ang II-mediated increases in blood pressure through vasoconstriction or an increased release of aldosterone from the zona glomerulosa of the adrenal gland [19,22]. Besides their antihypertensive effects, ARBs also have anti-inflammatory effects that contribute to their beneficial effects on the cardiovascular system. Signaling through the AT1R activates some pro-inflammatory pathways, including the NF–κB pathway, NADPH oxidase, and ROS production, in addition to upregulating pro-inflammatory mediators [19,23]. ARB’s anti-inflammatory effects are mediated by AT1R-dependent and -independent mechanisms [21,24,25].
Although the systemic RAS functions are mainly outside the CNS, more recent research has revealed the presence of all components of RAS within cells comprising the CNS [26,27,28,29]. These findings have opened up new avenues for therapeutic options for ARBs. A growing number of research studies have demonstrated the neuroprotective effects of ARBs in preclinical models of Alzheimer’s disease (AD), Parkinson’s disease, stroke, and TBI [21,30,31,32]. In addition to their neuroprotective effects, ARBs also have anxiolytic and anti-depressive properties and enhance cognitive function—effects that are probably mediated at least in part by the regulation of AT1R signaling in the brain [33,34]. Many of the neuroprotective effects of ARBs have been attributed to their anti-inflammatory effects in CNS tissues through AT1R-dependent and -independent mechanisms [21,35,36].
There are currently eight FDA approved ARBs [37,38]. Although all these drugs act as AT1R antagonists, they vary in their ability to modulate non-AT1R signaling pathways [39]. Losartan, telmisartan, candesartan, and irbesartan act as partial agonists at the anti-inflammatory PPARγ receptor [31,40]. Telmisartan activates PPARγ more potently than all the other ARBs [41]. Until recently, it was assumed that the AT1R-independent anti-inflammatory effects of ARBs were mediated through the activation of PPARγ [42]. However, telmisartan has been shown to activate AMPK in a variety of cell types, including microglia [43,44,45]. The anti-inflammatory actions of telmisartan in microglia were mediated by AMPK activation [45]. We have shown that both candesartan and telmisartan have efficacy in improving recovery in a mouse model of TBI, including reducing the amount of microglial activation after injury [32]. We were therefore curious as to whether candesartan, telmisartan, and other ARBs had similar mechanisms of anti-inflammatory action in microglia. Using LPS stimulation as a model of microglial activation, we compared the ability of ARBs to reduce microglial activation through AT1R-dependent and -independent pathways. We report that telmisartan is unique in its mechanism of anti-inflammatory action and its effects on microglia. These results provide insights into the action of telmisartan on this important CNS cell type and its therapeutic potential in disorders with an inflammatory component.
2. Materials and Methods
2.1. Materials
The AMPK inhibitor, compound C, and the PPARγ inhibitor, T0070907, were purchased from Sigma-Aldrich (St Louis, MO, USA). The CYTO-ID autophagy assay kit was purchased from Enzo Life Sciences (Farmingdale, NY, USA). Primers were synthesized by Integrated DNA Technologies (Coralville, IA, USA). Dulbecco’s modified Eagle’s medium (15-013-CV), Hanks’ balanced salt solution (21-022-CV), and an antibiotic–antimycotic solution (30-004-CL) were purchased from Corning Cellgro (Manassas, VA, USA). Glutamax was obtained from Gibco Thermo Fisher Scientific (Waltham, MA, USA). Donor equine serum was purchased from Hyclone Thermo Fisher Scientific (Waltham, MA, USA), and Benchmark Fetal Bovine serum was purchased from Gemini Bio-Products (Sacramento, CA, USA). Complete Mini protease inhibitor cocktail tablets (04 693 124 001) were purchased from Roche (Nutley, NJ, USA). The LC3B antibody (NB100-2220) was purchased from Novus Biologicals (Littleton, CO, USA). Antisera recognizing AMPK (2532S), p-AMPK (2535S), mTOR (2972S), and p-mTOR (2971S) were purchased from Cell Signaling Technology (Danvers, MA, USA). Anti-β-Actin (A1978), LPS, telmisartan, candesartan, losartan, irbesartan, azilsartan, valsartan, eprosartan, z-VAD-FMK, Hoechst dye, staurosporine, 3-methyladenine, and chloroquine were all purchased from Sigma-Aldrich (St Louis, MO, USA). H2DCFDA (D399) was purchased from Therma Fisher Scientific (Waltham, MA, USA). PVDF and nitrocellulose membranes were from BioRad, Hercules, CA, USA). Candesartan (AstraZeneca, CV-11974) was a gift from AstraZeneca (Sweden). Faststart universal SYBER Green was purchased from Roche (Indianapolis, IN, USA).
2.2. Cell Culture
2.2.1. Primary Microglia
All animal care and procedures were approved by the Institutional Animal Care and Use Committee of the Uniformed Services University. One-day-old Sasco/Sprague–Dawley rat pups (male and female) were purchased from Charles River Laboratories (Frederick, MD, USA). Mixed glial cultures were made as previously described by Mitchell et al. [46]. Briefly, brains were removed from the pups on day 2, the meninges were removed, and the cortices dissected into a culture medium (Dulbecco’s modified Eagle’s medium, containing 10% fetal bovine serum, 1% glutamax, and 1% antibiotic–antimycotic). Tissue was triturated twice with a 10 mL pipette, followed by successive triturations with an 18G needle (3X), a 22G needle (3X), and finally a 25G needle (1x). The resulting suspension was filtered using a 70 μm nylon mesh before pelleting at 168 g for 10 min at 25 °C. The cortical tissue homogenate was suspended in a culture medium and seeded into T75 flasks at half cortex/flask density. Cells were incubated in a culture medium under standard conditions (37 °C; 5% CO2), and the medium was replaced every 3 or 4 days. On day 14, the flasks were placed on an orbital shaker (MaxQ 2000 orbital shaker; Thermo Fisher Scientific, Rockford, IL, USA) and shaken for 1 h at 200 rpm to detach microglia. The medium containing microglia was collected and centrifuged at 671× g for 5 min at room temperature followed by counting and resuspension in a microglial culture medium (Dulbecco’s modified Eagle’s medium, containing 10% normal horse serum, 1x glutamax, and 1% antibiotic–antimycotic). Cells were plated in 96-well plates at a density of 5 × 105 cells/well for RNA isolation and analysis of NO and ROS in the media. A total of 12 h prior to treatment, the media were replaced with media containing 2.5% normal horse serum.
2.2.2. BV2 Cell Culture
BV2 microglia between passages of 6 and 20 were used. Cells were plated at 1.5 × 105 cells/well in 96-well plates at 200 µL/well and 2.5 × 105 cells/well in 6-well plates with 2 ml of media/well for 24 h using a BV2 culture medium (DMEM supplemented with 10% normal horse serum and 1x glutamax). Cells were serum starved overnight before treatment.
2.2.3. Reactive Oxygen Species (ROS) Assay
ROS was assessed by an H2DCFDA assay according to the manufacturer’s instructions (Thermo Fisher Scientific, Waltham, MA, USA). A total of 20 µM of an H2DCFDA solution was constituted in HBSS. A total of 100 µL of an H2DCFDA solution was added to each well and incubated for 30 min at 37 °C. Fluorescence intensity was determined with the Synergy H1 plate reader at the excitation wavelength of 470 nm and emission wavelength of 530 nm.
2.2.4. Griess Assay
Cells were plated in 96-well plates and treated as indicated. A total of 50 µL of the conditioned medium was removed and combined with 50 µL of the Griess assay reagent in a new plate (Signa-Aldrich, St. Louis, MO, USA). The reaction was incubated in the dark for 30 min at room temperature. The amount of NO generated was determined by reading the absorbance at 540 nm in the Synergy H1 plate reader (BioTek Instruments, Agilent, CA, USA).
2.2.5. RNA Isolation and Quantitative PCR
RNA was isolated using the Trizol reagent according to the manufacturer’s instructions (Thermo Fisher Scientific, Carlsbad, CA, USA). Briefly, treated cells were washed once with 1x PBS, followed by the addition of 500 µL of trizol/well. Phase separation was performed by the addition of chloroform followed by centrifugation at 14,000× g for 15 min. The aqueous phase was carefully transferred into new 1.5 mL eppendorf tubes and the RNA precipitated with isopropyl alcohol. The RNA was quantified by UV spectrometry and its integrity ascertained by gel electrophoresis. cDNA was synthesized from equal amounts of RNA with Superscript III reverse transcriptase (Thermo Fisher Scientific, Carlsbad, CA, USA). SYBR Green qPCR was performed in CFX96 (Bio-Rad, Hercules, CA, USA). Target gene expression was normalized to the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) using the delta threshold cycle (ΔΔCt) method [47] and analyzed with the Bio-Rad CFX Manager 2.0 software.
2.2.6. Western Blots
Cells were washed with ice-cold PBS followed by lysis with a RIPA buffer for 10 min at 4 °C. Lysates were centrifuged to remove the insoluble fraction and the supernatant collected for protein quantification by BCA assay. A 6x loading buffer was added and the samples denatured at 100 °C for 5 min. A total of 15 μg of protein was separated by SDS PAGE. Proteins were transferred onto membranes (PVDF-LC3B, nitrocellulose—all others) by semi-dry transfer (Transblot-turbo, BioRad, Hercules, CA, USA). Membranes were blocked in 5% milk/TBST for 1 h followed by probing with a primary antibody overnight. Membranes were rinsed in TBST, incubated with the appropriate secondary antisera linked to horseradish peroxidase for 1 h, and developed with chemiluminescence (Super Signal West Pico Chemiluminescent substrate, Thermo Fisher Scientific, Waltham, MA, USA). Images were acquired and analyzed with the Chemidoc Touch imaging system (BioRad, Hercules, CA, USA).
2.2.7. Autophagy Assay
BV2 microglia were plated in 96-well plates and treated as required. Cells were analyzed for autophagy using the CYTO-ID assay (Enzo, New York, NY, USA). The medium was removed after the required treatment, and cells were washed in 1x PBS. The CYTO-ID reagent was prepared using a 1:1000 dilution of both the CYTO-ID reagent and the Hoechst 33,342 nuclear stain in a 1x assay buffer. A total of 100 µL of the diluted CYTO-ID reagent was added to each well and incubated for 30 min at 37 °C. The dual detection reagent was removed, and the cells were washed once with PBS. Plates were read in the Synergy H1 fluorescent plate reader with an excitation wavelength of 480 nm and an emission of 530 nm for the CYTO-ID reagent and an excitation of 340 nm and an emission of 480 nm for the Hoechst dye (DAPI filter). Fluorescence intensity for CYTO-ID was normalized to that of Hoechst for each well.
2.2.8. LDH Assay
A total of 50 µL of the conditioned medium was removed and combined with an equal volume of the LDH reagent (Promega, Madison, WI, USA). Reactions were incubated for 30 min in the dark at room temperature before the addition of a stop solution. Absorbance at 490 nm was determined in the synergy H1 plate reader.
2.2.9. MTS Cell Proliferation Assay
To assess cell viability, 20 µL of the MTS reagent (Promega, Madison, WI, USA) was added to cells in 100 µL of a culture medium and incubated for 30 min followed by reading of absorbance in the Synergy H1 plate reader at 490 nM.
2.2.10. Trypan Blue Exclusion
Cells were grown in 6-well plates and treated as required. After treatment, the cells were washed with 1xPBS, trypsinized, and transferred into 15 mL tubes. The cells were centrifuged for 2 min to remove trypsin followed by resuspension in PBS. A total of 20 µL of cell suspension was combined with 20 µL of trypan blue (0.4%), and exclusion was assessed using the Biorad TC20 automated cell counter (BioRad, Hercules, CA, USA).
2.2.11. Mitochondrial Membrane Potential Assay
BV2 cells were plated in 6-well plates or 96-well plates as required. The cells were then treated with telmisartan for 6 h followed by the addition of TMRE (Thermo Fisher Scientific, Waltham, MA, USA) to 1 µM of the final concentration and incubated for 20 min. Cells from the 6-well plates were harvested by trypsinization for flow cytometry in the Acuri C6 flow cytometer (BD Bioscience, San Jose, CA, USA). Cells treated in the 96-well plates were read on the Synergy H1 plate reader at 549 nm excitation and 575 nm emission, and fluorescent images were taken afterwards using the Olympus microscope IX73.
2.2.12. Statistical Analysis
All data are expressed as mean ± SEM. Data were analyzed using the Prism software v 8.0 (Graphpad, La Jolla, CA, USA). A minimum of four independent wells were assayed in each treatment group, and the effects were repeated in at least two independent experiments. A representative experiment is shown in all figures. For the assessment of gene expression by qPCR experiments, the corrected expression was normalized to untreated samples and expressed as a fold change in the expression. Significant changes in reduced or induced gene expression relative to control wells were calculated by one-way ANOVA with Dunnet’s correction for multiple comparisons.
3. Results
3.1. Telmisartan Counteracts LPS-Mediated Inflammatory Changes More Strongly than Other Angiotensin II Receptor Blockers
We have previously shown that telmisartan and candesartan have anti-inflammatory actions in a mouse model of TBI [32]. However, it was not known whether other ARBs share this property. We therefore compared all 8 FDA-approved ARBs for their ability to reduce LPS-stimulated NO and ROS production in rat primary microglia or in the mouse microglial BV2 cell line. A total of 20 µM of telmisartan reduced LPS-induced NO to a greater extent than 100 µM of the other ARBs in either primary microglia (Figure 1A) or BV2 cells (Figure 1C).
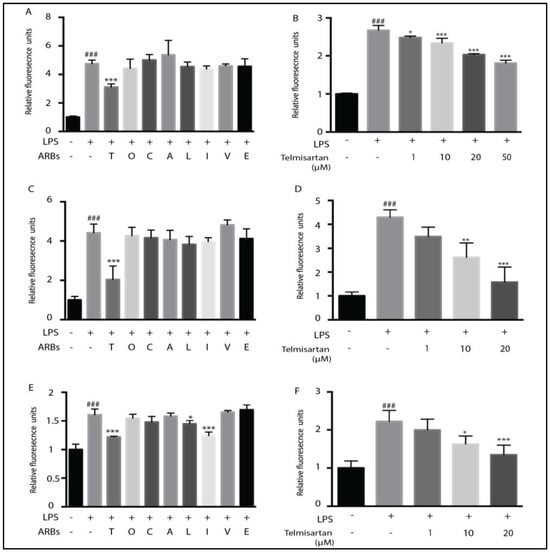
Figure 1.
Telmisartan is the most potent anti-inflammatory angiotensin receptor blocker. Primary microglia (A,B) or BV2 cells (C–F) were pretreated for 2 h with the indicated ARB, followed by treatment with LPS for 12 h (2.5 ng/mL of primary microglia and 100 ng/mL of BV2 cells). Media were collected for NO analysis by the Griess assay (A–D), and the cells were analyzed for the presence of reactive oxygen species (ROS) using the fluorescent indicator dye H2DCFDA (E,F). Telmisartan (T) (20 μM) significantly reduced NO release, whereas the remaining ARBs, (100 µM) Olmesartan (O), Candesartan (C), Azilsartan (A), Losartan (L), Irbsartan (I), Valsartan (V), and Eprosartan (E), had minimal effects (A,C). Telmisartan dose-dependently reduced NO in primary microglia (B) or BV2 cells (D). Telmisartan, losartan, and irbsartan significantly reduced ROS generation in BV2 microglia (E). Telmisartan dose-dependently reduced ROS in BV2 cells (F). (Data are representative of at least 3 repeated experiments; n = 4, mean ± s.e.m. *** p < 0.001, ** p < 0.01, * p < 0.05, compared with LPS-treated cells; ### p < 0.001, compared with untreated cells).
Telmisartan dose-dependently reduced LPS-mediated NO release. In primary microglia, this effect was detectable with 1 µM of telmisartan, increasing up to 50 µM of telmisartan (Figure 1B). In BV2 cells, 10 µM of telmisartan was sufficient to reduce NO production by LPS significantly, and 20 µM of telmisartan reduced NO production to levels of unstimulated cells (Figure 1D). LPS-induced ROS generation in BV2 cells was similarly inhibited by 20 µM of telmisartan (Figure 1E,F). However, irbesartan and losartan also reduced LPS-stimulated ROS production in BV2 cells (Figure 1E). Telmisartan dose-dependently reduced LPS-mediated iNOS and IL-1β mRNA induction in primary microglia and BV2 cells (Figure 2). Thus, telmisartan has a specific anti-inflammatory action in microglia that is not shared by other ARBs.
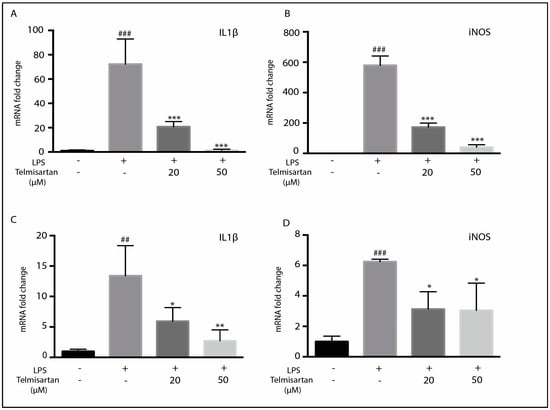
Figure 2.
Telmisartan reduces mRNA of pro-inflammatory mediators. Primary microglia (A,B) or BV2 cells (C,D) were pretreated with telmisartan at the indicated dose for 2 h followed by LPS (2.5 ng/mL of primary microglia and 100 ng/mL of BV2 cells) for 12 h. The cells were then harvested and RNA-isolated for qPCR analysis. Telmisartan dose-dependently reduced IL1β (A,C) and iNOS (B,D) mRNA in BV2 and primary microglia. (Data are representative of at least 3 repeated experiments; n = 4, mean ± s.e.m. *** p < 0.001, ** p < 0.01, * p < 0.05, compared with LPS-treated cells; ### p < 0.001, ## p < 0.01, compared with untreated cells).
3.2. Telmisartan Activates the AMPK/mTOR Pathway
Telmisartan was shown to partially promote the anti-inflammatory polarization of microglia by activating the AMPK pathway [45]. However, the ability of other ARBs to activate AMPK is not known [45]. We therefore compared the ability of telmisartan and other ARBs to induce AMPK phosphorylation in BV2 cells. As was previously reported, 10 µM of telmisartan induced AMPK phosphorylation in BV2 cells, but candesartan, valsartan, irbesartan, and olmesartan did not significantly alter AMPK phosphorylation at this dose (Figure 3A). The activation of AMPK by telmisartan was dose dependent (Figure 3B). As activated AMPK inhibits mTOR phosphorylation [48], we assessed whether telmisartan treatment reduced mTOR phosphorylation in BV2 cells. Telmisartan dose-dependently reduced the amount of phosphorylated mTOR detected (Figure 3C).
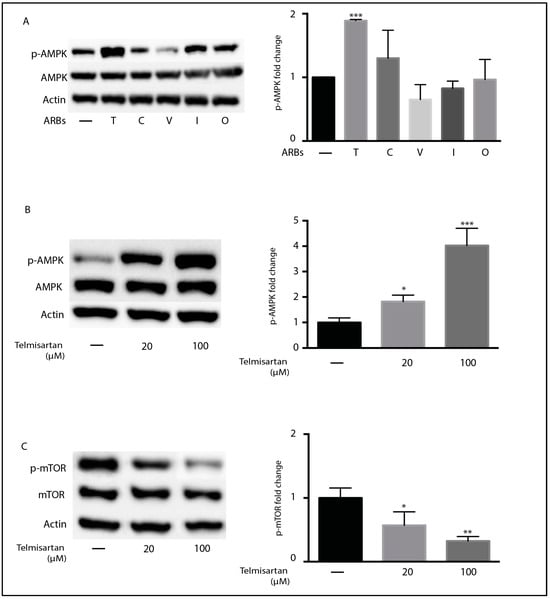
Figure 3.
Telmisartan activates the AMPK/mTOR pathway in BV2 microglia: (A) BV2 cells were treated with the indicated ARBs (100 µM) for 3 h before the harvesting and isolation of proteins. Western blots indicate the effect of treatment on the phosphorylation of AMPK (A,B) and mTOR (C). Graphs show the quantification of Western blots. (A) Telmisartan (T) but not candesartan (C), olmesartan (O), valsartan (V), or irbesartan (I) significantly induced the phosphorylation of AMPK. Telmisartan treatment resulted in the dose-dependent phosphorylation of AMPK (B) and reduction in mTOR phosphorylation (C). (Data are representative of at least 2 repeat experiments. n = 3, mean ± s.e.m. *** p < 0.001,** p < 0.01, * p < 0.05, relative to untreated cells).
3.3. Telmisartan’s Anti-Inflammatory Activity Is Partially Inhibited by Compound C
Some ARBs, in addition to acting as antagonists at AT1R, also act as partial agonists at PPARγ [40]. As activation of PPARγ has anti-inflammatory activity in microglia [49], it was assumed that PPARγ effects may mediate some anti-inflammatory activity. Indeed, Telmisartan is the strongest partial PPARγ agonist of the FDA-approved ARBs [40,41]. To determine if PPARγ may mediate the anti-inflammatory effects of telmisartan, we treated cells with a PPARγ antagonist (T0070907) before stimulating with LPS. However, pretreatment of microglia with two different concentrations of T0070907 did not block telmisartan’s ability to reduce the LPS-mediated production of ROS or NO (Figure 4A,C), suggesting that telmisartan’s activation of PPARγ was not responsible for its anti-inflammatory activity. To determine whether AMPK activity was responsible for telmisartan’s anti-inflammatory effects, we pretreated BV2 cells with the AMPK inhibitor compound C (CC) before adding telmisartan and then LPS. Inhibition of AMPK with compound C partially inhibited telmisartan’s ability to reduce LPS-generated ROS production (Figure 4B) but did not prevent telmisartan’s ability to reduce LPS-mediated NO production (Figure 4D). These data suggest that telmisartan’s anti-inflammatory ability involves the activation of AMPK but not PPARγ.
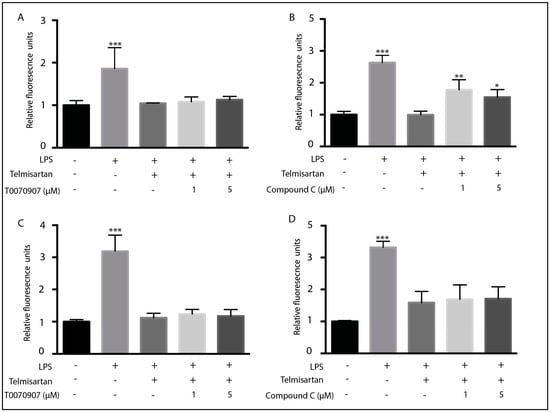
Figure 4.
Inhibition of AMPK by compound C reduced reactive oxygen species but not nitric oxide release from microglia: BV2 microglia were pretreated with (A,C) the PPARγ inhibitor T0070907 (1 μm) or (B,D) the AMPK inhibitor compound C for 1 h followed by telmisartan (20 µM) for 2 h. Cells were then treated with LPS (100 ng/mL) for 12 h. Cells were analyzed for ROS content using H2DCFDA (A,B), and media were collected for analysis of NO content by the Griess assay (C,D). Compound C (B), but not T0070907 (A), prevented the telmisartan-dependent reduction in ROS in LPS-treated BV2 microglia. However, neither compound C nor T0070907 altered the telmisartan-mediated reduction in the LPS-induced NO release (C,D). (Data are representative of at least 3 repeat experiments; n = 4, mean ± s.e.m. *** p < 0.001, ** p < 0.01, * p < 0.05 compared to control cells).
3.4. Telmisartan Increases Mitochondrial Membrane Potential in BV2 Cells
AMPK plays a critical role in mitochondrial biogenesis, fission, and signaling [50]. We therefore assessed whether telmisartan treatment resulted in an alteration of the mitochondrial membrane potential in BV2 cells. The mitochondrial membrane potential was assessed with the tetramethylrhodamine ethyl ester (TMRE) dye that accumulates in active mitochondria in direct proportion to their membrane potential (ΔΨm) [51]. Telmisartan treatment increased the mitochondrial membrane potential in BV2 cells after 6 h of treatment (Figure 5). The fluorescent intensity of cells incubated with TMRE increased with telmisartan treatment as determined by fluorescent intensity (Figure 5A), fluorescent microscopy (Figure 5B) and flow cytometry (Figure 5C). These data show that telmisartan treatment, at least initially, enhances the mitochondrial membrane potential.
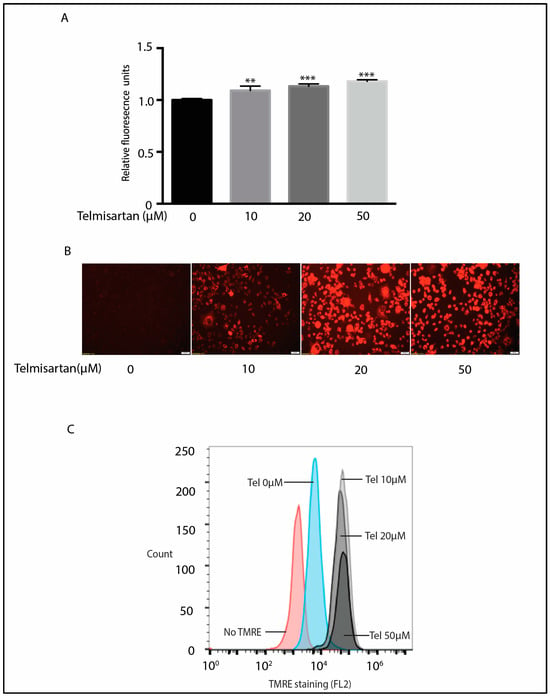
Figure 5.
Telmisartan increases mitochondrial membrane potential in BV2 cells. (A) BV2 microglia were treated with increasing doses of telmisartan for 6 h followed by incubation with TMRE (1 μm) for 20 min. Telmisartan increased TMRE fluorescence (n = 3, mean ± s.e.m. *** p < 0.001, ** p < 0.01). (B) Representative fluorescent images of telmisartan-treated BV2 cells after treatment with TMRE show an increased mitochondrial membrane potential with telmisartan treatment. (C) BV2 cells were treated with increasing doses of telmisartan for 6 h followed by 20 min of TMRE (1 µM) treatment. Flow cytometry shows that telmisartan increased the intensity of TMRE-positive cells. (Data are representative of 3 repeated experiments).
3.5. Telmisartan Induces Autophagy in BV2 Microglia
As AMPK activation with an associated inhibition of mTOR is associated with the induction of autophagy, we next determined whether telmisartan treatment resulted in increased autophagy in BV2 cells. Autophagy is a cellular process whereby damaged cytoplasmic components are degraded in order to preserve cellular homeostasis [52,53]. We examined the ratio of LC3BII to LC3BI, a marker of initiation of autophagosome and autolysosome formation [54]. A total of 10 h of telmisartan treatment led to an increase in the LC3BII/1 ratio (Figure 6A). Co-treatment with chloroquine, a drug that blocks autophagic flux and therefore allows for the accumulation of autophagosomes, enhanced this LC3BII/1 ratio (Figure 6A). Dose-dependent telmisartan-induced autophagy was also detected with the autophagy detection kit, CytoID (Figure 6C).
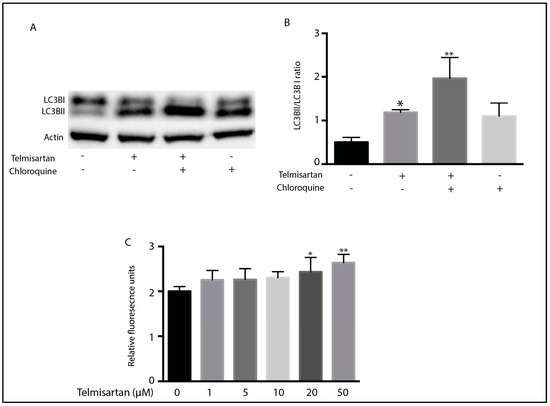
Figure 6.
Telmisartan induces markers of autophagy in BV2 microglia: BV2 cells were treated for 10 h with telmisartan (20 µM) with or without chloroquine (50 µM) before harvesting and protein extraction. (A) Representative Western blot of LC3B shows that telmisartan increased the ratio of LC3BII to LC3BI. This increase was augmented by chloroquine treatment. (B) Quantification of LC3B Western blot showing the ratio of LC3BII to LC3BI. (C) Autophagy was assessed in BV2 cells with the CytoID kit. Telmisartan dose-dependently increased the fluorescence of BV2 cells incubated with the CytoID dye, indicating increased autophagosome formation. (Data are representative of 3 repeated experiments; n = 4, mean ± s.e.m, ** p < 0.01, * p < 0.05 relative to untreated cells).
3.6. Telmisartan Dose-Dependently Reduce Viability of Microglia
Telmisartan has previously been shown to induce apoptosis in adult T-cell leukemia cells [55] and human endometrial cancer cells [56]. However, the ability of ARBs to alter microglial viability is unknown. We therefore determined whether any of the eight ARBs altered microglial viability through assaying for lactate dehydrogenase (LDH) release as a measure of drug-induced cytotoxicity. Telmisartan was unique in its ability to increase LDH release from primary microglial cultures or BV2 microglia (Figure 7A,C). Telmisartan dose-dependently induced LDH release in both cell types, with 10 µM of telmisartan producing a significant induction of LDH release (Figure 7B,D). Telmisartan’s reduction in BV2 microglial viability was confirmed by both the MTS cell proliferation assay (Figure 7E) and trypan blue exclusion (Figure 7F).
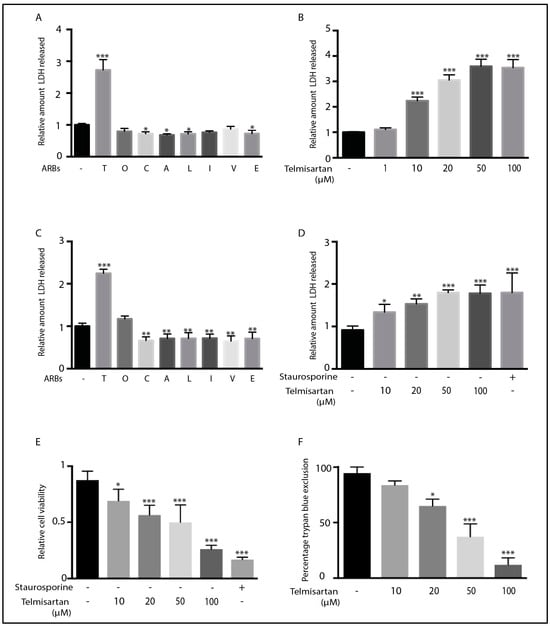
Figure 7.
Telmisartan induces death of BV2 and primary microglia: Primary microglia (A,B) and BV2 cells (C–F) were treated with different ARBs (100 µM) for 12 h before an assessment of LDH release and cell viability. Telmisartan was the only ARB to significantly induce LDH release in either primary microglia (A) or BV2 cells (C). Telmisartan dose-dependently increased LDH release in primary microglia (B) and BV2 cells (D). Staurosporine (1 μM) was added as a positive control for cell death (D,E). Telmisartan also dose-dependently reduced cell viability and proliferation, as indicated by the amount of reduced MTS detected (E) and trypan blue exclusion (F) in BV2 cells. (Data are representative of at least 3 repeat experiments; n = 4, mean ± s.e.m. *** p < 0.001, ** p < 0.01, * p < 0.05 compared to untreated cells).
3.7. Inhibition of Autophagy and Apoptosis Reduced Telmisartan-Induced Cell Death in BV2 Cells
To determine whether telmisartan is cytotoxic to microglia through the induction of autophagy or apoptosis, we pretreated primary microglia with increasing doses of the autophagy inhibitor 3MA or the pan-Caspase inhibitor Z-VAD-FMK prior to telmisartan treatment. The telmisartan-mediated LDH release was almost completely inhibited by Z-VAD-FMK (Figure 8B) and partially inhibited with 2.5 µM of 3MA (Figure 8A). These data suggest that telmisartan is cytotoxic to microglia through the induction of apoptosis. They also suggest that telmisartan-induced autophagy may contribute to microglial-induced cell death.
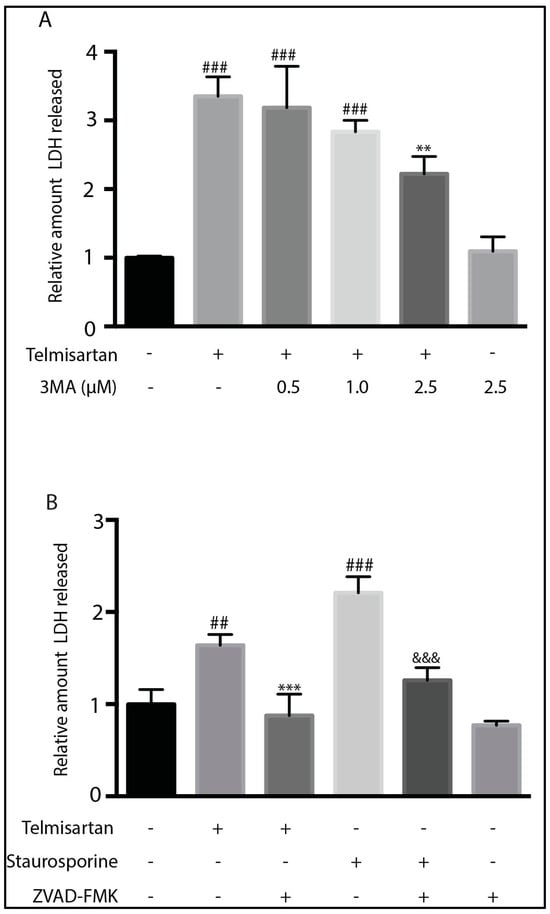
Figure 8.
Inhibition of autophagy or caspases reduced telmisartan-mediated cell death in BV2 cells. (A). BV2 cells were pretreated with 3-methyladenine (3MA) for 1 h followed by telmisartan (50 μM) for 12 h before an assessment of LDH release. 2.5 µM of 3MA treatment partially reduced the telmisartan-mediated LDH release (mean ± s.e.m, n = 3, ### p < 0.001, compared to untreated cells, ** p < 0.01, compared to telmisartan treated cells). (B). BV2 cells were pretreated with Z-VAD-FMK (20 μM) for 1 h followed by either telmisartan (50 μM) or Staurosporine (0.5 μM) for 12 h. Either telmisartan or staurosporine treatment increased cell death, as determined by LDH release, compared to no treatment (### p < 0.001, ## p < 0.01). Z-VAD-FMK significantly reduced either telmisartan-(*** p < 0.001) or staurosporine (&&& p < 0.001)-mediated LDH release. (Data represent 3 experiments (mean ± s.e.m, n = 3)).
4. Discussion
Losartan, candesartan, and telmisartan have efficacy in preclinical models of TBI [32,57,58]. Telmisartan and candesartan are able to reduce neuroinflammation, reduce amyloid burden, and improve cognitive performance in rodent AD models [59,60,61]. Retrospective studies have shown that patients taking ARBs for hypertension have a reduced incidence of AD, and there is a growing consensus that ARBs may have efficacy in preventing or treating AD [31,35,58,62,63,64]. Given the interest in these drugs to treat neurodegenerative diseases and neurotrauma, an understanding on their actions in specific cell types is warranted. Here, we show that telmisartan has unique mechanisms for reducing pro-inflammatory mediators in microglia. We also demonstrate that telmisartan is able to induce autophagy and cell death in microglia in contrast to other ARBs. This effect may, in part, account for its more potent anti-inflammatory effects.
Although ARBs were designed as antagonists of AT1Rs, it is likely that their direct anti-inflammatory effects on microglia are not mediated through this mechanism. It is uncertain whether microglia express AT1Rs in culture or in vivo [65], and others have not found AT1R expressed in BV2 microglia [66,67]. Further, we found telmisartan to have the same anti-inflammatory effects in microglia cultured from AT1R knockout mice. We did not find angiotensinogen mRNA, encoding the upstream precursor for the AT1R ligand, Ang II, in our primary microglial cultures. We therefore suggest that the ARB-mediated anti-inflammatory effects in our microglial cultures are independent of AT1R antagonism. However, AT1R blockade may play a significant role in vivo in mediating the anti-inflammatory effects of ARBs due to complex interactions between different cell types together with potentially different microglial phenotypes in vivo than in culture [68].
ARBs can also modulate AT1R-independent signaling, including through PPARγ [41,69,70], PPARα [71], PPARδ [44], PTEN [72], and AMPK [45,73]. To date, telmisartan is a stronger inducer of these alternate pathways than other ARBs: something our data also support (Figure 1 and Figure 3). Losartan, irbesartan, and candesartan also act as partial PPARγ agonists in vitro [69], and we have shown that candesartan’s anti-inflammatory actions in a mouse model of TBI can be reduced with a PPARγ antagonist [25,32]. Although PPARγ agonists reduce inflammation [74,75], inhibition of the PPARγ pathway with T0070907 did not interfere with telmisartan’s ability to reduce ROS and NO induction in LPS-treated BV2 cells (Figure 4). In contrast, PPARγ knockdown with siRNA partially prevented telmisartan’s ability to reduce IL-1β and TNFα mRNA in LPS-activated microglia [45]. The discrepancy between our data and those of Xu et al. [45] could be due to the different outcome measures used or the different methods of interfering with PPARγ signaling. We can conclude that telmisartan’s ability to activate PPARγ signaling is at most only a partial contributor to its anti-inflammatory actions.
In this study, we showed that telmisartan, but not candesartan, olmesartan, valsartan, or irbesartan, activates AMPK in microglia (Figure 3). AMPK is involved in cellular responses to stress, nutrient deprivation, and energy homeostasis [76]. It has also been associated with anti-inflammatory states in immune cells [45,57,77]. AMPK activation is associated with macrophage polarization towards an anti-inflammatory phenotype and can block the LPS-mediated induction of proinflammatory cytokines [78,79]. In our case, compound C inhibition of AMPK partially ameliorated telmisartan’s ability to reduce ROS but not NO induction by LPS in microglia, suggesting a linkage between telmisartan’s activation of AMPK and its anti-inflammatory action. Xu et al. also showed that compound C inhibited telmisartan’s ability to reduce IL-1β and TNFα mRNA in LPS-activated microglia [45]. Thus, telmisartan’s activation of the AMPK pathway may partially account for some of its anti-inflammatory effects in microglia.
Several downstream pathways are regulated by AMPK [76]. However, no downstream or cellular effector of AMPK activation has been assessed in telmisartan-treated microglia. mTOR is an important downstream target negatively regulated by AMPK [48]. We showed that telmisartan reduced mTOR phosphorylation in BV2 microglia (Figure 3C). The reduced phosphorylation of mTOR reduces its activity [48]. mTOR mediates cell survival and proliferation with overall anabolic effects [80,81] in contrast to the catabolic effects of AMPK [82]. The role of mTOR in regulating inflammation is controversial, with rapamycin-mediated inhibition leading to either the reduction or stimulation of inflammatory mediators, dependent on context [83]. In microglia, mTOR inhibition results in reduced pro-inflammatory cytokine release [84,85], which is consistent with our data. Thus, increased AMPK phosphorylation and decreased mTOR phosphorylation should reduce inflammatory signaling in response to telmisartan.
The commonly used anti-diabetic drug metformin activates AMPK by increasing cytoplasmic AMP through an inhibition of complex I of the mitochondrial electron transport chain [53,86]. Telmisartan has also been shown to regulate mitochondrial function, suggesting a possible mechanism that may link telmisartan treatment to AMPK activation. Telmisartan increases mitochondrial activity in vascular smooth muscle cells [87], but inhibits the mitochondrial membrane polarization in T-cell leukemic cells [55]. Our data show that telmisartan increased rather than decreased the mitochondrial membrane potential (Figure 5), indicating that telmisartan does not induce AMPK phosphorylation by inhibiting mitochondrial function. Telmisartan has also been shown to induce AMPK activation through the calcium/calmodulin-dependent protein kinase β (CaMKKβ) in microglia [45], suggesting an alternate mechanism through which telmisartan can activate AMPK.
We show telmisartan-induced autophagy in BV2 microglia (Figure 6). Telmisartan can induce autophagy in vascular smooth muscle endothelial cells [48] and T-cells [55]. As the AMPK–mTOR pathway is linked to the induction of autophagy [88], our data are consistent with telmisartan activating an AMPK–mTOR–autophagy pathway. AMPK directly promotes autophagy by phosphorylating autophagy-associated proteins, such as ULK1 and Beclin1-Vps34, in addition to mTOR [89]. The induction of microglial autophagy could serve many functions including enhanced phagocytosis, the regulation of apoptosis, and the regulation of metabolism [90]. Additionally, the induction of autophagy in microglia is linked to an enhanced anti-inflammatory response, with a microglial-specific inhibition of autophagy, promoting a proinflammatory phenotype [91,92,93]. Thus, telmisartan’s anti-inflammatory actions could be partly mediated through autophagy induction.
It is possible that telmisartan’s induction of autophagy is also linked to its beneficial action in neurodegenerative diseases [62]. Telmisartan has beneficial effects on rodent models of AD [60,94]. These most probably result from a combined action of telmisartan on neurons and glial cells. In these murine models telmisartan does have specific microglial anti-inflammatory activity [60,94] and can increase the autophagic digestion of Aβ [94], but it has not previously been shown that telmisartan directly increases microglial autophagy. Therefore, our data suggest that increased microglial autophagy may be one mechanism through which telmisartan treatment leads to decreased Aβ accumulation in the brain. To our knowledge, there are no clinical studies that have examined the role of individual ARBs in reducing the development of AD. Thus, we do not know whether telmisartan is better than other ARBs in this respect. Telmisartan and candesartan, classified together as a “blood–brain barrier-crossing ARBs”, gave better protection against memory loss in older adults versus “non-blood–brain barrier-crossing ARBs” when used as chronic hypertension medication [95].
Our data show that increasing telmisartan doses can lead to reduced proliferation and increased death of both primary microglia and BV2 microglia (Figure 7A,C), which is surprising in light of telmisartan’s generally protective actions with a favorable safety profile [96]. Telmisartan’s toxicity was dose-dependent, with doses as low as 10 µM causing significant toxicity, as assessed by LDH release (Figure 7B,D). More extensive cell death was observed at doses of 50 µM and 100 µM. It is difficult to translate drug doses used in cell cultures to those used in vivo, due to the complexity of the many factors in vivo that can influence drug concentrations and effects. It is therefore possible that these results are an anomaly of an individual, isolated cell culture, and any cell death telmisartan may cause in vivo is negligible.
Interestingly, we did not detect toxicity of low doses of telmisartan in the presence of LPS, suggesting that this is not a mechanism that mediates telmisartan’s protective, anti-inflammatory actions. None of the other ARBs showed a similar toxicity at 100 µM (Figure 7A,C), illustrating the unique action of telmisartan on microglia. Telmisartan can induce apoptosis in a variety of cell types, although these are mainly transformed cells, so this activity is considered beneficial anti-tumor activity [55,97,98,99]. Telmisartan’s toxicity was partially ameliorated by 3-methyladenine, an inhibitor of early autophagy (Figure 8A), and prevented by pretreatment with the pan-caspase inhibitor Z-VAD-FMK (Figure 8B). These data suggest that telmisartan activates caspases to cause apoptotic cell death and that the induction of autophagy partially mediates this. There is known to be a link between autophagy and apoptosis in microglia. Autophagy promoted hypoxia-induced primary microglial cell death, and mTOR-regulated autophagy was involved in microglial apoptosis after hypoxia/reperfusion [70,100]. Thus, telmisartan’s activation of the AMPK–mTOR–autophagy pathway may contribute to its toxicity in microglia.
In conclusion, we have shown that telmisartan, unlike other ARBs, is a potent inhibitor of microglial activation in vitro. We showed that the PPARγ pathway was not involved in the reduction in ROS and NO in LPS-activated microglia by telmisartan. However, the decrease in ROS by telmisartan in LPS-activated microglia was partially AMPK dependent. We also show, for the first time, that telmisartan activates autophagy in microglia and reduces microglial viability (Figure 9). These data suggest that telmisartan has unique properties relative to other ARBs, which may enable specific therapeutic uses, but caution may also be warranted when used clinically.
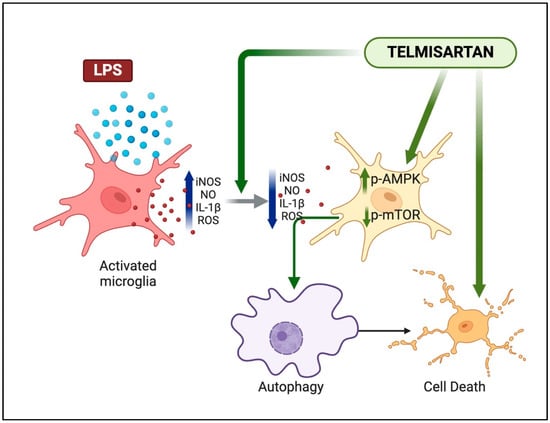
Figure 9.
Summary illustration of telmisartan actions on microglia. Telmisartan can reduce the LPS-mediated activation of proinflammatory mediators and activate the AMPK–mTOR–autophagy pathway. At higher concentrations, telmisartan can also induce microglial apoptosis.
Author Contributions
K.O.A., Z.C.J. and N.S. designed and performed experiments and analyzed the data; K.O.A., S.V. and A.J.S. made substantial contributions to the conception and design of the study and performed data analysis and interpretation; and K.O.A., S.V. and A.J.S. wrote and edited the paper and compiled figures. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the Centre for Neuroscience and Regenerative Medicine, a pre-doctoral fellowship (K.O.A.), and a standard grant (306135 6.01 60855) to A.J.S.
Institutional Review Board Statement
All experiments involving animals in this paper were approved by the Institutional Animal Care and Use Committee of the Uniformed Services University (PHA-15-736 approved on 16 July 2015) and performed according to predetermined protocols.
Informed Consent Statement
Not applicable.
Data Availability Statement
Raw data shown in graph form in this paper are available from the authors upon request.
Acknowledgments
The authors thank Andrew Snow for his guidance in using flow cytometry. We thank all members of the Symes laboratory for helpful discussions. The opinions and assertions contained herein are the private opinions of the authors and are not to be construed as reflecting the views of the Uniformed Services University of the Health Sciences or the US Department of Defense.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Nichols, M.R.; St-Pierre, M.K.; Wendeln, A.C.; Makoni, N.J.; Gouwens, L.K.; Garrad, E.C.; Sohrabi, M.; Neher, J.J.; Tremblay, M.E.; Combs, C.K. Inflammatory mechanisms in neurodegeneration. J. Neurochem. 2019, 149, 562–581. [Google Scholar] [CrossRef] [PubMed]
- De Marchi, F.; Munitic, I.; Vidatic, L.; Papic, E.; Racki, V.; Nimac, J.; Jurak, I.; Novotni, G.; Rogelj, B.; Vuletic, V.; et al. Overlapping Neuroimmune Mechanisms and Therapeutic Targets in Neurodegenerative Disorders. Biomedicines 2023, 11, 2793. [Google Scholar] [CrossRef] [PubMed]
- Strogulski, N.R.; Portela, L.V.; Polster, B.M.; Loane, D.J. Fundamental Neurochemistry Review: Microglial immunometabolism in traumatic brain injury. J. Neurochem. 2023, 167, 129–153. [Google Scholar] [CrossRef] [PubMed]
- Lull, M.E.; Block, M.L. Microglial activation and chronic neurodegeneration. Neurotherapeutics 2010, 7, 354–365. [Google Scholar] [CrossRef] [PubMed]
- Shadfar, S.; Hwang, C.J.; Lim, M.S.; Choi, D.Y.; Hong, J.T. Involvement of inflammation in Alzheimer’s disease pathogenesis and therapeutic potential of anti-inflammatory agents. Arch. Pharmacal Res. 2015, 38, 2106–2119. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Xu, Y.; Sun, L.; Hashimoto, M.; Wei, J. Microglial response to aging and neuroinflammation in the development of neurodegenerative diseases. Neural Regen. Res. 2024, 19, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Kochanek, P.M.; Jackson, T.C.; Ferguson, N.M.; Carlson, S.W.; Simon, D.W.; Brockman, E.C.; Ji, J.; Bayir, H.; Poloyac, S.M.; Wagner, A.K.; et al. Emerging therapies in traumatic brain injury. Semin. Neurol. 2015, 35, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Tobeh, N.S.; Bruce, K.D. Emerging Alzheimer’s disease therapeutics: Promising insights from lipid metabolism and microglia-focused interventions. Front. Aging Neurosci. 2023, 15, 1259012. [Google Scholar] [CrossRef]
- Wu, Q.; Zou, C. Microglial Dysfunction in Neurodegenerative Diseases via RIPK1 and ROS. Antioxidants 2022, 11, 2201. [Google Scholar] [CrossRef]
- Lively, S.; Schlichter, L.C. Microglia Responses to Pro-inflammatory Stimuli (LPS, IFNgamma+TNFalpha) and Reprogramming by Resolving Cytokines (IL-4, IL-10). Front. Cell Neurosci. 2018, 12, 215. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.W.; McGeachy, M.J.; Bayir, H.; Clark, R.S.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Chan, Y.L.; Nguyen, L.T.; Mao, Y.; de Rosa, A.; Beh, I.T.; Chee, C.; Oliver, B.; Herok, G.; Saad, S.; et al. Moderate traumatic brain injury is linked to acute behaviour deficits and long term mitochondrial alterations. Clin. Exp. Pharmacol. Physiol. 2016, 43, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Jiang, J.; Tan, Y.; Chen, S. Microglia in neurodegenerative diseases: Mechanism and potential therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 359. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. A polarizing question: Do M1 and M2 microglia exist? Nat. Neurosci. 2016, 19, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Faden, A.I.; Loane, D.J. Chronic neurodegeneration after traumatic brain injury: Alzheimer disease, chronic traumatic encephalopathy, or persistent neuroinflammation? Neurotherapeutics 2015, 12, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Wangler, L.M.; Godbout, J.P. Microglia moonlighting after traumatic brain injury: Aging and interferons influence chronic microglia reactivity. Trends Neurosci. 2023, 46, 926–940. [Google Scholar] [CrossRef] [PubMed]
- Karnik, S.S.; Unal, H.; Kemp, J.R.; Tirupula, K.C.; Eguchi, S.; Vanderheyden, P.M.; Thomas, W.G. International Union of Basic and Clinical Pharmacology. XCIX. Angiotensin Receptors: Interpreters of Pathophysiological Angiotensinergic Stimuli [corrected]. Pharmacol. Rev. 2015, 67, 754–819. [Google Scholar] [CrossRef]
- Johnson, S.A.; Spurney, R.F. Twenty years after ACEIs and ARBs: Emerging treatment strategies for diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2015, 309, F807–F820. [Google Scholar] [CrossRef]
- Villapol, S.; Janatpour, Z.C.; Affram, K.O.; Symes, A.J. The Renin Angiotensin System as a Therapeutic Target in Traumatic Brain Injury. Neurotherapeutics 2023, 20, 1565–1591. [Google Scholar] [CrossRef] [PubMed]
- Barreras, A.; Gurk-Turner, C. Angiotensin II receptor blockers. Proceedings 2003, 16, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Nagai, N.; Oike, Y.; Izumi-Nagai, K.; Urano, T.; Kubota, Y.; Noda, K.; Ozawa, Y.; Inoue, M.; Tsubota, K.; Suda, T.; et al. Angiotensin II type 1 receptor-mediated inflammation is required for choroidal neovascularization. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2252–2259. [Google Scholar] [CrossRef] [PubMed]
- Pang, T.; Benicky, J.; Wang, J.; Orecna, M.; Sanchez-Lemus, E.; Saavedra, J.M. Telmisartan ameliorates lipopolysaccharide-induced innate immune response through peroxisome proliferator-activated receptor-gamma activation in human monocytes. J. Hypertens. 2012, 30, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Villapol, S.; Yaszemski, A.K.; Logan, T.T.; Sanchez-Lemus, E.; Saavedra, J.M.; Symes, A.J. Candesartan, an Angiotensin II AT1-Receptor Blocker and PPAR-gamma Agonist, Reduces Lesion Volume and Improves Motor and Memory Function After Traumatic Brain Injury in Mice. Neuropsychopharmacology 2012, 37, 2817–2829. [Google Scholar] [CrossRef] [PubMed]
- Von Bohlen und Halbach, O.; Albrecht, D. The CNS renin-angiotensin system. Cell Tissue Res. 2006, 326, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Farag, E.; Sessler, D.I.; Ebrahim, Z.; Kurz, A.; Morgan, J.; Ahuja, S.; Maheshwari, K.; John Doyle, D. The renin angiotensin system and the brain: New developments. J. Clin. Neurosci. 2017, 46, 1–8. [Google Scholar] [CrossRef]
- Samani, N.J.; Swales, J.D.; Brammar, W.J. Expression of the renin gene in extra-renal tissues of the rat. Biochem. J. 1988, 253, 907–910. [Google Scholar] [CrossRef]
- Mizuno, K.; Hashimoto, S.; Ojima, M.; Kunii, N.; Tani, M.; Niimura, S.; Watari, H.; Fukuchi, S. Immunoreactive renin in human brain: Distribution and properties. Jpn. Circ. J. 1985, 49, 1005–1011. [Google Scholar] [CrossRef][Green Version]
- Benicky, J.; Sanchez-Lemus, E.; Honda, M.; Pang, T.; Orecna, M.; Wang, J.; Leng, Y.; Chuang, D.M.; Saavedra, J.M. Angiotensin II AT(1) receptor blockade ameliorates brain inflammation. Neuropsychopharmacology 2011, 36, 857–870. [Google Scholar] [CrossRef]
- Villapol, S.; Saavedra, J.M. Neuroprotective effects of angiotensin receptor blockers. Am. J. Hypertens. 2015, 28, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Villapol, S.; Balarezo, M.G.; Affram, K.; Saavedra, J.M.; Symes, A.J. Neurorestoration after traumatic brain injury through angiotensin II receptor blockage. Brain 2015, 138, 3299–3315. [Google Scholar] [CrossRef] [PubMed]
- Urmila, A.; Rashmi, P.; Nilam, G.; Subhash, B. Recent Advances in the Endogenous Brain Renin-Angiotensin System and Drugs Acting on It. J. Renin Angiotensin Aldosterone Syst. 2021, 2021, 9293553. [Google Scholar] [CrossRef]
- Chrissobolis, S.; Luu, A.N.; Waldschmidt, R.A.; Yoakum, M.E.; D’Souza, M.S. Targeting the renin angiotensin system for the treatment of anxiety and depression. Pharmacol. Biochem. Behav. 2020, 199, 173063. [Google Scholar] [CrossRef]
- Gouveia, F.; Camins, A.; Ettcheto, M.; Bicker, J.; Falcao, A.; Cruz, M.T.; Fortuna, A. Targeting brain Renin-Angiotensin System for the prevention and treatment of Alzheimer’s disease: Past, present and future. Ageing Res. Rev. 2022, 77, 101612. [Google Scholar] [CrossRef]
- Elsaafien, K.; de Kloet, A.D.; Krause, E.G.; Sumners, C. Brain Angiotensin Type-1 and Type-2 Receptors in Physiological and Hypertensive Conditions: Focus on Neuroinflammation. Curr. Hypertens. Rep. 2020, 22, 48. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, J.M. Angiotensin Receptor Blockers Are Not Just for Hypertension Anymore. Physiology 2021, 36, 160–173. [Google Scholar] [CrossRef]
- Chen, Y.J.; Li, L.J.; Tang, W.L.; Song, J.Y.; Qiu, R.; Li, Q.; Xue, H.; Wright, J.M. First-line drugs inhibiting the renin angiotensin system versus other first-line antihypertensive drug classes for hypertension. Cochrane Database Syst. Rev. 2018, 11, CD008170. [Google Scholar] [CrossRef]
- Michel, M.C.; Foster, C.; Brunner, H.R.; Liu, L. A systematic comparison of the properties of clinically used angiotensin II type 1 receptor antagonists. Pharmacol. Rev. 2013, 65, 809–848. [Google Scholar] [CrossRef]
- Erbe, D.V.; Gartrell, K.; Zhang, Y.L.; Suri, V.; Kirincich, S.J.; Will, S.; Perreault, M.; Wang, S.; Tobin, J.F. Molecular activation of PPARgamma by angiotensin II type 1-receptor antagonists. Vasc. Pharmacol. 2006, 45, 154–162. [Google Scholar] [CrossRef]
- Benson, S.C.; Pershadsingh, H.A.; Ho, C.I.; Chittiboyina, A.; Desai, P.; Pravenec, M.; Qi, N.; Wang, J.; Avery, M.A.; Kurtz, T.W. Identification of telmisartan as a unique angiotensin II receptor antagonist with selective PPARgamma-modulating activity. Hypertension 2004, 43, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Ernsberger, P.; Koletsky, R.J. Metabolic actions of angiotensin receptor antagonists: PPAR-gamma agonist actions or a class effect? Curr. Opin. Pharmacol. 2007, 7, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.J.; Cho, D.H. Activation of AMPK/proteasome/MLCK degradation signaling axis by telmisartan inhibits VSMC contractility and vessel contraction. Biochem. Biophys. Res. Commun. 2020, 524, 853–860. [Google Scholar] [CrossRef]
- Feng, X.; Luo, Z.; Ma, L.; Ma, S.; Yang, D.; Zhao, Z.; Yan, Z.; He, H.; Cao, T.; Liu, D.; et al. Angiotensin II receptor blocker telmisartan enhances running endurance of skeletal muscle through activation of the PPAR-delta/AMPK pathway. J. Cell. Mol. Med. 2011, 15, 1572–1581. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xu, Y.; Wang, Y.; Wang, Y.; He, L.; Jiang, Z.; Huang, Z.; Liao, H.; Li, J.; Saavedra, J.M.; et al. Telmisartan prevention of LPS-induced microglia activation involves M2 microglia polarization via CaMKKbeta-dependent AMPK activation. Brain Behav. Immun. 2015, 50, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, K.; Shah, J.P.; Tsytsikova, L.V.; Campbell, A.M.; Affram, K.; Symes, A.J. LPS antagonism of TGF-beta signaling results in prolonged survival and activation of rat primary microglia. J. Neurochem. 2014, 129, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Susarla, B.T.; Laing, E.D.; Yu, P.; Katagiri, Y.; Geller, H.M.; Symes, A.J. Smad proteins differentially regulate transforming growth factor-beta-mediated induction of chondroitin sulfate proteoglycans. J. Neurochem. 2011, 119, 868–878. [Google Scholar] [CrossRef]
- Li, B.H.; Liao, S.Q.; Yin, Y.W.; Long, C.Y.; Guo, L.; Cao, X.J.; Liu, Y.; Zhou, Y.; Gao, C.Y.; Zhang, L.L.; et al. Telmisartan-induced PPARgamma activity attenuates lipid accumulation in VSMCs via induction of autophagy. Mol. Biol. Rep. 2015, 42, 179–186. [Google Scholar] [CrossRef]
- Bernardo, A.; Minghetti, L. PPAR-gamma agonists as regulators of microglial activation and brain inflammation. Curr. Pharm. Des. 2006, 12, 93–109. [Google Scholar] [CrossRef]
- Trefts, E.; Shaw, R.J. AMPK: Restoring metabolic homeostasis over space and time. Mol. Cell 2021, 81, 3677–3690. [Google Scholar] [CrossRef]
- Crowley, L.C.; Christensen, M.E.; Waterhouse, N.J. Measuring Mitochondrial Transmembrane Potential by TMRE Staining. Cold Spring Harb. Protoc. 2016, 2016, pdb-prot087361. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.A. Neuronal autophagy: A housekeeper or a fighter in neuronal cell survival? Exp. Neurobiol. 2012, 21, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Moreau, K.; Puri, C.; Renna, M.; Rubinsztein, D.C. Measurement of autophagic activity in mammalian cells. Curr. Protoc. Cell Biol. 2012, 15, 15.16.1–15.16.25. [Google Scholar] [CrossRef] [PubMed]
- Kozako, T.; Soeda, S.; Yoshimitsu, M.; Arima, N.; Kuroki, A.; Hirata, S.; Tanaka, H.; Imakyure, O.; Tone, N.; Honda, S.; et al. Angiotensin II type 1 receptor blocker telmisartan induces apoptosis and autophagy in adult T-cell leukemia cells. FEBS Open Bio 2016, 6, 442–460. [Google Scholar] [CrossRef] [PubMed]
- Koyama, N.; Nishida, Y.; Ishii, T.; Yoshida, T.; Furukawa, Y.; Narahara, H. Telmisartan induces growth inhibition, DNA double-strand breaks and apoptosis in human endometrial cancer cells. PLoS ONE 2014, 9, e93050. [Google Scholar] [CrossRef] [PubMed]
- Bar-Klein, G.; Cacheaux, L.P.; Kamintsky, L.; Prager, O.; Weissberg, I.; Schoknecht, K.; Cheng, P.; Kim, S.Y.; Wood, L.; Heinemann, U.; et al. Losartan prevents acquired epilepsy via TGF-beta signaling suppression. Ann. Neurol. 2014, 75, 864–875. [Google Scholar] [CrossRef]
- Timaru-Kast, R.; Gotthardt, P.; Luh, C.; Huang, C.; Hummel, R.; Schafer, M.K.E.; Thal, S.C. Angiotensin II Receptor 1 Blockage Limits Brain Damage and Improves Functional Outcome After Brain Injury in Aged Animals Despite Age-Dependent Reduction in AT1 Expression. Front. Aging Neurosci. 2019, 11, 63. [Google Scholar] [CrossRef]
- Kurata, T.; Lukic, V.; Kozuki, M.; Wada, D.; Miyazaki, K.; Morimoto, N.; Ohta, Y.; Deguchi, K.; Yamashita, T.; Hishikawa, N.; et al. Long-term effect of telmisartan on Alzheimer’s amyloid genesis in SHR-SR after tMCAO. Transl. Stroke Res. 2015, 6, 107–115. [Google Scholar] [CrossRef]
- Torika, N.; Asraf, K.; Cohen, H.; Fleisher-Berkovich, S. Intranasal telmisartan ameliorates brain pathology in five familial Alzheimer’s disease mice. Brain Behav. Immun. 2017, 64, 80–90. [Google Scholar] [CrossRef]
- Torika, N.; Asraf, K.; Apte, R.N.; Fleisher-Berkovich, S. Candesartan ameliorates brain inflammation associated with Alzheimer’s disease. CNS Neurosci. Ther. 2018, 24, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Kehoe, P.G. The Coming of Age of the Angiotensin Hypothesis in Alzheimer’s Disease: Progress Toward Disease Prevention and Treatment? J. Alzheimers Dis. 2018, 62, 1443–1466. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, J.M. Evidence to Consider Angiotensin II Receptor Blockers for the Treatment of Early Alzheimer’s Disease. Cell Mol. Neurobiol. 2016, 36, 259–279. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Kim, S.; Jo, Y.; Kim, Y.; Ye, B.S.; Yu, Y.M. Neuroprotective effect of angiotensin II receptor blockers on the risk of incident Alzheimer’s disease: A nationwide population-based cohort study. Front. Aging Neurosci. 2023, 15, 1137197. [Google Scholar] [CrossRef] [PubMed]
- McKinley, M.J.; Albiston, A.L.; Allen, A.M.; Mathai, M.L.; May, C.N.; McAllen, R.M.; Oldfield, B.J.; Mendelsohn, F.A.; Chai, S.Y. The brain renin-angiotensin system: Location and physiological roles. Int. J. Biochem. Cell Biol. 2003, 35, 901–918. [Google Scholar] [CrossRef] [PubMed]
- Torika, N.; Asraf, K.; Danon, A.; Apte, R.N.; Fleisher-Berkovich, S. Telmisartan Modulates Glial Activation: In Vitro and In Vivo Studies. PLoS ONE 2016, 11, e0155823. [Google Scholar] [CrossRef] [PubMed]
- Elkahloun, A.G.; Rodriguez, Y.; Alaiyed, S.; Wenzel, E.; Saavedra, J.M. Telmisartan Protects a Microglia Cell Line from LPS Injury Beyond AT1 Receptor Blockade or PPARgamma Activation. Mol. Neurobiol. 2019, 56, 3193–3210. [Google Scholar] [CrossRef]
- Carson, M.J.; Crane, J.; Xie, A.X. Modeling CNS microglia: The quest to identify predictive models. Drug Discov. Today Dis. Models 2008, 5, 19–25. [Google Scholar] [CrossRef][Green Version]
- Schupp, M.; Janke, J.; Clasen, R.; Unger, T.; Kintscher, U. Angiotensin type 1 receptor blockers induce peroxisome proliferator-activated receptor-gamma activity. Circulation 2004, 109, 2054–2057. [Google Scholar] [CrossRef]
- Chen, C.M.; Wu, C.T.; Yang, T.H.; Chang, Y.A.; Sheu, M.L.; Liu, S.H. Green Tea Catechin Prevents Hypoxia/Reperfusion-Evoked Oxidative Stress-Regulated Autophagy-Activated Apoptosis and Cell Death in Microglial Cells. J. Agric. Food Chem. 2016, 64, 4078–4085. [Google Scholar] [CrossRef]
- Yin, S.N.; Liu, M.; Jing, D.Q.; Mu, Y.M.; Lu, J.M.; Pan, C.Y. Telmisartan increases lipoprotein lipase expression via peroxisome proliferator-activated receptor-alpha in HepG2 cells. Endocr. Res. 2014, 39, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.F.; Li, J.; Ma, C.; Huang, C.; Li, Z.Q. Telmisartan ameliorates Abeta oligomer-induced inflammation via PPARgamma/PTEN pathway in BV2 microglial cells. Biochem. Pharmacol. 2020, 171, 113674. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Shen, F.; Yao, J.; Su, S.; Zhao, Z. Angiotensin II receptor type 1 blocker candesartan improves morphine tolerance by reducing morphine-induced inflammatory response and cellular activation of BV2 cells via the PPARgamma/AMPK signaling pathway. Mol. Med. Rep. 2022, 26, 318. [Google Scholar] [CrossRef]
- Kapadia, R.; Yi, J.H.; Vemuganti, R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front. Biosci. 2008, 13, 1813–1826. [Google Scholar] [CrossRef]
- Woster, A.P.; Combs, C.K. Differential ability of a thiazolidinedione PPARgamma agonist to attenuate cytokine secretion in primary microglia and macrophage-like cells. J. Neurochem. 2007, 103, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Carling, D. AMPK signalling in health and disease. Curr. Opin. Cell Biol. 2017, 45, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Salminen, A.; Hyttinen, J.M.; Kaarniranta, K. AMP-activated protein kinase inhibits NF-kappaB signaling and inflammation: Impact on healthspan and lifespan. J. Mol. Med. 2011, 89, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Peixoto, C.A.; Oliveira, W.H.; Araujo, S.; Nunes, A.K.S. AMPK activation: Role in the signaling pathways of neuroinflammation and neurodegeneration. Exp. Neurol. 2017, 298, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Sag, D.; Carling, D.; Stout, R.D.; Suttles, J. Adenosine 5′-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J. Immunol. 2008, 181, 8633–8641. [Google Scholar] [CrossRef]
- Betz, C.; Hall, M.N. Where is mTOR and what is it doing there? J. Cell Biol. 2013, 203, 563–574. [Google Scholar] [CrossRef]
- Howell, J.J.; Ricoult, S.J.; Ben-Sahra, I.; Manning, B.D. A growing role for mTOR in promoting anabolic metabolism. Biochem. Soc. Trans. 2013, 41, 906–912. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Bridges, D.; Nakada, D.; Skiniotis, G.; Morrison, S.J.; Lin, J.D.; Saltiel, A.R.; Inoki, K. Inhibition of AMPK catabolic action by GSK3. Mol. Cell 2013, 50, 407–419. [Google Scholar] [CrossRef] [PubMed]
- Haidinger, M.; Poglitsch, M.; Geyeregger, R.; Kasturi, S.; Zeyda, M.; Zlabinger, G.J.; Pulendran, B.; Horl, W.H.; Saemann, M.D.; Weichhart, T. A versatile role of mammalian target of rapamycin in human dendritic cell function and differentiation. J. Immunol. 2010, 185, 3919–3931. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Sun, F.; Wang, J.; Mao, X.; Xie, L.; Yang, S.H.; Su, D.M.; Simpkins, J.W.; Greenberg, D.A.; Jin, K. mTOR signaling inhibition modulates macrophage/microglia-mediated neuroinflammation and secondary injury via regulatory T cells after focal ischemia. J. Immunol. 2014, 192, 6009–6019. [Google Scholar] [CrossRef] [PubMed]
- Tateda, S.; Kanno, H.; Ozawa, H.; Sekiguchi, A.; Yahata, K.; Yamaya, S.; Itoi, E. Rapamycin suppresses microglial activation and reduces the development of neuropathic pain after spinal cord injury. J. Orthop. Res. 2017, 35, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Yamamoto, K.; Ohishi, M.; Takeshita, H.; Hongyo, K.; Kawai, T.; Takeda, M.; Kamide, K.; Kurtz, T.W.; Rakugi, H. Telmisartan modulates mitochondrial function in vascular smooth muscle cells. Hypertens. Res. 2013, 36, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Wang, S.; Li, H.; Yuan, M.; Fan, H.; Cai, Z. Role of AMPK in autophagy. Front. Physiol. 2022, 13, 1015500. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, H.; Chu, Y.H.; Tang, Y.; Pang, X.W.; Qin, C.; Tian, D.S. Microglial autophagy in cerebrovascular diseases. Front. Aging Neurosci. 2022, 14, 1023679. [Google Scholar] [CrossRef]
- Hegdekar, N.; Sarkar, C.; Bustos, S.; Ritzel, R.M.; Hanscom, M.; Ravishankar, P.; Philkana, D.; Wu, J.; Loane, D.J.; Lipinski, M.M. Inhibition of autophagy in microglia and macrophages exacerbates innate immune responses and worsens brain injury outcomes. Autophagy 2023, 19, 2026–2044. [Google Scholar] [CrossRef]
- Choi, I.; Wang, M.; Yoo, S.; Xu, P.; Seegobin, S.P.; Li, X.; Han, X.; Wang, Q.; Peng, J.; Zhang, B.; et al. Autophagy enables microglia to engage amyloid plaques and prevents microglial senescence. Nat. Cell Biol. 2023, 25, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Bussi, C.; Ramos, J.M.; Arroyo, D.S.; Gaviglio, E.A.; Gallea, J.I.; Wang, J.M.; Celej, M.S.; Iribarren, P. Autophagy down regulates pro-inflammatory mediators in BV2 microglial cells and rescues both LPS and alpha-synuclein induced neuronal cell death. Sci. Rep. 2017, 7, 43153. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.X.; Wei, B.; Cao, H.M.; Duan, R.; Deng, Y.; Lian, H.W.; Zhang, Y.D.; Jiang, T. Telmisartan Alleviates Alzheimer’s Disease-Related Neuropathologies and Cognitive Impairments. J. Alzheimers Dis. 2023, 94, 919–933. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.K.; Moriarty, F.; Manly, J.J.; Larson, E.B.; Evans, D.A.; Rajan, K.B.; Hudak, E.M.; Hassan, L.; Liu, E.; Sato, N.; et al. Blood-Brain Barrier Crossing Renin-Angiotensin Drugs and Cognition in the Elderly: A Meta-Analysis. Hypertension 2021, 78, 629–643. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, H.; Mancia, G. The safety profile of telmisartan as monotherapy or combined with hydrochlorothiazide: A retrospective analysis of 50 studies. Blood Press. Suppl. 2008, 1, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.T.; Niu, H.S.; Chen, L.J.; Cheng, J.T.; Tong, Y.C. Increase of human prostate cancer cell (DU145) apoptosis by telmisartan through PPAR-delta pathway. Eur. J. Pharmacol. 2016, 775, 35–42. [Google Scholar] [CrossRef]
- Wang, C.; Wang, W.B. Telmisartan Induces Osteosarcoma Cells Growth Inhibition and Apoptosis Via Suppressing mTOR Pathway. Open Life Sci. 2018, 13, 242–249. [Google Scholar] [CrossRef]
- Tsujiya, Y.; Hasegawa, A.; Yamamori, M.; Okamura, N. Telmisartan-Induced Cytotoxicity via G(2)/M Phase Arrest in Renal Cell Carcinoma Cell Lines. Biol. Pharm. Bull. 2021, 44, 1878–1885. [Google Scholar] [CrossRef]
- Yang, Z.; Zhao, T.Z.; Zou, Y.J.; Zhang, J.H.; Feng, H. Hypoxia Induces autophagic cell death through hypoxia-inducible factor 1alpha in microglia. PLoS ONE 2014, 9, e96509. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).