Role for Astroglia-Derived BDNF and MSK1 in Homeostatic Synaptic Plasticity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Slice and Cell Preparation
2.2. Electrophysiological Recordings
2.3. High-Resolution Scanning of Synaptic Boutons
2.4. Multiphoton Fluorescent Ca2+-Imaging in Astrocytes
2.5. Data Analysis
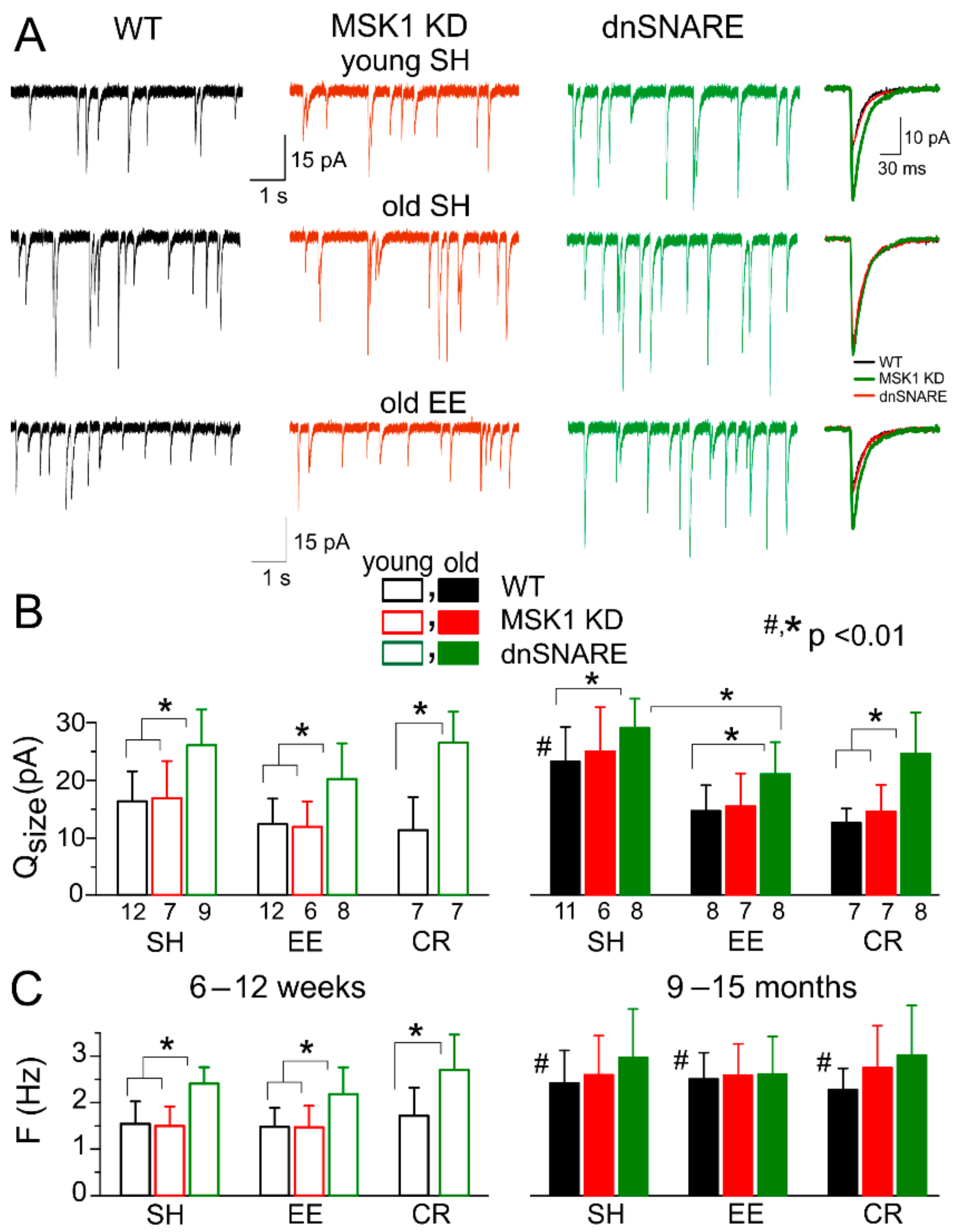
3. Results
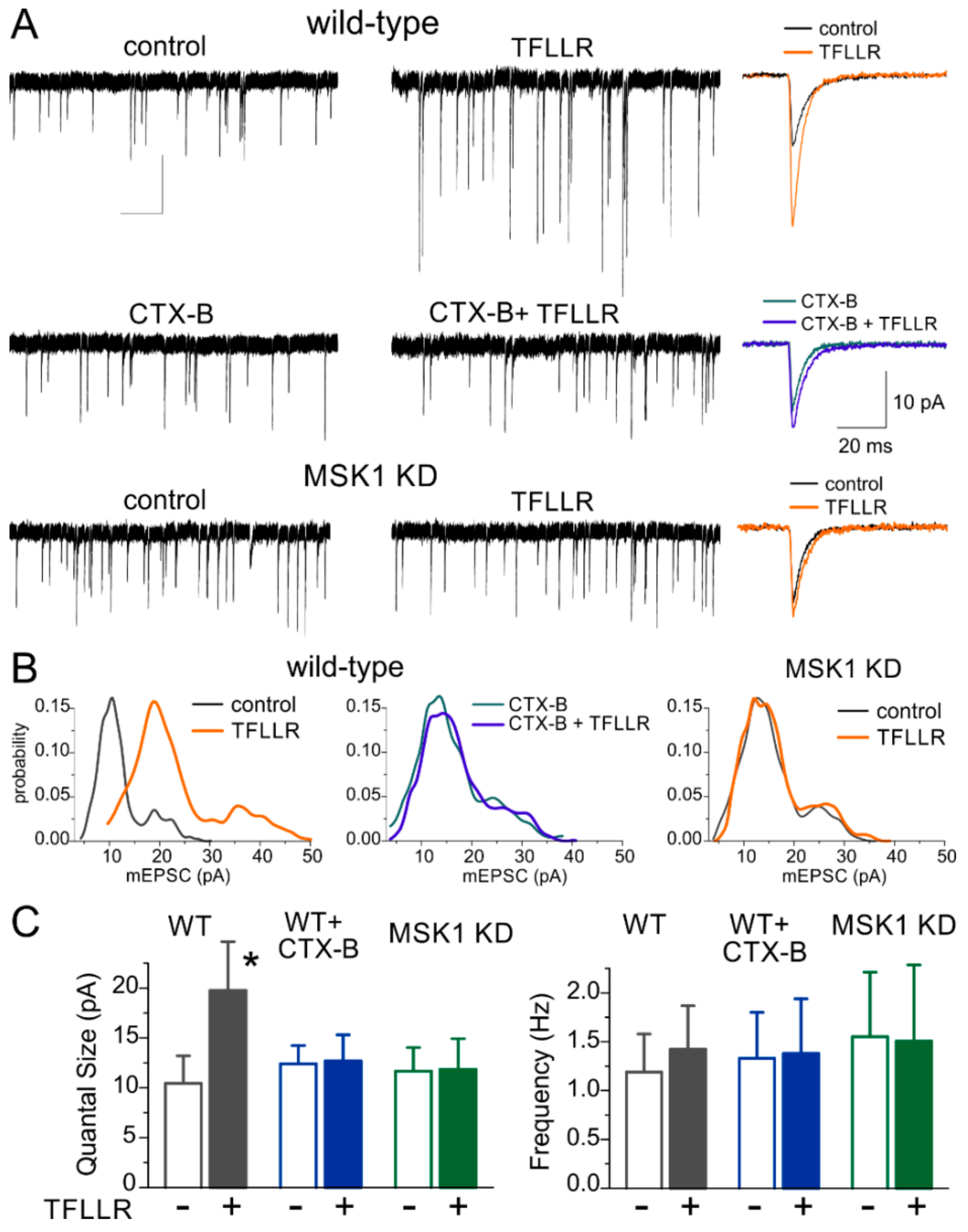
3.1. Astroglia-Induced Homeostatic Synaptic Scaling in Cultured Neurons
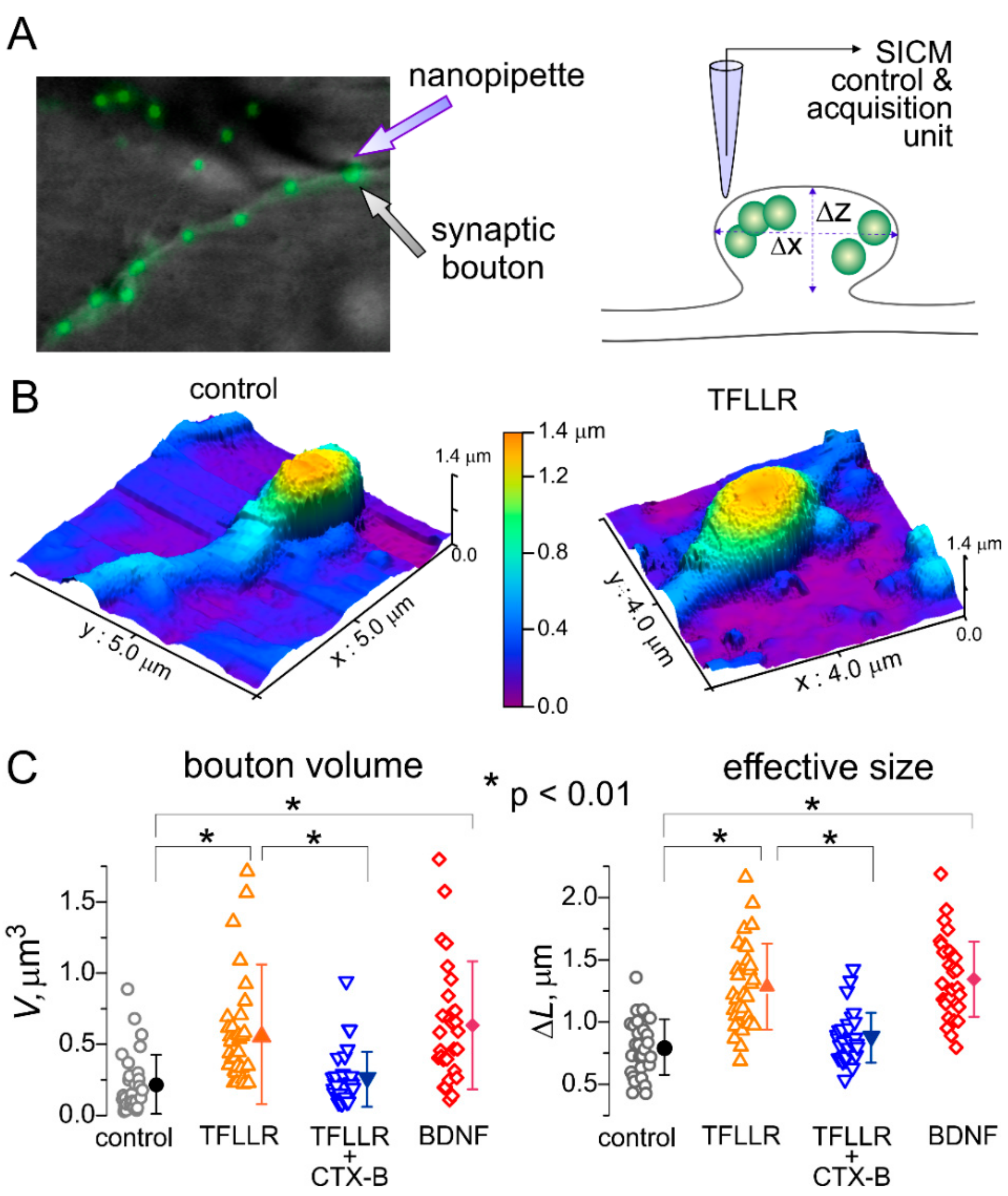
3.2. Astroglia-Induced Homeostatic Changes in Synaptic Morphology
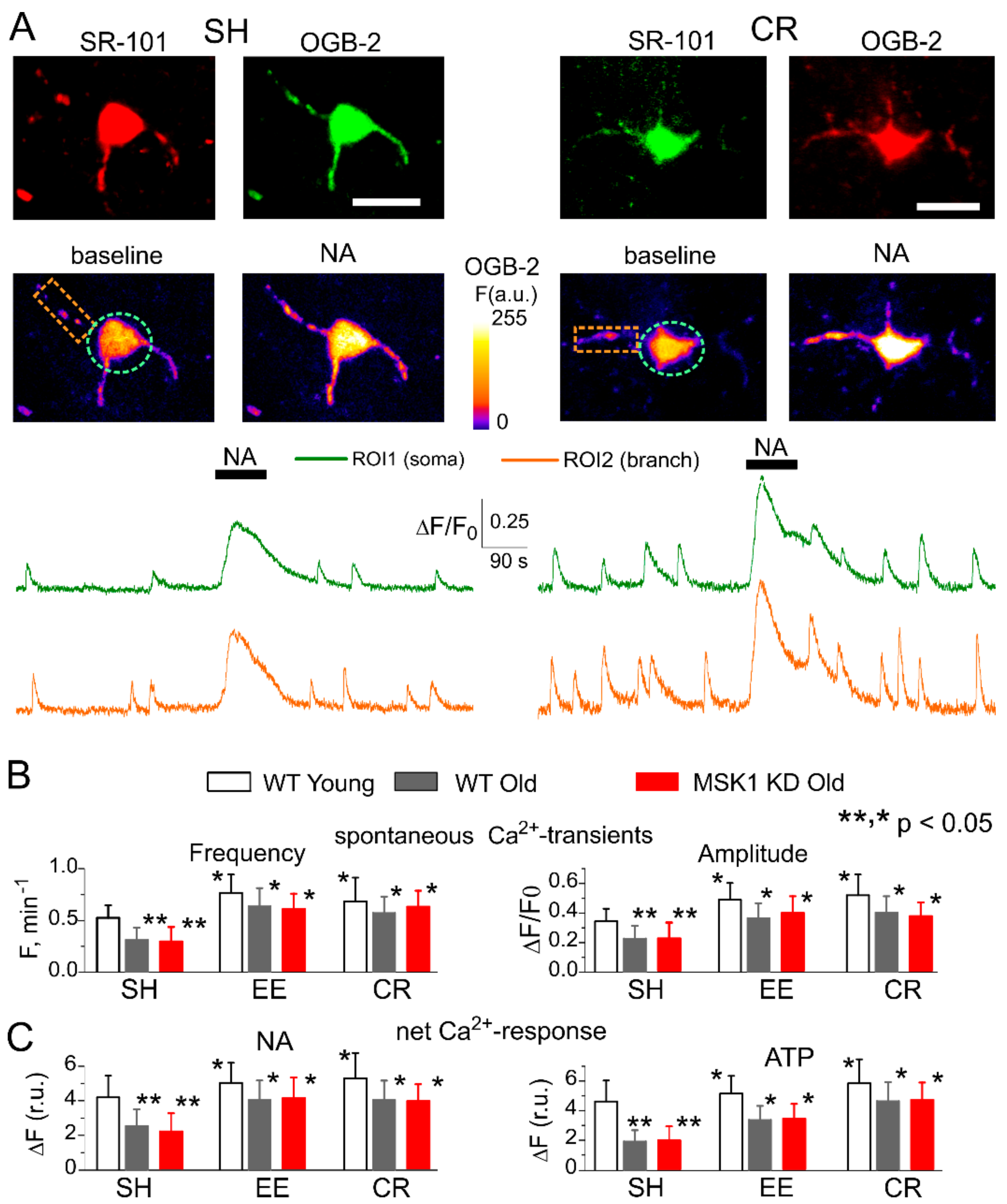
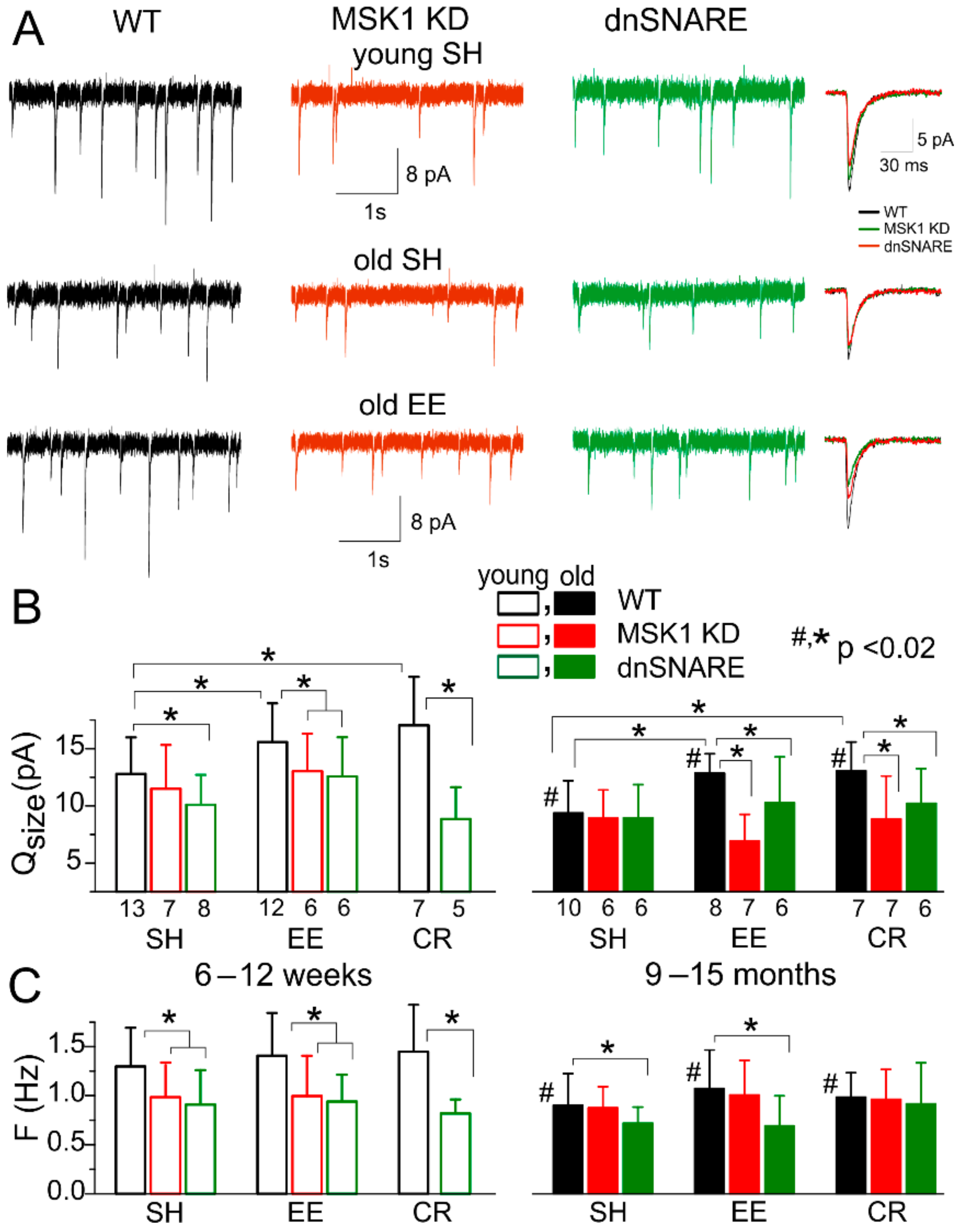
3.3. Astrocytes Participate in Homeostatic Plasticity In Vivo
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Baroncelli, L.; Braschi, C.; Spolidoro, M.; Begenisic, T.; Sale, A.; Maffei, L. Nurturing brain plasticity: Impact of environmental enrichment. Cell Death Differ. 2010, 17, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Nelson, S.B.; Turrigiano, G.G. Strength through diversity. Neuron 2008, 60, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Nithianantharajah, J.; Hannan, A.J. Enriched environments, experience-dependent plasticity and disorders of the nervous system. Nat. Rev. Neurosci. 2006, 7, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Hillman, C.H.; Erickson, K.I.; Kramer, A.F. Be smart, exercise your heart: Exercise effects on brain and cognition. Nat. Rev. Neurosci. 2008, 9, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Mercken, E.M.; Carboneau, B.A.; Krzysik-Walker, S.M.; de Cabo, R. Of mice and men: The benefits of caloric restriction, exercise, and mimetics. Ageing Res. Rev. 2012, 11, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Merzenich, M.M.; Van Vleet, T.M.; Nahum, M. Brain plasticity-based therapeutics. Front. Hum. Neurosci. 2014, 8, 385. [Google Scholar] [CrossRef] [PubMed]
- Nithianantharajah, J.; Hannan, A.J. The neurobiology of brain and cognitive reserve: Mental and physical activity as modulators of brain disorders. Prog. Neurobiol. 2009, 89, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Van Praag, H. Exercise and the brain: Something to chew on. Trends Neurosci. 2009, 32, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Correa, S.A.; Hunter, C.J.; Palygin, O.; Wauters, S.C.; Martin, K.J.; McKenzie, C.; McKelvey, K.; Morris, R.G.; Pankratov, Y.; Arthur, J.S.; et al. MSK1 regulates homeostatic and experience-dependent synaptic plasticity. J. Neurosci. 2012, 32, 13039–13051. [Google Scholar] [CrossRef] [PubMed]
- Turrigiano, G.G.; Leslie, K.R.; Desai, N.S.; Rutherford, L.C.; Nelson, S.B. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 1998, 391, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Cowansage, K.K.; LeDoux, J.E.; Monfils, M.H. Brain-derived neurotrophic factor: A dynamic gatekeeper of neural plasticity. Curr. Mol. Pharmacol. 2010, 3, 12–29. [Google Scholar] [CrossRef] [PubMed]
- Rothman, S.M.; Griffioen, K.J.; Wan, R.; Mattson, M.P. Brain-derived neurotrophic factor as a regulator of systemic and brain energy metabolism and cardiovascular health. Ann. N. Y. Acad. Sci. 2012, 1264, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Yu, J.T.; Tan, L. Brain-Derived Neurotrophic Factor in Alzheimer’s Disease: Risk, Mechanisms, and Therapy. Mol. Neurobiol. 2015, 52, 1477–1493. [Google Scholar] [CrossRef] [PubMed]
- Stenovec, M.; Lasic, E.; Bozic, M.; Bobnar, S.T.; Stout, R.F., Jr.; Grubisic, V.; Parpura, V.; Zorec, R. Ketamine Inhibits ATP-Evoked Exocytotic Release of Brain-Derived Neurotrophic Factor from Vesicles in Cultured Rat Astrocytes. Mol. Neurobiol. 2016, 53, 6882–6896. [Google Scholar] [CrossRef] [PubMed]
- Vignoli, B.; Battistini, G.; Melani, R.; Blum, R.; Santi, S.; Berardi, N.; Canossa, M. Peri-Synaptic Glia Recycles Brain-Derived Neurotrophic Factor for LTP Stabilization and Memory Retention. Neuron 2016, 92, 873–887. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Arellano, J.J.; Parpura, V.; Zorec, R.; Verkhratsky, A. Astrocytes in physiological aging and Alzheimer’s disease. Neuroscience 2015. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Nedergaard, M. Astroglial cradle in the life of the synapse. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.J.; Terzieva, S.; Olabarria, M.; Lanza, R.G.; Verkhratsky, A. Enriched environment and physical activity reverse astrogliodegeneration in the hippocampus of AD transgenic mice. Cell Death Dis. 2013, 4, e678. [Google Scholar] [CrossRef]
- Lalo, U.; Bogdanov, A.; Pankratov, Y. Diversity of Astroglial Effects on Aging- and Experience-Related Cortical Metaplasticity. Front. Mol. Neurosci. 2018, 11, 239. [Google Scholar] [CrossRef] [PubMed]
- Lalo, U.; Palygin, O.; Rasooli-Nejad, S.; Andrew, J.; Haydon, P.G.; Pankratov, Y. Exocytosis of ATP from astrocytes modulates phasic and tonic inhibition in the neocortex. PLoS Biol. 2014, 12, e1001747. [Google Scholar] [CrossRef] [PubMed]
- Lalo, U.; Palygin, O.; Verkhratsky, A.; Grant, S.G.; Pankratov, Y. ATP from synaptic terminals and astrocytes regulates NMDA receptors and synaptic plasticity through PSD-95 multi-protein complex. Sci. Rep. 2016, 6, 33609. [Google Scholar] [CrossRef] [PubMed]
- Pankratov, Y.; Lalo, U. Role for astroglial alpha1-adrenoreceptors in gliotransmission and control of synaptic plasticity in the neocortex. Front. Cell. Neurosci. 2015, 9, 230. [Google Scholar] [CrossRef] [PubMed]
- Rasooli-Nejad, S.; Palygin, O.; Lalo, U.; Pankratov, Y. Cannabinoid receptors contribute to astroglial Ca2+-signalling and control of synaptic plasticity in the neocortex. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20140077. [Google Scholar] [CrossRef] [PubMed]
- Lalo, U.; Pankratov, Y. Exploring the Ca2+-dependent synaptic dynamics in vibro-dissociated cells. Cell Calcium 2017, 64, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Novak, P.; Li, C.; Shevchuk, A.I.; Stepanyan, R.; Caldwell, M.; Hughes, S.; Smart, T.G.; Gorelik, J.; Ostanin, V.P.; Lab, M.J.; et al. Nanoscale live-cell imaging using hopping probe ion conductance microscopy. Nat. Methods 2009, 6, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Sikora, A.; Rodak, A.; Unold, O.; Klapetek, P. The development of the spatially correlated adjustment wavelet filter for atomic force microscopy data. Ultramicroscopy 2016, 171, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Zhong, P.; Liu, Y.; Hu, Y.; Wang, T.; Zhao, Y.P.; Liu, Q.S. BDNF interacts with endocannabinoids to regulate cocaine-induced synaptic plasticity in mouse midbrain dopamine neurons. J. Neurosci. 2015, 35, 4469–4481. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, E.; Kanhema, T.; Soule, J.; Tiron, A.; Dagyte, G.; da Silva, B.; Bramham, C.R. Sustained Arc/Arg3.1 synthesis controls long-term potentiation consolidation through regulation of local actin polymerization in the dentate gyrus in vivo. J. Neurosci. 2007, 27, 10445–10455. [Google Scholar] [CrossRef] [PubMed]
- Waung, M.W.; Pfeiffer, B.E.; Nosyreva, E.D.; Ronesi, J.A.; Huber, K.M. Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron 2008, 59, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Korchev, Y.E.; Negulyaev, Y.A.; Edwards, C.R.; Vodyanoy, I.; Lab, M.J. Functional localization of single active ion channels on the surface of a living cell. Nat. Cell Biol. 2000, 2, 616–619. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Galluzzi, L.; Freije, J.M.; Madeo, F.; Kroemer, G. Metabolic Control of Longevity. Cell 2016, 166, 802–821. [Google Scholar] [CrossRef] [PubMed]
- Paukert, M.; Agarwal, A.; Cha, J.; Doze, V.A.; Kang, J.U.; Bergles, D.E. Norepinephrine controls astroglial responsiveness to local circuit activity. Neuron 2014, 82, 1263–1270. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, R.; Jahn, H.M.; Courjaret, R.; Messemer, N.; Kirchhoff, F.; Deitmer, J.W. The inhibitory input to mouse cerebellar Purkinje cells is reciprocally modulated by Bergmann glial P2Y1 and AMPA receptor signaling. Glia 2016, 64, 1265–1280. [Google Scholar] [CrossRef] [PubMed]
- Lalo, U.; Palygin, O.; North, R.A.; Verkhratsky, A.; Pankratov, Y. Age-dependent remodelling of ionotropic signalling in cortical astroglia. Aging Cell 2011, 10, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Lalo, U.; Rasooli-Nejad, S.; Pankratov, Y. Exocytosis of gliotransmitters from cortical astrocytes: Implications for synaptic plasticity and aging. Biochem. Soc. Trans. 2014, 42, 1275–1281. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lalo, U.; Bogdanov, A.; Moss, G.W.J.; Frenguelli, B.G.; Pankratov, Y. Role for Astroglia-Derived BDNF and MSK1 in Homeostatic Synaptic Plasticity. Neuroglia 2018, 1, 381-394. https://doi.org/10.3390/neuroglia1020026
Lalo U, Bogdanov A, Moss GWJ, Frenguelli BG, Pankratov Y. Role for Astroglia-Derived BDNF and MSK1 in Homeostatic Synaptic Plasticity. Neuroglia. 2018; 1(2):381-394. https://doi.org/10.3390/neuroglia1020026
Chicago/Turabian StyleLalo, Ulyana, Alexander Bogdanov, Guy W. J. Moss, Bruno G. Frenguelli, and Yuriy Pankratov. 2018. "Role for Astroglia-Derived BDNF and MSK1 in Homeostatic Synaptic Plasticity" Neuroglia 1, no. 2: 381-394. https://doi.org/10.3390/neuroglia1020026
APA StyleLalo, U., Bogdanov, A., Moss, G. W. J., Frenguelli, B. G., & Pankratov, Y. (2018). Role for Astroglia-Derived BDNF and MSK1 in Homeostatic Synaptic Plasticity. Neuroglia, 1(2), 381-394. https://doi.org/10.3390/neuroglia1020026