Development and Assessment of a Diagnostic DNA Oligonucleotide Microarray for Detection and Typing of Meningitis-Associated Bacterial Species


Abstract
:
1. Introduction
2. Materials and Methods
2.1. Culture of Bacterial Pathogen Strains and Nucleic Acid Purification
2.2. Collection of Human Donor Blood and Patient Cerebrospinal Fluid Samples and Purification of Nucleic Acids
2.3. Design of Oligonucleotide Microarray Probes
2.4. Printing of Microarray Oligonucleotide Probes in Glass Slide Format
2.5. Amplification and Cy3-Labelling of Purified Nucleic Acids and Hybridization to the Meningitis Glass Slide Array
2.5.1. Hybridization to Pan-Pathogen Arrays Using Randomly-Amplified Cy3 Labelled Targets and the Manual Hybridization Method
2.5.2. Hybridization to Pan-Pathogen Arrays Using Randomly-Amplified Cy3 Labelled Targets Using the Advalytix Slidebooster™ Hybridization Station.
2.6. Data Processing and Analysis
2.7. Reconfiguration of the Pan-Pathogen Array in Alere ArrayTube™ Format
2.8. Development of a Targeted DNA Amplification and Labelling System Using VisualOMP Primer Design Software
2.9. Amplification and Labelling of Target DNAs and ArrayTube Hybridizations
2.10. Real-Time Qualitative PCR Assays
3. Results
3.1. Hybridization of Randomly Amplified DNA Targets to the Meningitis Pan-Pathogen Glass Slide Printed Array
3.1.1. Hybridization Profiling of Nonmeningococcal Bacterial Pathogens
3.1.2. Hybridization Profiling of Meningococcal Bacterial Pathogens of Known Serogroup
3.1.3. Hybridization Profiling of Meningococcal Bacterial Pathogens of Unknown Serogroup
3.1.4. Hybridization Profiling of Nonmeningococcal Neisseria spp.
3.2. Purification of DNAs from Patient CSF, Labelling and Array Hybridizations
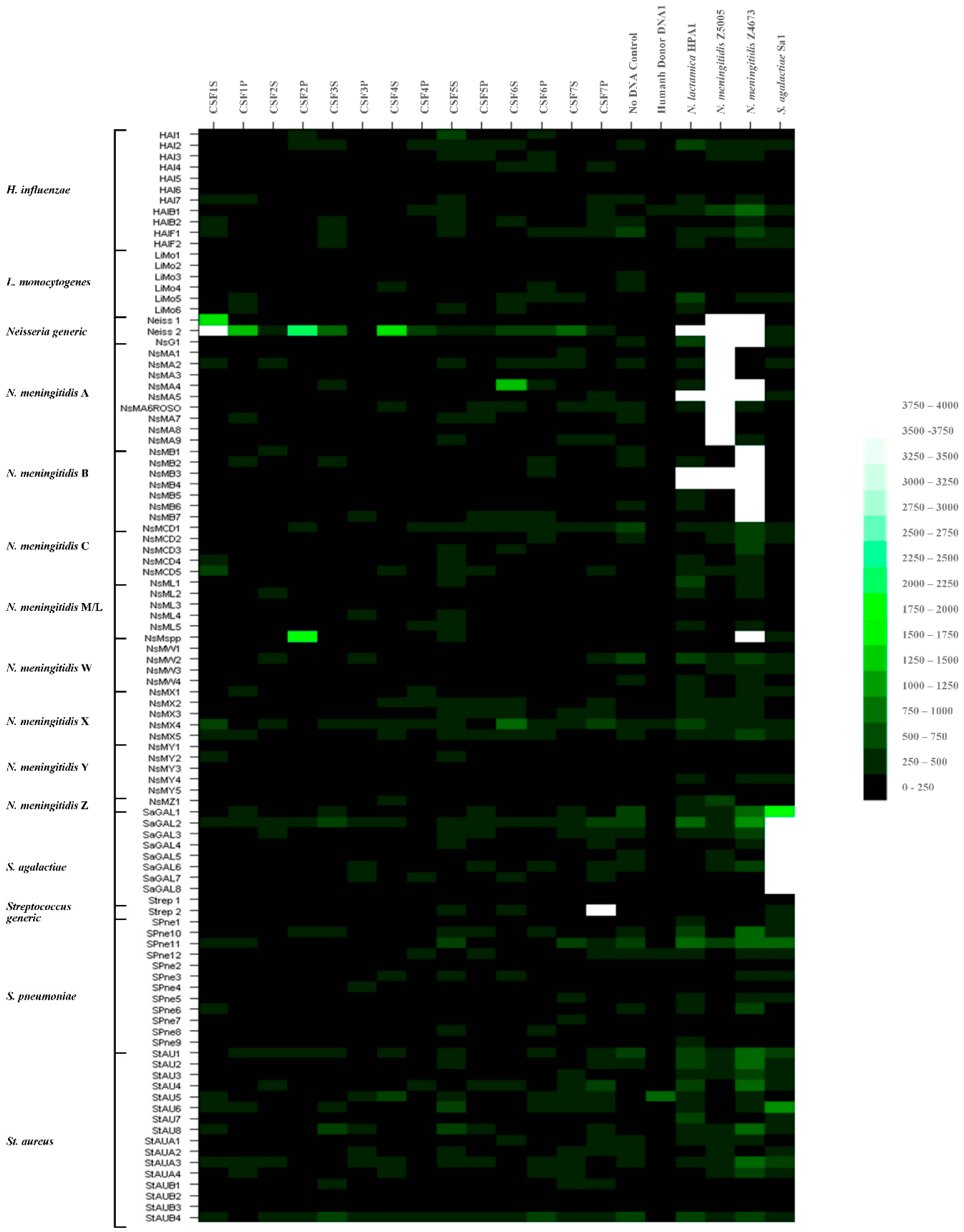
3.2.1. Hybridization Profiling of Patient CSF Target DNAs and Putative Pathogen Identification to the Meningitis Array in Glass Slide Format
3.2.2. Further Development of Methods for Pathogen Identification in Patient CSF Samples to the Meningitis Array in Glass Slide Format
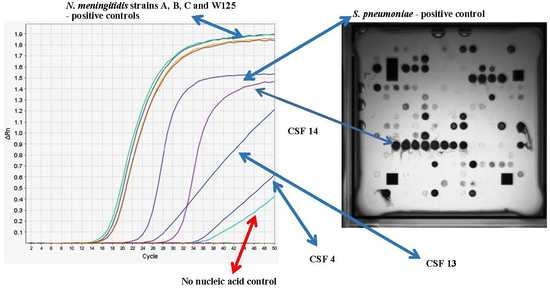
3.3. Determination of the Infectious Pathogen in Patient CSF Samples Using Multiplex RT-PCR
3.4. Redevelopment of the Meningitis Array on the Alere ArrayTubeTM Platform
3.4.1. Singleplex and Multiplex Target Amplification Using Purified Pathogen Nucleic Acids and Hybridization to the Meningitis Array in ArrayTubeTM Format
3.4.2. Multiplex Target Amplification Using Purified Pathogen Nucleic Acids and Hybridization to the Meningitis Array in ArrayTube™ Format Using Patient CSF Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Davison, K.L.; Ramsay, M.E. The epidemiology of acute meningitis in children in England and Wales. Arch. Dis. Child. 2003, 88, 662–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitch, M.T.; van de Beek, D. Emergency diagnosis and treatment of adult meningitis. Lancet Infect. Dis. 2007, 7, 191–200. [Google Scholar] [CrossRef]
- Mount, H.R.; Boyle, S.D. Aseptic and Bacterial Meningitis: Evaluation, Treatment, and Prevention. Am. Fam. Physician 2017, 96, 314–322. [Google Scholar] [PubMed]
- Lee, B.E.; Davies, H.D. Aseptic meningitis. Curr. Opin. Infect. Dis. 2007, 20, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Shukla, B.; Aguilera, E.A.; Salazar, L.; Wootton, S.H.; Kaewpoowat, Q.; Hasbun, R. Aseptic meningitis in adults and children: Diagnostic and management challenges. J. Clin. Virol. 2017, 94, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Siafakas, N.; Markoulatos, P.; Levidiotou-Stefanou, S. Molecular identification of enteroviruses responsible for an outbreak of aseptic meningitis; implications in clinical practice and epidemiology. Mol. Cell. Probes 2004, 18, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Overturf, G.D. Defining bacterial meningitis and other infections of the central nervous system. Pediatr. Crit. Care Med. 2005, 6 (Suppl. 3), S14–S18. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, C.; Nassif, X. Meningococcal pathogenesis: At the boundary between the pre- and post-genomic eras. Curr. Opin. Microbiol. 2001, 4, 47–52. [Google Scholar] [CrossRef]
- Bodilsen, J.; Brandt, C.T.; Sharew, A.; Dalager-Pedersen, M.; Benfield, T.; Schønheyder, H.C.; Nielsen, H. Early versus late diagnosis in community-acquired bacterial meningitis: A retrospective cohort study. Clin. Microbiol. Infect. 2017, 24, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, Y.L.; Stephens, D.S. Epidemiology and pathogenesis of Neisseria meningitidis. Microbes Infect. 2000, 2, 687–700. [Google Scholar] [CrossRef]
- Enright, M.C.; Knox, K.; Griffiths, D.; Crook, D.W.; Spratt, B.G. Molecular typing of bacteria directly from cerebrospinal fluid. Eur. J. Clin. Microbiol. Infect. Dis. 2000, 19, 627–630. [Google Scholar] [CrossRef] [PubMed]
- Caws, M.; Wilson, S.M.; Clough, C.; Drobniewski, F. Role of IS6110-targeted PCR, culture, biochemical, clinical, and immunological criteria for diagnosis of tuberculous meningitis. J. Clin. Microbiol. 2000, 38, 3150–3155. [Google Scholar] [PubMed]
- Theodoridou, M.N.; Vasilopoulou, V.A.; Atsali, E.E.; Pangalis, A.M.; Mostrou, G.J.; Syriopoulou, V.P.; Hadjichristodoulou, C.S. Meningitis registry of hospitalized cases in children: Epidemiological patterns of acute bacterial meningitis throughout a 32-year period. BMC Infect. Dis. 2007, 7, 101. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, M.C.; Tunkel, A.R.; van de Beek, D. Epidemiology, diagnosis, and antimicrobial treatment of acute bacterial meningitis. Clin. Microbiol. Rev. 2010, 23, 467–492. [Google Scholar] [CrossRef] [PubMed]
- van Deuren, M.; Brandtzaeg, P.; van der Meer, J.W. Update on meningococcal disease with emphasis on pathogenesis and clinical management. Clin. Microbiol. Rev. 2000, 13, 144–166. [Google Scholar] [CrossRef] [PubMed]
- van Deuren, M.; van Dijke, B.J.; Koopman, R.J.; Horrevorts, A.M.; Meis, J.F.; Santman, F.W.; van der Meer, J.W. Rapid diagnosis of acute meningococcal infections by needle aspiration or biopsy of skin lesions. Br. Med. J. 1993, 306, 1229–1232. [Google Scholar] [CrossRef]
- Hampshire Hospitals NHS Foundation Trust. Empirical Treatment of Suspected Bacterial Meningitis; Hampshire Hospitals NHS Foundation Trust: Hampshire, UK, 2018; Available online: www.hampshirehospitals.nhs.uk/uploaded_files/GP%20section/microbiology_guidelines/central_nervous_system_bnhft_2010.pdf (accessed on 8 October 2018).
- Cunha, B.A. The clinical and laboratory diagnosis of acute meningitis and acute encephalitis. Expert Opin. Med. Diagn. 2013, 7, 343–364. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, K.; Reilly, S.; White, D.; Stuart, J. Early treatment with parenteral penicillin in meningococcal disease. Br. Med. J. 1992, 305, 143–147. [Google Scholar] [CrossRef]
- Cartwright, K.; Strang, J.; Gossain, S.; Begg, N. Early treatment of meningococcal disease. Br. Med. J. 1992, 305, 774. [Google Scholar] [CrossRef]
- Corless, C.E.; Guiver, M.; Borrow, R.; Edwards-Jones, V.; Fox, A.J.; Kaczmarski, E.B. Simultaneous detection of Neisseria meningitidis, Haemophilus influenzae, and Streptococcus pneumoniae in suspected cases of meningitis and septicemia using real-time PCR. J. Clin. Microbiol. 2001, 39, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Guiver, M.; Borrow, R.; Marsh, J.; Gray, S.J.; Kaczmarski, E.B.; Howells, D.; Boseley, P.; Fox, A.J. Evaluation of the Applied Biosystems automated Taqman polymerase chain reaction system for the detection of meningococcal DNA. FEMS Immunol. Med. Microbiol. 2000, 28, 173–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corless, C.E.; Guiver, M.; Borrow, R.; Edwards-Jones, V.; Fox, A.J.; Kaczmarski, E.B.; Mutton, K.J. Development and evaluation of a real-time RT-PCR for the detection of enterovirus and parechovirus RNA in CSF and throat swab samples. J. Med. Virol. 2002, 67, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Dumaidi, K.; Al-Jawabreh, A. Molecular detection and genotyping of enteroviruses from CSF samples of patients with suspected sepsis-like illness and/or aseptic meningitis from 2012 to 2015 in West Bank, Palestine. PLoS ONE 2017, 12, e0172357. [Google Scholar] [CrossRef] [PubMed]
- Guney, C.; Ozkaya, E.; Yapar, M.; Gumus, I.; Kubar, A.; Doganci, L. Laboratory diagnosis of enteroviral infections of the central nervous system by using a nested RT-polymerase chain reaction (PCR) assay. Diagn. Microbiol. Infect. Dis. 2003, 47, 557–562. [Google Scholar] [CrossRef]
- McGill, F.; Griffiths, M.J.; Solomon, T. Viral meningitis: Current issues in diagnosis and treatment. Curr. Opin. Infect. Dis. 2017, 30, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Borrow, R.; Claus, H.; Chaudhry, U.; Guiver, M.; Kaczmarski, E.B.; Frosch, M.; Fox, A.J. siaD PCR ELISA for confirmation and identification of serogroup Y and W135 meningococcal infections. FEMS Microbiol. Lett. 1998, 159, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Borrow, R.; Claus, H.; Guiver, M.; Smart, L.; Jones, D.M.; Kaczmarski, E.B.; Frosch, M.; Fox, A.J. Non-culture diagnosis and serogroup determination of meningococcal B and C infection by a sialyltransferase (siaD) PCR ELISA. Epidemiol. Infect. 1997, 118, 111–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, C.; Clarke, S.C. Identification of Neisseria meningitidis serogroups Y and W135 by siaD nucleotide sequence analysis. J. Clin. Microbiol. 2003, 41, 2697–2699. [Google Scholar] [CrossRef] [PubMed]
- Sadler, F.; Fox, A.; Neal, K.; Dawson, M.; Cartwright, K.; Borrow, R. Genetic analysis of capsular status of meningococcal carrier isolates. Epidemiol. Infect. 2003, 130, 59–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohkusu, K.; Nash, K.A.; Inderlied, C.B. Molecular characterisation of Haemophilus influenzae type a and untypeable strains isolated simultaneously from cerebrospinal fluid and blood: Novel use of quantitative real-time PCR based on the cap copy number to determine virulence. Clin. Microbiol. Infect. 2005, 11, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Jolley, K.A.; Brehony, C.; Maiden, M.C. Molecular typing of meningococci: Recommendations for target choice and nomenclature. FEMS Microbiol. Rev. 2007, 31, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Vogel, U. European efforts to harmonize typing of meningococci. Int. J. Med. Microbiol. 2011, 301, 659–662. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.H.; Saeed, A.I. Microarrays: An overview. Methods Mol. Biol. 2007, 353, 265–300. [Google Scholar] [PubMed]
- Ye, R.W.; Wang, T.; Bedzyk, L.; Croker, KM. Applications of DNA microarrays in microbial systems. J. Microbiol. Methods 2001, 47, 257–272. [Google Scholar] [CrossRef]
- Zammatteo, N.; Hamels, S.; De Longueville, F.; Alexandre, I.; Gala, J.L.; Brasseur, F.; Remacle, J. New chips for molecular biology and diagnostics. Biotechnol. Annu. Rev. 2002, 8, 85–101. [Google Scholar] [PubMed]
- Berthet, N.; Dickinson, P.; Filliol, I.; Reinhardt, A.K.; Batejat, C.; Vallaeys, T.; Kong, K.A.; Davies, C.; Lee, W.; Zhang, S.; et al. Massively parallel pathogen identification using high-density microarrays. Microb. Biotechnol. 2008, 1, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Hanson, E.H.; Niemeyer, D.M.; Folio, L.; Agan, B.K.; Rowley, R.K. Potential use of microarray technology for rapid identification of central nervous system pathogens. Mil. Med. 2004, 169, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Bodrossy, L.; Sessitsch, A. Oligonucleotide microarrays in microbial diagnostics. Curr. Opin. Microbiol. 2004, 7, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Loy, A.; Bodrossy, L. Highly parallel microbial diagnostics using oligonucleotide microarrays. Clin. Chim. Acta 2006, 363, 106–119. [Google Scholar] [CrossRef] [PubMed]
- Palacios, G.; Quan, P.L.; Jabado, O.J.; Conlan, S.; Hirschberg, D.L.; Liu, Y.; Zhai, J.; Renwick, N.; Hui, J.; Hegyi, H.; et al. Panmicrobial oligonucleotide array for diagnosis of infectious diseases. Emerg. Infect. Dis. 2007, 13, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Schrenzel, J. Clinical relevance of new diagnostic methods for bloodstream infections. Int. J. Antimicrob. Agents 2007, 30 (Suppl. 1), S2–S6. [Google Scholar] [CrossRef] [PubMed]
- Chiang, Y.C.; Yang, C.Y.; Li, C.; Ho, Y.C.; Lin, C.K.; Tsen, H.Y. Identification of Bacillus spp.; Escherichia coli, Salmonella spp.; Staphylococcus spp. and Vibrio spp. with 16S ribosomal DNA-based oligonucleotide array hybridization. Int. J. Food Microbiol. 2006, 107, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Maynard, C.; Berthiaume, F.; Lemarchand, K.; Harel, J.; Payment, P.; Bayardelle, P.; Masson, L.; Brousseau, R. Waterborne pathogen detection by use of oligonucleotide-based microarrays. Appl. Environ. Microbiol. 2005, 71, 8548–8557. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.W.; Zhang, L.; Jin, L.Q.; Jin, M.; Shen, Z.Q.; An, S.; Chao, F.H.; Li, J.W. Development and application of an oligonucleotide microarray for the detection of food-borne bacterial pathogens. Appl. Microbiol. Biotechnol. 2007, 76, 225–233. [Google Scholar] [CrossRef] [PubMed]
- Dawson, E.D.; Moore, C.L.; Dankbar, D.M.; Mehlmann, M.; Townsend, M.B.; Smagala, J.A.; Smith, C.B.; Cox, N.J.; Kuchta, R.D.; Rowlen, K.L. Identification of A/H5N1 influenza viruses using a single gene diagnostic microarray. Anal. Chem. 2007, 79, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Hsia, C.C.; Chizhikov, V.E.; Yang, A.X.; Selvapandiyan, A.; Hewlett, I.; Duncan, R.; Puri, R.K.; Nakhasi, H.L.; Kaplan, G.G. Microarray multiplex assay for the simultaneous detection and discrimination of hepatitis B, hepatitis C, and human immunodeficiency type-1 viruses in human blood samples. Biochem. Biophys. Res. Commun. 2007, 356, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Neverov, A.A.; Riddell, M.A.; Moss, W.J.; Volokhov, D.V.; Rota, P.A.; Lowe, L.E.; Chibo, D.; Smit, S.B.; Griffin, D.E.; Chumakov, K.M.; et al. Genotyping of measles virus in clinical specimens on the basis of oligonucleotide microarray hybridization patterns. J. Clin. Microbiol. 2006, 44, 3752–3759. [Google Scholar] [CrossRef] [PubMed]
- Nordstrom, H.; Johansson, P.; Li, Q.G.; Lundkvist, A.; Nilsson, P.; Elgh, F. Microarray technology for identification and distinction of hantaviruses. J. Med. Virol. 2004, 72, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Cleven, B.E.; Palka-Santini, M.; Gielen, J.; Meembor, S.; Krönke, M.; Krut, O. Identification and characterization of bacterial pathogens causing bloodstream infections by DNA microarray. J. Clin. Microbiol. 2006, 44, 2389–2397. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.Z.; Wen, S.Y.; Chen, S.H.; Lin, F.; Wang, S.Q. Detection and identification of intestinal pathogens in clinical specimens using DNA microarrays. Mol. Cell. Probes 2006, 20, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.Z.; Xu, X.J.; Chen, S.H.; Wen, S.Y.; Ma, X.E.; Zhang, Z.; Lin, F.; Wang, S.Q. Detection and identification of enterohemorrhagic Escherichia coli O157:H7 and Vibrio cholerae O139 using oligonucleotide microarray. Infect. Agent Cancer 2007, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- Wiesinger-Mayr, H.; Vierlinger, K.; Pichler, R.; Kriegner, A.; Hirschl, A.M.; Presterl, E.; Bodrossy, L.; Noehammer, C. Identification of human pathogens isolated from blood using microarray hybridisation and signal pattern recognition. BMC Microbiol. 2007, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Li, J.W.; Shen, Z.Q.; Wang, X.W.; Jin, M. High-throughput identification of clinical pathogenic fungi by hybridization to an oligonucleotide microarray. J. Clin. Microbiol. 2006, 44, 3299–3305. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Orlandi, P.A.; Stenger, D.A. Simultaneous detection of four human pathogenic microsporidian species from clinical samples by oligonucleotide microarray. J. Clin. Microbiol. 2005, 43, 4121–4128. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Vora, G.J.; Stenger, D.A. Detection and genotyping of Entamoeba histolytica, Entamoeba dispar, Giardia lamblia, and Cryptosporidium parvum by Oligonucleotide Microarray. J. Clin. Microbiol. 2004, 42, 3262–3271. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Y.; Alizadeh, A.A.; Rouskin, S.; Merker, J.D.; Yeh, E.; Yagi, S.; Schnurr, D.; Patterson, B.K.; Ganem, D.; DeRisi, J.L. Diagnosis of a critical respiratory illness caused by human metapneumovirus by use of a pan-virus microarray. J. Clin. Microbiol. 2007, 45, 2340–2343. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Y.; Rouskin, S.; Koshy, A.; Urisman, A.; Fischer, K.; Yagi, S.; Schnurr, D.; Eckburg, P.B.; Tompkins, L.S.; Blackburn, B.G.; et al. Microarray detection of human parainfluenzavirus 4 infection associated with respiratory failure in an immunocompetent adult. Clin. Infect. Dis. 2006, 43, e71–e76. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.Y.; Urisman, A.; Greenhow, T.L.; Rouskin, S.; Yagi, S.; Schnurr, D.; Wright, C.; Drew, W.L.; Wang, D.; Weintrub, P.S.; et al. Utility of DNA microarrays for detection of viruses in acute respiratory tract infections in children. J. Pediatr. 2008, 153, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Urisman, A.; Fischer, K.F.; Chiu, C.Y.; Kistler, A.L.; Beck, S.; Wang, D.; DeRisi, J.L. E-Predict: A computational strategy for species identification based on observed DNA microarray hybridization patterns. Genome Biol. 2005, 6, R78. [Google Scholar] [CrossRef] [PubMed]
- Corless, C.E.; Kaczmarski, E.; Borrow, R.; Guiver, M. Molecular characterization of Neisseria meningitidis isolates using a resequencing DNA microarray. J. Mol. Diagn. 2008, 10, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Pannucci, J.; Cai, H.; Pardington, P.E.; Williams, E.; Okinaka, R.T.; Kuske, C.R.; Cary, R.B. Virulence signatures: Microarray-based approaches to discovery and analysis. Biosens. Bioelectron. 2004, 20, 706–718. [Google Scholar] [CrossRef] [PubMed]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Peñaranda, S.; Bankamp, B.; Maher, K.; Chen, M.H.; et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003, 300, 1394–1399. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.M.; Yang, R.Q.; Tao, S.C.; Li, Z.; Zhang, Q.; Gao, H.F.; Zhang, Z.W.; Du, J.Y.; Zhu, P.X.; Ren, L.L.; et al. The design and application of DNA chips for early detection of SARS-CoV from clinical samples. J. Clin. Virol. 2005, 33, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Marks, D. (Ed.) Guidance on the Use of Clinical Samples for a Range of Purposes That Are Not within the Remit of Research Ethics Committees (RECs), 3rd ed.; Royal College of Pathologists: London, UK, 2015; Available online: https://www.rcpath.org/asset/E3FB6B13-F947-4E28-AF9DAEE2FD2CF9D8/ (accessed on 3 October 2018).
- Burton, J.E.; Oshota, O.J.; North, E.; Hudson, M.J.; Polyanskaya, N.; Brehm, J.; Lloyd, G.; Silman, N.J. Development of a multi-pathogen oligonucleotide microarray for detection of Bacillus anthracis. Mol. Cell. Probes 2005, 19, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- DNASTAR Homepage. Available online: http://www.dnastar.com (accessed on 11 September 2018).
- BioEdit Homepage. Available online: http://www.mbio.ncsu.edu/BioEdit/bioedit.html (accessed on 11 September 2018).
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Burton, J.E.; Oshota, O.J.; Silman, N.J. Differential identification of Bacillus anthracis from environmental Bacillus species using microarray analysis. J. Appl. Microbiol. 2006, 101, 754–763. [Google Scholar] [CrossRef] [PubMed]
- Bohlander, S.K.; Espinosa, R.; Le Beau, M.M.; Rowley, J.D.; Díaz, M.O. A method for the rapid sequence-independent amplification of microdissected chromosomal material. Genomics 1992, 13, 1322–1324. [Google Scholar] [CrossRef]
- Corrigan, D.K.; Schulze, H.; Ciani, I.; Henihan, G.; Mount, A.R.; Bachmann, T.T. Improving performance of a rapid electrochemical MRSA assay: Optimisation of assay conditions to achieve enhanced discrimination of clinically important DNA sequences under ambient conditions. J. Electroanal. Chem. 2017, 786, 58–62. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2017. [Google Scholar]
- DeLuca, D.S.; Marina, O.; Ray, S.; Zhang, G.L.; Wu, C.J.; Brusic, V. Data processing and analysis for protein microarrays. Methods Mol. Biol. 2011, 723, 337–347. [Google Scholar] [PubMed]
- Watson, M.; Dukes, J.; Abu-Median, A.B.; King, D.P.; Britton, P. DetectiV: Visualization, normalization and significance testing for pathogen-detection microarray data. Genome Biol. 2007, 8, R190. [Google Scholar] [CrossRef] [PubMed]
- Basic Local Alignment Search Tool Homepage. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi (accessed on 11 September 2018).
- Batchelor, M.; Hopkins, K.L.; Liebana, E.; Slickers, P.; Ehricht, R.; Mafura, M.; Aarestrup, F.; Mevius, D.; Clifton-Hadley, F.A.; Woodward, M.J.; et al. Development of a miniaturised microarray-based assay for the rapid identification of antimicrobial resistance genes in Gram-negative bacteria. Int. J. Antimicrob. Agents 2008, 31, 440–451. [Google Scholar] [CrossRef] [PubMed]
- de Zoysa, A.; Edwards, K.; Gharbia, S.; Underwood, A.; Charlett, A.; Efstratiou, A. Non-culture detection of Streptococcus agalactiae (Lancefield group B Streptococcus) in clinical samples by real-time PCR. J. Med. Microbiol. 2012, 61, 1086–1090. [Google Scholar] [CrossRef] [PubMed]
- Ahmad-Saeed, N.S. Detection of Difficult to Culture Organisms Associated with Paediatric Bone and Joint Infections using Species Specific Real-Time Polymerase Chain Reaction. PhD Thesis, University of Southampton, Southampton, UK, 2018. in press. [Google Scholar]
- Saeed, K.; Ahmad, N.; Pallett, A.; Guiver, M.; Marsh, P. Specific staphylococcal polymerase chain reaction can be a complementary tool for identifying causative organisms and guiding antibiotic management in orthopaedic infections. Curr. Orthop. Pract. 2010, 21, 628–631. [Google Scholar] [CrossRef]
- McHugh, M.P.; Gray, S.J.; Kaczmarski, E.B.; Guiver, M. Reduced turnaround time and improved diagnosis of invasive serogroup B Neisseria meningitidis and Streptococcus pneumoniae infections using a lyophilized quadruplex quantitative PCR. J. Med. Microbiol. 2015, 64, 1321–1328. [Google Scholar] [CrossRef] [PubMed]
- Choudhuri, D.; Huda, T.; Theodoratou, E.; Nair, H.; Zgaga, L.; Falconer, R.; Luksic, I.; Johnson, H.L.; Zhang, J.S.; El Arifeen, S.; et al. An evaluation of emerging vaccines for childhood meningococcal disease. BMC Public Health 2011, 11 (Suppl. 3), S29. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Lazaro, D.; Hernández, M.; Scortti, M.; Esteve, T.; Vázquez-Boland, J.A.; Pla, M. Quantitative detection of Listeria monocytogenes and Listeria innocua by real-time PCR: Assessment of hly, iap, and lin02483 targets and AmpliFluor technology. Appl. Environ. Microbiol. 2004, 70, 1366–1377. [Google Scholar] [CrossRef] [PubMed]
- Kierzek, R.; Turner, D.H.; Kierzek, E. Microarrays for identifying binding sites and probing structure of RNAs. Nucleic Acids Res. 2015, 43, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Falla, T.J.; Crook, D.W.; Brophy, L.N.; Maskell, D.; Kroll, J.S.; Moxon, E.R. PCR for capsular typing of Haemophilus influenzae. J. Clin. Microbiol. 1994, 32, 2382–2386. [Google Scholar] [PubMed]
- Harrison, O.B.; Claus, H.; Jiang, Y.; Bennett, J.S.; Bratcher, H.B.; Jolley, K.A.; Corton, C.; Care, R.; Poolman, J.T.; Zollinger, W.D. Description and nomenclature of Neisseria meningitidis capsule locus. Emerg. Infect. Dis. 2013, 19, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Swiderek, H.; Claus, H.; Frosch, M.; Vogel, U. Evaluation of custom-made DNA microarrays for multilocus sequence typing of Neisseria meningitidis. Int. J. Med. Microbiol. 2005, 295, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.; Wang, Z.; Vora, G.J.; Thornton, J.A.; Schnur, J.M.; Thach, D.C.; Blaney, K.M.; Ligler, A.G.; Malanoski, A.P.; Santiago, J.; et al. Broad-spectrum respiratory tract pathogen identification using resequencing DNA microarrays. Genome Res. 2006, 16, 527–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, W.; Liu, B.; Cao, B.; Beutin, L.; Krüger, U.; Liu, H.; Li, Y.; Liu, Y.; Feng, L.; Wang, L. DNA microarray-based identification of serogroups and virulence gene patterns of Escherichia coli isolates associated with porcine postweaning diarrhea and edema disease. Appl. Environ. Microbiol. 2007, 73, 4082–4088. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Wu, F.; Li, D.; Beutin, L.; Chen, M.; Cao, B.; Wang, L. Development of a serogroup-specific DNA microarray for identification of Escherichia coli strains associated with bovine septicemia and diarrhea. Vet. Microbiol. 2010, 142, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, M.; Kong, F.; Gilbert, G.L.; Cao, B.; Wang, L.; Feng, L. Development of a DNA microarray to identify the Streptococcus pneumoniae serotypes contained in the 23-valent pneumococcal polysaccharide vaccine and closely related serotypes. J. Microbiol. Methods 2007, 68, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Wang, Q.; Li, Y.; Kong, F.; Gilbert, G.L.; Cao, B.; Wang, L.; Feng, L. Use of a serotype-specific DNA microarray for identification of group B Streptococcus (Streptococcus agalactiae). J. Clin. Microbiol. 2006, 44, 1447–1452. [Google Scholar] [CrossRef] [PubMed]
- Hamels, S.; Gala, J.L.; Dufour, S.; Vannuffel, P.; Zammatteo, N.; Remacle, J. Consensus PCR and microarray for diagnosis of the genus Staphylococcus, species, and methicillin resistance. Biotechniques 2001, 31, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
- Oh, T.J.; Kim, C.J.; Woo, S.K.; Kim, T.S.; Jeong, D.J.; Kim, M.S.; Lee, S.; Cho, H.S.; An, S. Development and clinical evaluation of a highly sensitive DNA microarray for detection and genotyping of human papillomaviruses. J. Clin. Microbiol. 2004, 42, 3272–3280. [Google Scholar] [CrossRef] [PubMed]
- Ma, M.; Wang, H.; Yu, Y.; Zhang, D.; Liu, S. Detection of antimicrobial resistance genes of pathogenic Salmonella from swine with DNA microarray. J. Vet. Diagn. Investig. 2007, 19, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.X.; Zhang, Z.W.; Wang, C.; Yang, H.W.; Jiang, D.; Zhang, Q.; Mitchelson, K.; Cheng, J. Use of a DNA microarray for simultaneous detection of antibiotic resistance genes among staphylococcal clinical isolates. J. Clin. Microbiol. 2007, 45, 3514–3521. [Google Scholar] [CrossRef] [PubMed]
- Dunning Hotopp, J.C.; Grifantini, R.; Kumar, N.; Tzeng, Y.L.; Fouts, D.; Frigimelica, E.; Draghi, M.; Giuliani, M.M.; Rappuoli, R.; Stephens, D.S.; et al. Comparative genomics of Neisseria meningitidis: Core genome, islands of horizontal transfer and pathogen-specific genes. Microbiology 2006, 152, 3733–3749. [Google Scholar] [CrossRef] [PubMed]
- Schoen, C.; Joseph, B.; Claus, H.; Vogel, U.; Frosch, M. Living in a changing environment: insights into host adaptation in Neisseria meningitidis from comparative genomics. Int. J. Med. Microbiol. 2007, 297, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Snyder, L.A.; Saunders, N.J. The majority of genes in the pathogenic Neisseria species are present in non-pathogenic Neisseria lactamica, including those designated as ‘virulence genes’. BMC Genom. 2006, 7, 128. [Google Scholar] [CrossRef] [PubMed]
- Stabler, R.A.; Marsden, G.L.; Witney, A.A.; Li, Y.; Bentley, S.D.; Tang, C.M.; Hinds, J. Identification of pathogen-specific genes through microarray analysis of pathogenic and commensal Neisseria species. Microbiology 2005, 151, 2907–2922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winther-Larsen, H.C.; Hegge, F.T.; Wolfgang, M.; Hayes, S.F.; van Putten, J.P.; Koomey, M. Neisseria gonorrhoeae PilV, a type IV pilus-associated protein essential to human epithelial cell adherence. Proc. Natl. Acad. Sci. USA 2001, 98, 15276–15281. [Google Scholar] [CrossRef] [PubMed]
- Gala, J.-L.; Centre for Applied Molecular Technologies, Université Catholique de Louvain, Place de l’Université, Louvain-la-Neuve, Belgium. Personal communication, 2005.
- Tonjum, T.; Koomey, M. The pilus colonization factor of pathogenic neisserial species: Organelle biogenesis and structure/function relationships—A review. Gene 1997, 192, 155–163. [Google Scholar] [CrossRef]
- Findlow, H.; Vogel, U.; Mueller, J.E.; Curry, A.; Njanpop-Lafourcade, B.M.; Claus, H.; Gray, S.J.; Yaro, S.; Traoré, Y.; Sangaré, L.; et al. Three cases of invasive meningococcal disease caused by a capsule null locus strain circulating among healthy carriers in Burkina Faso. J. Infect. Dis. 2007, 195, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Hammerschmidt, S.; Müller, A.; Sillmann, H.; Mühlenhoff, M.; Borrow, R.; Fox, A.; van Putten, J.; Zollinger, W.D.; Gerardy-Schahn, R.; Frosch, M. Capsule phase variation in Neisseria meningitidis serogroup B by slipped-strand mispairing in the polysialyltransferase gene (siaD): Correlation with bacterial invasion and the outbreak of meningococcal disease. Mol. Microbiol. 1996, 20, 1211–1220. [Google Scholar] [CrossRef] [PubMed]
- Stephens, D.S.; Greenwood, B.; Brandtzaeg, P. Epidemic meningitis, meningococcaemia, and Neisseria meningitidis. Lancet 2007, 369, 2196–2210. [Google Scholar] [CrossRef]
- Korczak, B.; Frey, J.; Schrenzel, J.; Pluschke, G.; Pfister, R.; Ehricht, R.; Kuhnert, P. Use of diagnostic microarrays for determination of virulence gene patterns of Escherichia coli K1, a major cause of neonatal meningitis. J. Clin. Microbiol. 2005, 43, 1024–1031. [Google Scholar] [CrossRef] [PubMed]
- Monecke, S.; Ehricht, R. Rapid genotyping of methicillin-resistant Staphylococcus aureus (MRSA) isolates using miniaturised oligonucleotide arrays. Clin. Microbiol. Infect. 2005, 11, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Perreten, V.; Vorlet-Fawer, L.; Slickers, P.; Ehricht, R.; Kuhnert, P.; Frey, J. Microarray-based detection of 90 antibiotic resistance genes of gram-positive bacteria. J. Clin. Microbiol. 2005, 43, 2291–2302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sachse, K.; Hotzel, H.; Slickers, P.; Ellinger, T.; Ehricht, R. DNA microarray-based detection and identification of Chlamydia and Chlamydophila spp. Mol. Cell. Probes 2005, 19, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Card, R.; Zhang, J.; Das, P.; Cook, C.; Woodford, N.; Anjum, MF. Evaluation of an expanded microarray for detecting antibiotic resistance genes in a broad range of gram-negative bacterial pathogens. Antimicrob. Agents Chemother. 2013, 57, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Subba Rao, P.V.; McCartney-Francis, N.L.; Metcalfe, D.D. An avidin–biotin microELISA for rapid measurement of total and allergen-specific human IgE. J. Immunol. Methods 1983, 57, 71–85. [Google Scholar] [CrossRef]
- Townsend, M.B.; Dawson, E.D.; Mehlmann, M.; Smagala, J.A.; Dankbar, D.M.; Moore, C.L.; Smith, C.B.; Cox, N.J.; Kuchta, R.D.; Rowlen, K.L. Experimental evaluation of the FluChip diagnostic microarray for influenza virus surveillance. J. Clin. Microbiol. 2006, 44, 2863–2871. [Google Scholar] [CrossRef] [PubMed]
- Malanoski, A.P.; Lin, B.; Stenger, D.A. A model of base-call resolution on broad-spectrum pathogen detection resequencing DNA microarrays. Nucleic Acids Res. 2008, 36, 3194–3201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allred, A.F.; Renshaw, H.; Weaver, S.; Tesh, R.B.; Wang, D. VIPR HMM: A hidden Markov model for detecting recombination with microbial detection microarrays. Bioinformatics 2012, 28, 2922–2929. [Google Scholar] [CrossRef] [PubMed]
- Malanoski, A.P.; Lin, B.; Wang, Z.; Schnur, J.M.; Stenger, D.A. Automated identification of multiple micro-organisms from resequencing DNA microarrays. Nucleic Acids Res. 2006, 34, 5300–5311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alere Technologies GmbH. Available online: https://alere-technologies.com/products/ lab-solutions.html (accessed on 11 September 2018).
- Seki, M.; Kilgore, P.E.; Kim, E.J.; Ohnishi, M.; Hayakawa, S.; Kim, D.W. Loop-Mediated Isothermal Amplification Methods for Diagnosis of Bacterial Meningitis. Front. Pediatr. 2018, 6, 57. [Google Scholar] [CrossRef] [PubMed]



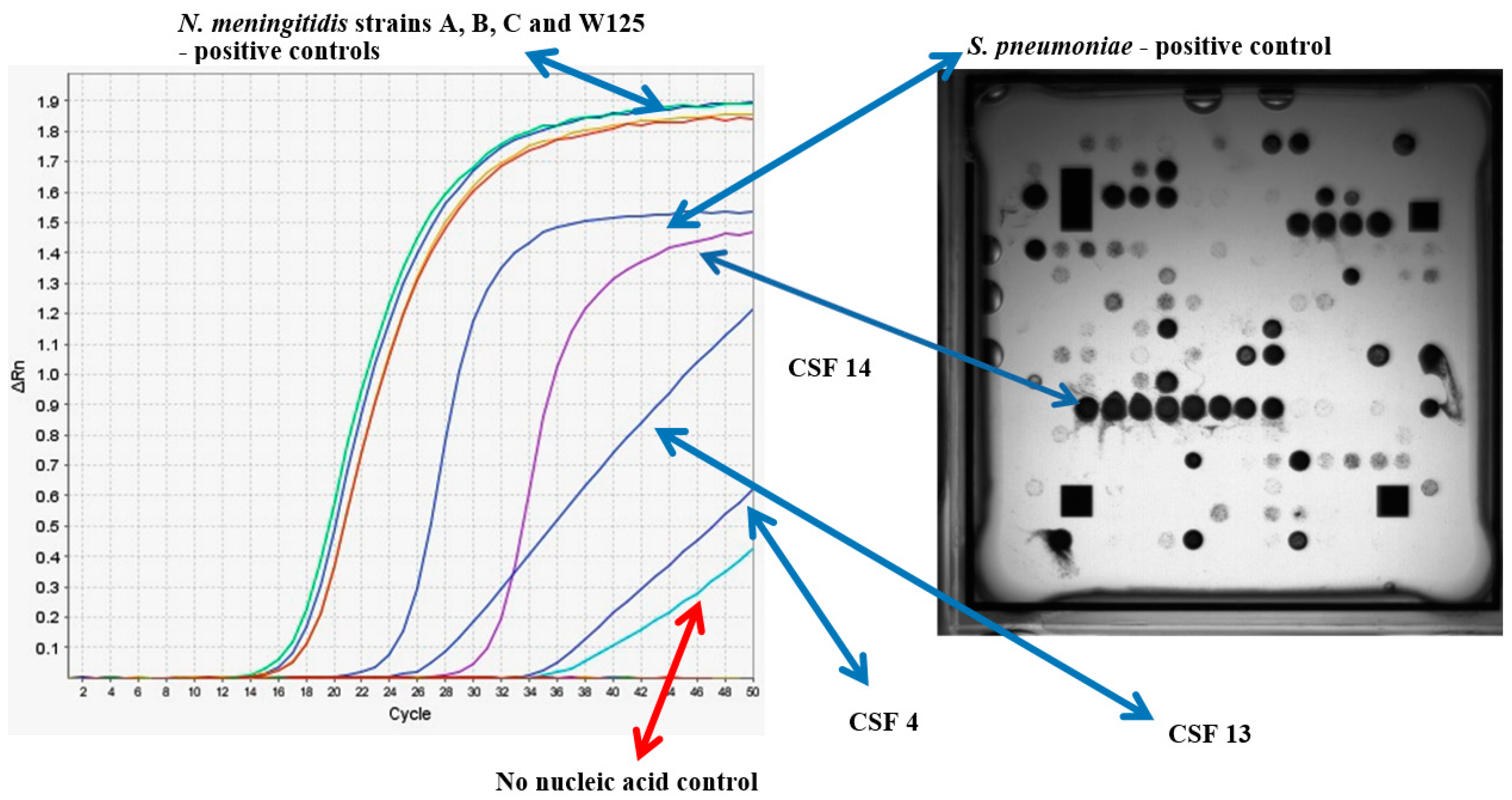


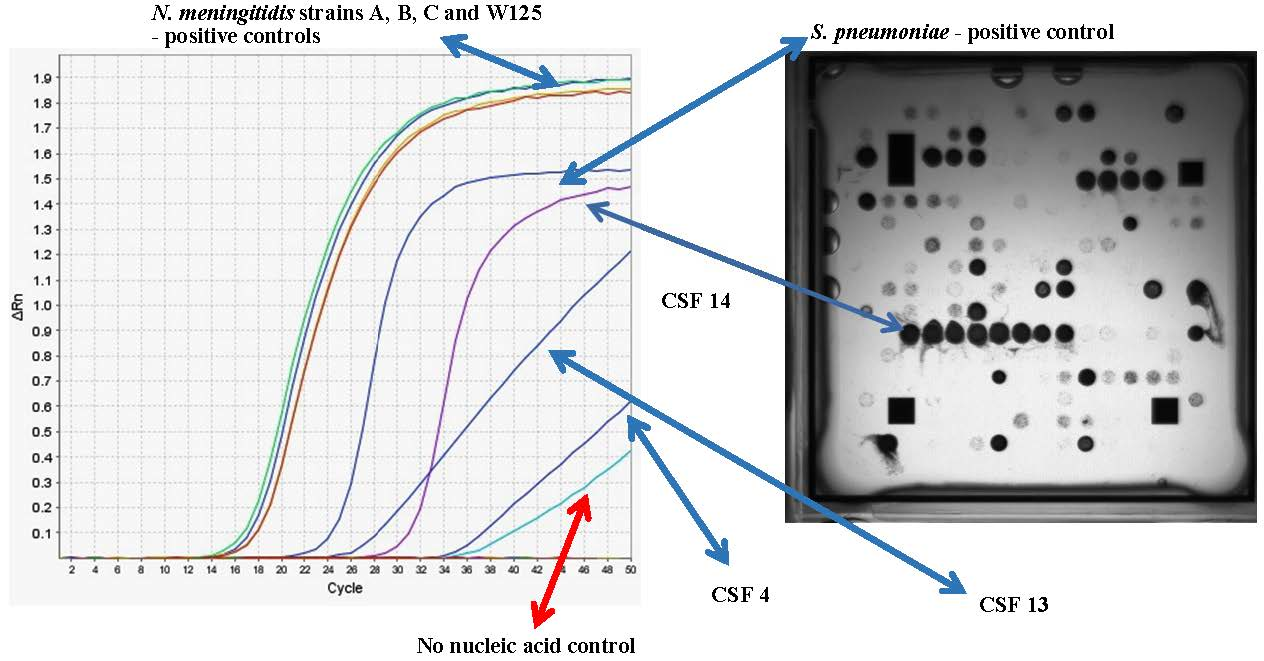{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
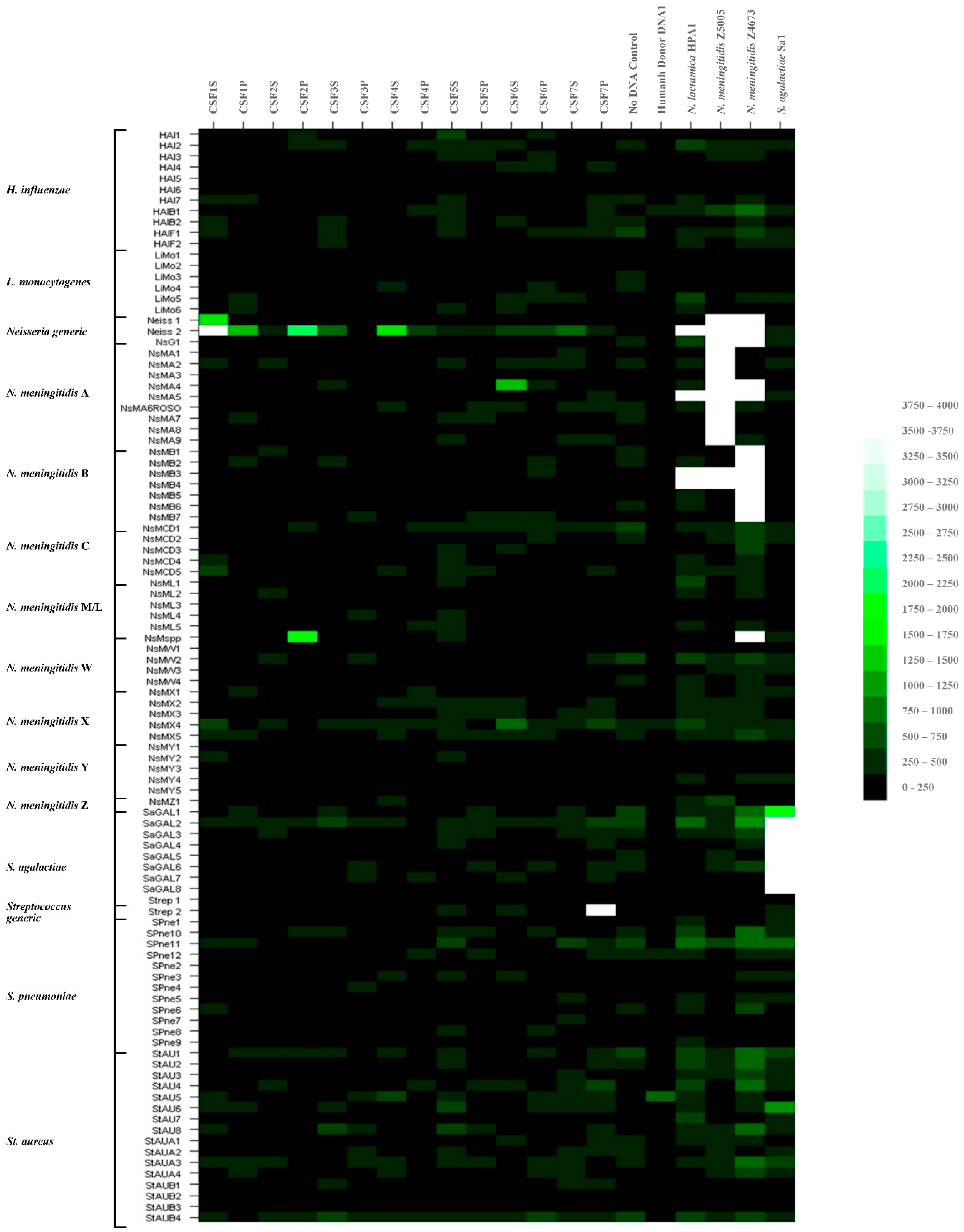
| Patient | Sample Designation | Sample Fraction | Culture | RT-PCR Ct Values for Neisseria meningitides (ctrA) * | Microarray Signal for Neisseria spp. | |
|---|---|---|---|---|---|---|
| 1 | CSF1S | Cerebrospinal Fluid | supernatant | - | 36 | + |
| 1 | CSF1P | Cerebrospinal Fluid | pellet | - | 37 | + |
| 2 | CSF2S | Cerebrospinal Fluid | supernatant | - | 35 | - |
| 2 | CSF2P | Cerebrospinal Fluid | pellet | - | 31 | + |
| 3 | CSF3S | Cerebrospinal Fluid | supernatant | - | ND | - |
| 3 | CSF3P | Cerebrospinal Fluid | pellet | - | ND | - |
| 4 | CSF4S | Cerebrospinal Fluid | supernatant | - | ND | + |
| 4 | CSF4P | Cerebrospinal Fluid | pellet | - | 38 | - |
| 5 | CSF5S | Cerebrospinal Fluid | supernatant | - | ND | - |
| 5 | CSF5P | Cerebrospinal Fluid | pellet | - | ND | - |
| 6 | CSF6S | Cerebrospinal Fluid | supernatant | - | 36 | - |
| 6 | CSF6P | Cerebrospinal Fluid | pellet | - | ND | - |
| 7 | CSF7S | Cerebrospinal Fluid | supernatant | - | ND | - |
| 7 | CSF7P | Cerebrospinal Fluid | pellet | - | ND | - |
| Sample Number | M/F | Date of Sample | Syndromic Meningitis PCR (CSF Unless Stated) | Bacterial Culture | White Blood Cells 109/L | CRP mg/L | Other/Clinical/Travel History |
|---|---|---|---|---|---|---|---|
| 4 | F | 25 June 2011 | Ent, HSV1/2, VZV-ve | no growth | 4 | <1 | |
| 11 | M | 4 July 2011 | N. meningitidis-ve | no growth | 11.7 | 16 | |
| 13 | F | 1 July 2011 | Ent, HSV1/2, VZV-ve | N/A | N/A | N/A | |
| 14 | F | 5 July 2011 | Ent, HSV1/2, VZV-ve | S. pneumoniae | 9.5 | 185 | clinical details: “pyrexia-strep meningitis”; β haemolytic Streptococcus not isolated |
| 21 | F | 22 July 2011 | Ent, HSV1/2, VZV-ve | N/A | N/A | N/A | |
| 24 | |||||||
| 29 | F | 21 July 2011 | Borrelia burgdorferi-ve | N/A | N/A | N/A | |
| 34 | M | 28 July 2011 | Ent, HSV1/2, VZV-ve | N/A | N/A | N/A | |
| 51 | F | 30 March 2011 | Ent, HSV1/2, VZV, N. meningitidis-ve | no growth | 7.8 | 13 | |
| 54 | F | 4 April 2011 | Ent, HSV1/2, VZV, N. meningitidis-ve | no growth | 18.6 | <1 | headache, photophobia, recent trip to Gambia |
| 61 * | M | 13 April 2011 | AdV, RotaVirus, Ent, HSV1/2, N. meningitidis-ve | no growth | 10 | 6 | |
| 63 * | M | 13 April 2011 | AdV, RotaVirus, Ent, HSV1/2, N. meningitidis-ve | no growth | 64 | 6 | |
| 70 | F | 20 April 2011 | Ent, HSV1/2, VZV-ve | no growth | 6 | 10 | |
| 72 | U | 14 April 2011 | B. burgdorferi-ve | N/A | N/A | N/A | |
| 76 | M | 27 April 2011 | Ent, HSV1/2, VZV-ve | no growth | 13.3 | 2 | |
| 90 | F | 12 May 2011 | Ent, HSV1/2, VZV-ve | mixed perineal flora | 14.3 | 55 | Pyrexia of unknown origin |
| 95 | M | 16 May 2011 | Ent, HSV1/2, VZV-ve | no growth | 6.8 | ||
| 127 | M | 23 June 2011 | Ent, HSV1/2, VZV, N. meningitidis-ve | no growth | <1 | ||
| 145 | F | 14 July 2011 | Parvo, CMV, EBV-ve (CSF). N. meningitidis-ve (blood) Varicella IgG detected (blood) | N. meningitidis in blood bottle | 60 | Ref lab report: Type B, subtype P1.7/P1.1/NT; porA seq: 7-1/1/35-1 | |
| 147 | F | 4 June 2011 | Ent, HSV1/2, VZV, N. meningitidis-ve | no growth | <1 | 7 |
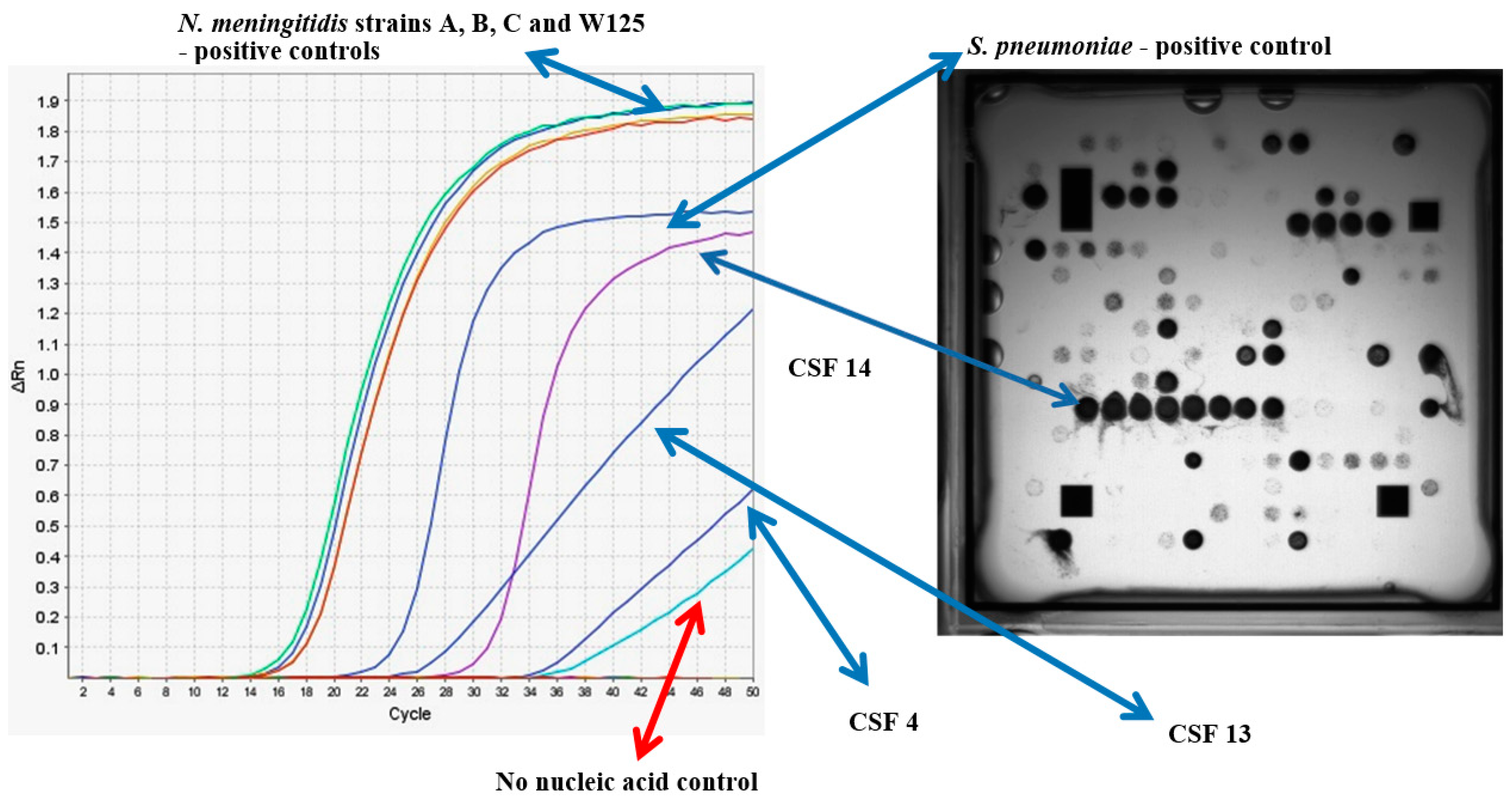
| Sample Number | Bacterial RT-PCR Result | Ct Value | Microarray Hybridization Result Multiplex | Probable Strain Designation |
|---|---|---|---|---|
| 4 | Neisseria meningitidis | 43.72 | N. meningitidis | N. meningitidis A |
| 11 | Staphylococcus epidermidis | 43.28 | N. meningitidis/Staphylococcus spp. | unknown |
| 13 | Neisseria meningitidis | 39.88 | N. meningitidis | N. meningitidis A |
| 14 | Streptococcus pneumoniae | 31.85 | S. pneumoniae * | |
| 21 | Haemophilus influenzae | 37.34 | ND | |
| 24 | Group B Streptococci | 36.39 | ND | |
| 29 | Staphylococcus epidermidis | 36.48 | ND | |
| 34 | Staphylococcus epidermidis | 38.8 | S. pneumoniae * | |
| 51 | Staphylococcus epidermidis | 39.37 | L. monocytogenes * | |
| 54 | Staphylococcus epidermidis | 38.79 | S. pneumoniae | |
| 61 | Staphylococcus epidermidis | 37.59 | N. meningitidis | N. meningitidis A * |
| 63 | Staphylococcus epidermidis | 40.66 | S. pneumoniae/Staphylococcus spp. * | |
| 70 | Haemophilus influenzae | 40.36 | ND | |
| 72 | Neisseria meningitidis | 36.00 | N. meningitidis | N. meningitidis A |
| 76 | Staphylococcus epidermidis & Staphylococcus aureus | 37.87, 45.13 | ND | |
| 90 | Staphylococcus epidermidis | 39.02 | ND | |
| 95 | Haemophilus influenzae | 36.02 | S. pneumonia * | |
| 127 | Staphylococcus epidermidis | 40.19 | Neisseria spp. | |
| 145 | Neisseria meningitidis | 19.38 | N. meningitidis/M. tuberculosis | N. meningitidis B |
| 147 | Staphylococcus epidermidis | 40.36 | ND |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bannister, S.A.; Kidd, S.P.; Kirby, E.; Shah, S.; Thomas, A.; Vipond, R.; Elmore, M.J.; Telfer Brunton, A.; Marsh, P.; Green, S.; et al. Development and Assessment of a Diagnostic DNA Oligonucleotide Microarray for Detection and Typing of Meningitis-Associated Bacterial Species. High-Throughput 2018, 7, 32. https://doi.org/10.3390/ht7040032
Bannister SA, Kidd SP, Kirby E, Shah S, Thomas A, Vipond R, Elmore MJ, Telfer Brunton A, Marsh P, Green S, et al. Development and Assessment of a Diagnostic DNA Oligonucleotide Microarray for Detection and Typing of Meningitis-Associated Bacterial Species. High-Throughput. 2018; 7(4):32. https://doi.org/10.3390/ht7040032
Chicago/Turabian StyleBannister, Stephanie A., Stephen P. Kidd, Elizabeth Kirby, Sonal Shah, Anvy Thomas, Richard Vipond, Michael J. Elmore, Andrew Telfer Brunton, Peter Marsh, Steve Green, and et al. 2018. "Development and Assessment of a Diagnostic DNA Oligonucleotide Microarray for Detection and Typing of Meningitis-Associated Bacterial Species" High-Throughput 7, no. 4: 32. https://doi.org/10.3390/ht7040032
APA StyleBannister, S. A., Kidd, S. P., Kirby, E., Shah, S., Thomas, A., Vipond, R., Elmore, M. J., Telfer Brunton, A., Marsh, P., Green, S., Silman, N. J., & Kempsell, K. E. (2018). Development and Assessment of a Diagnostic DNA Oligonucleotide Microarray for Detection and Typing of Meningitis-Associated Bacterial Species. High-Throughput, 7(4), 32. https://doi.org/10.3390/ht7040032