Molecular Lesions of Insulator CTCF and Its Paralogue CTCFL (BORIS) in Cancer: An Analysis from Published Genomic Studies


Abstract
:1. Introduction
2. Methods
3. Results
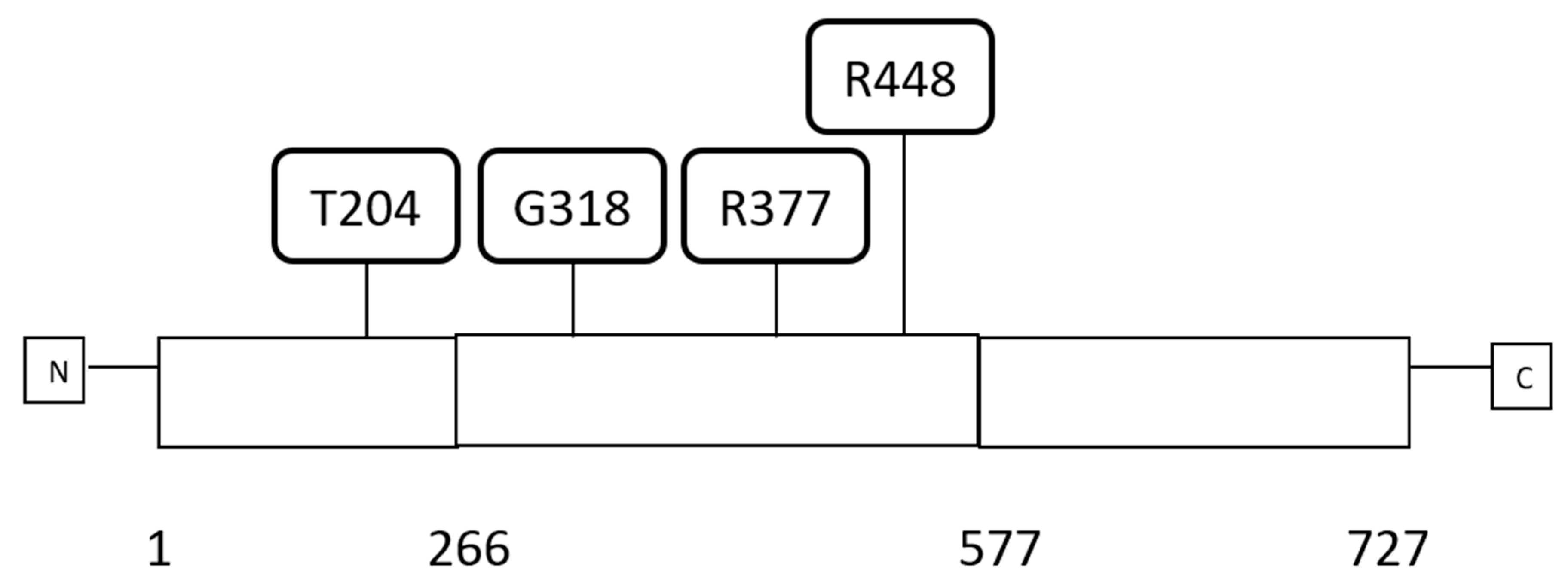
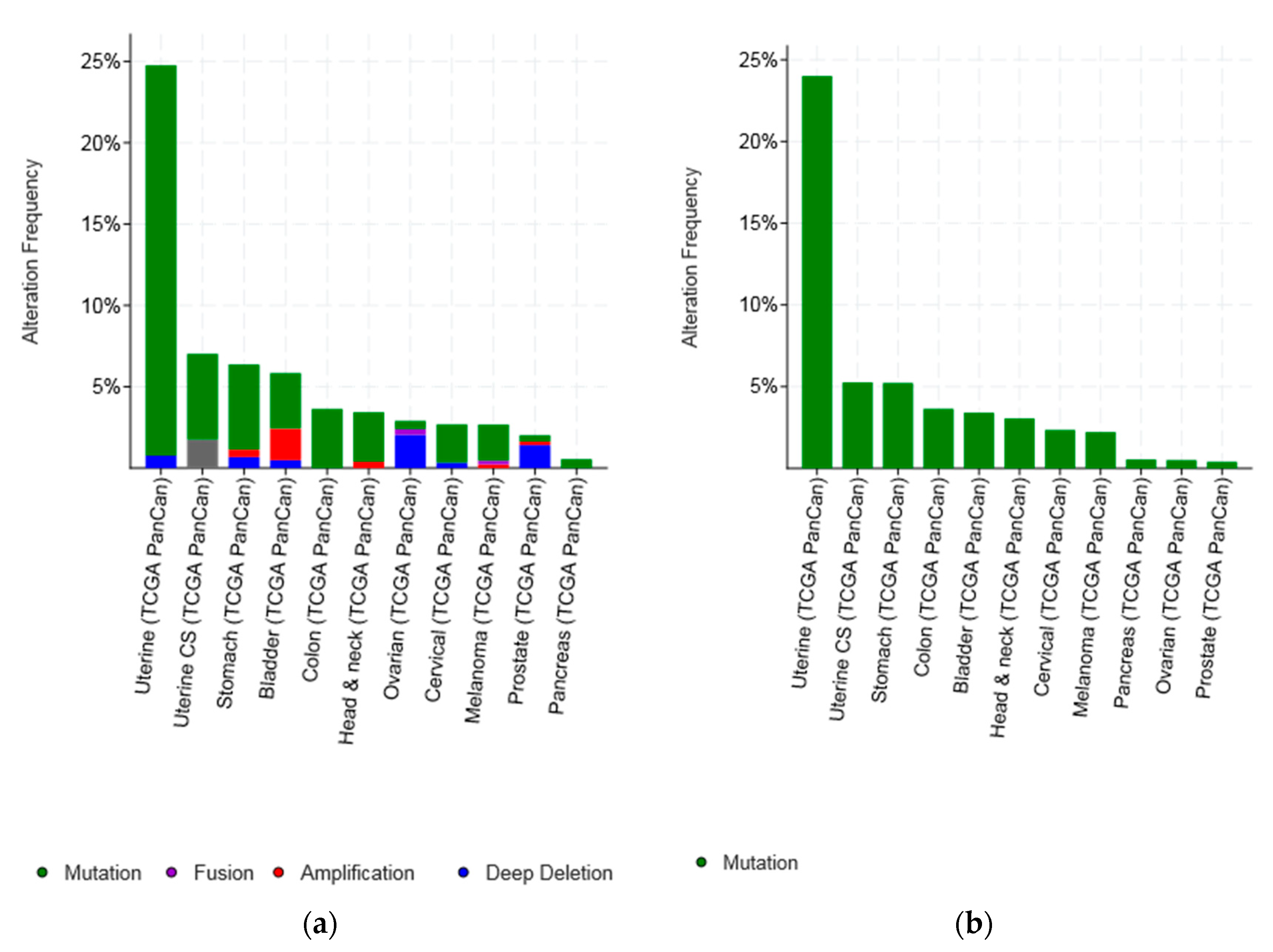
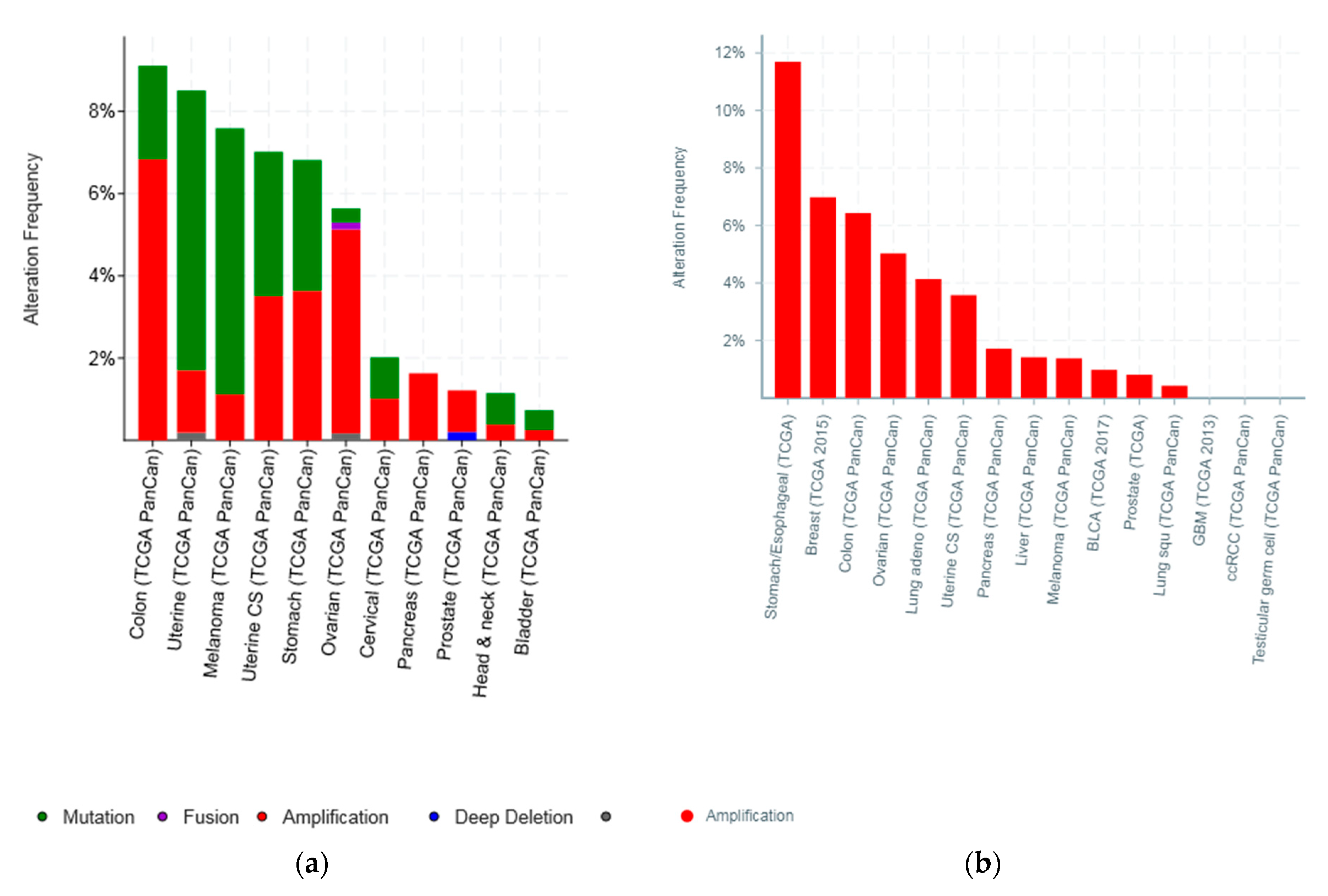
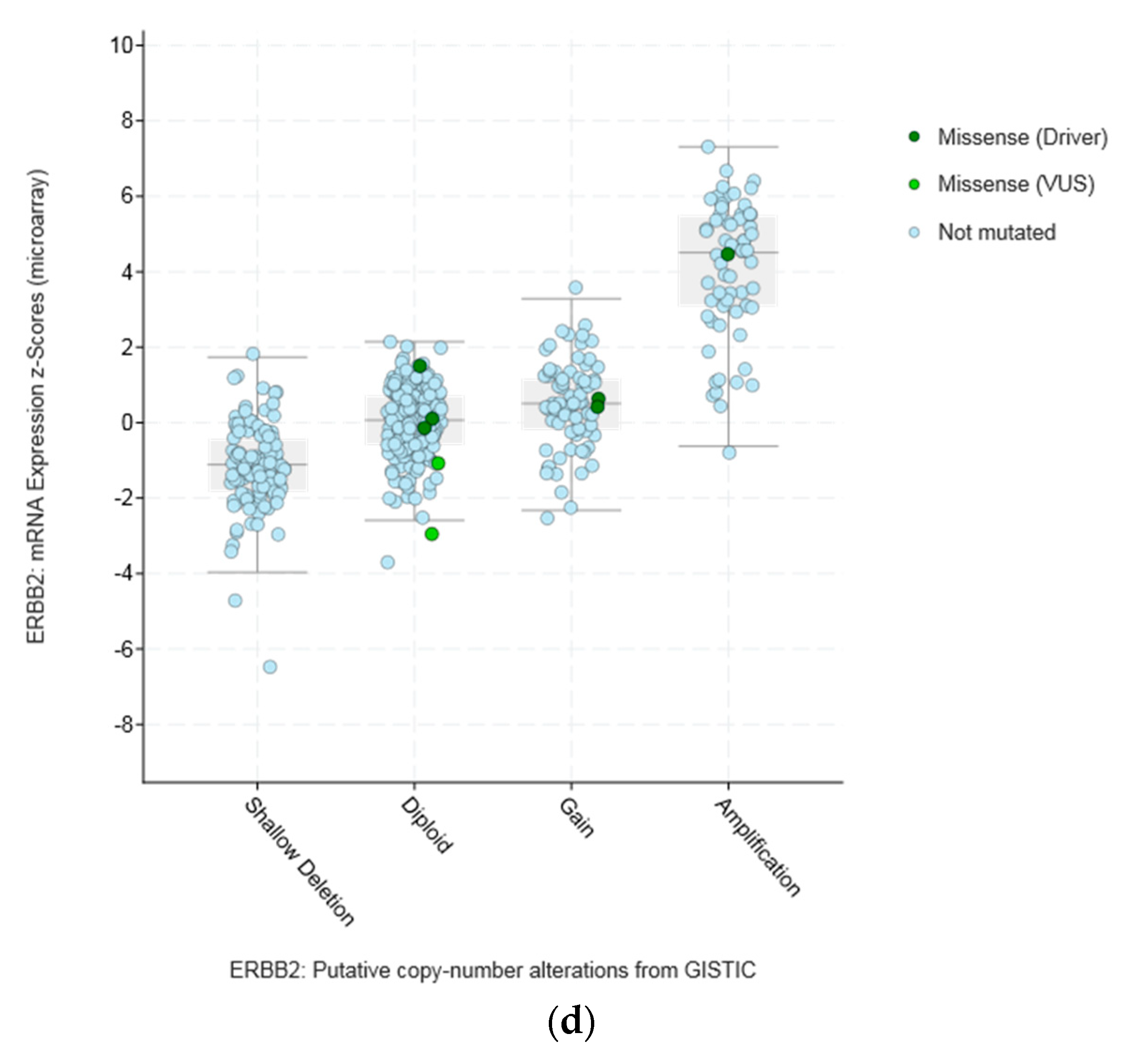
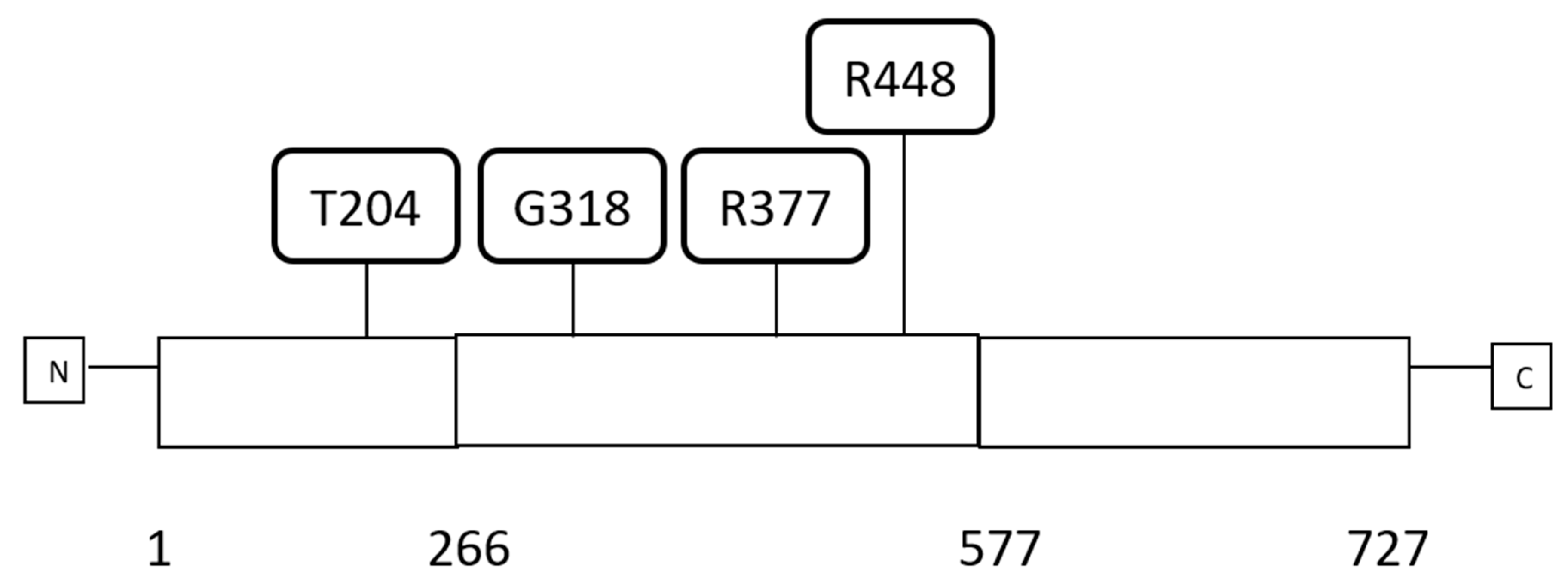
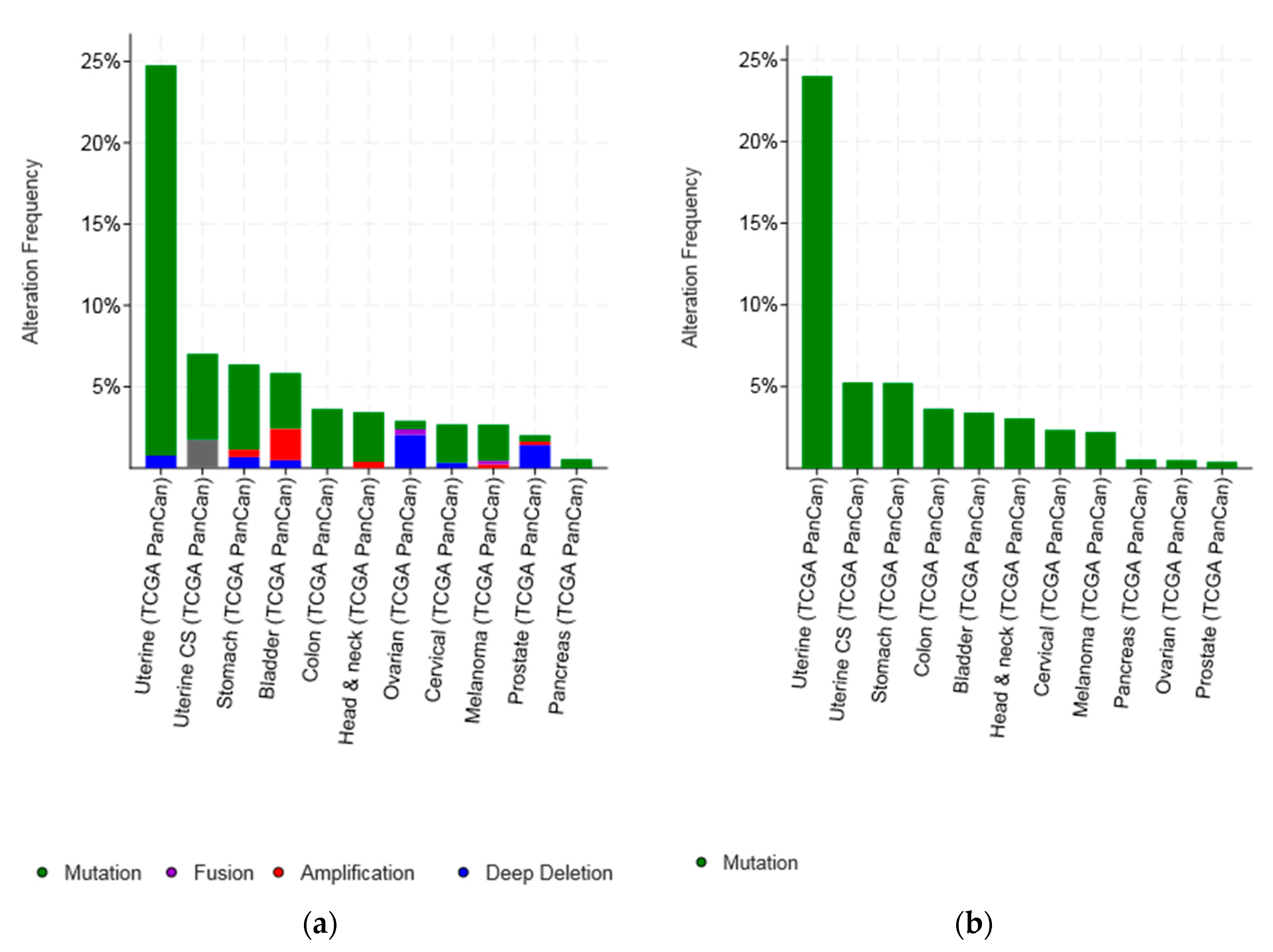
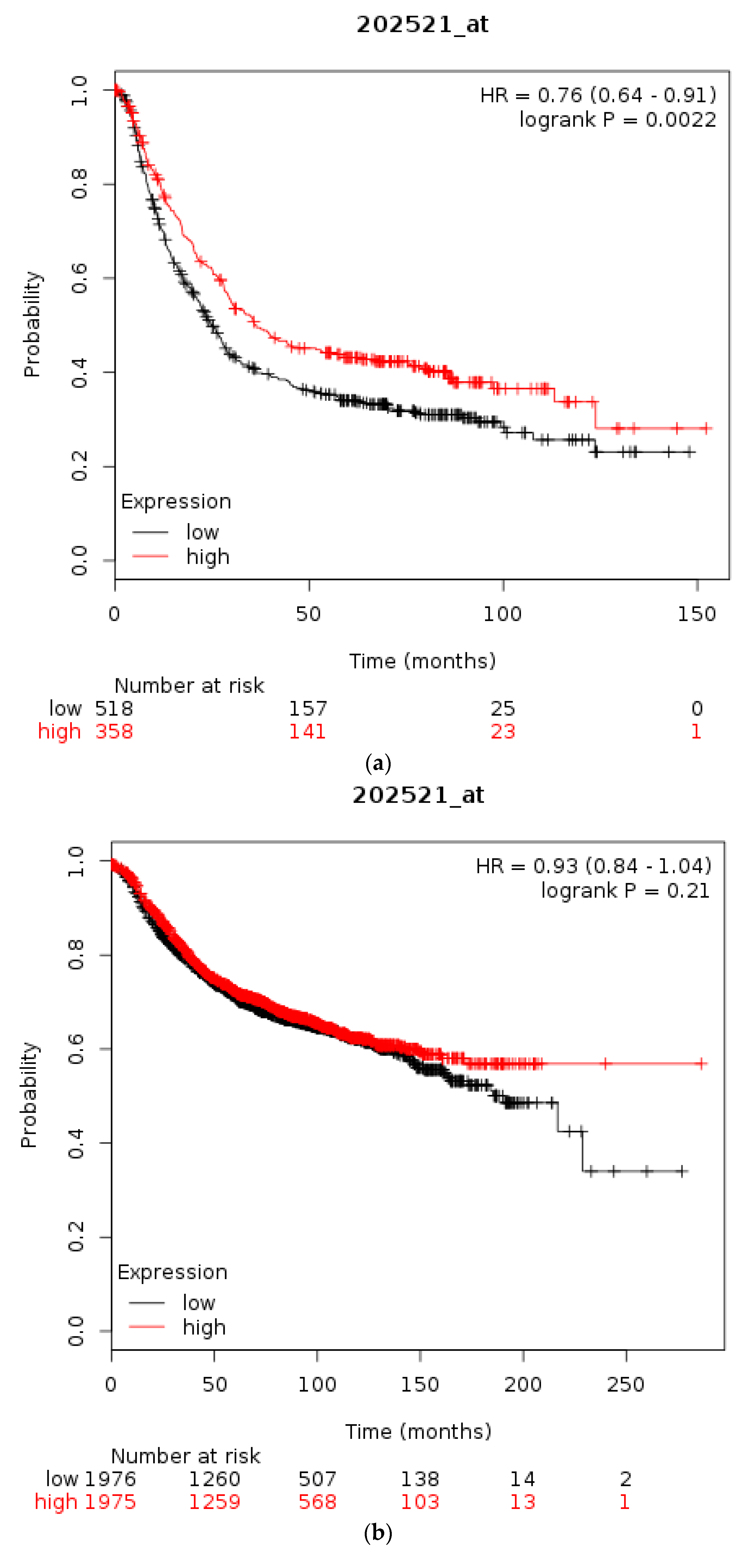
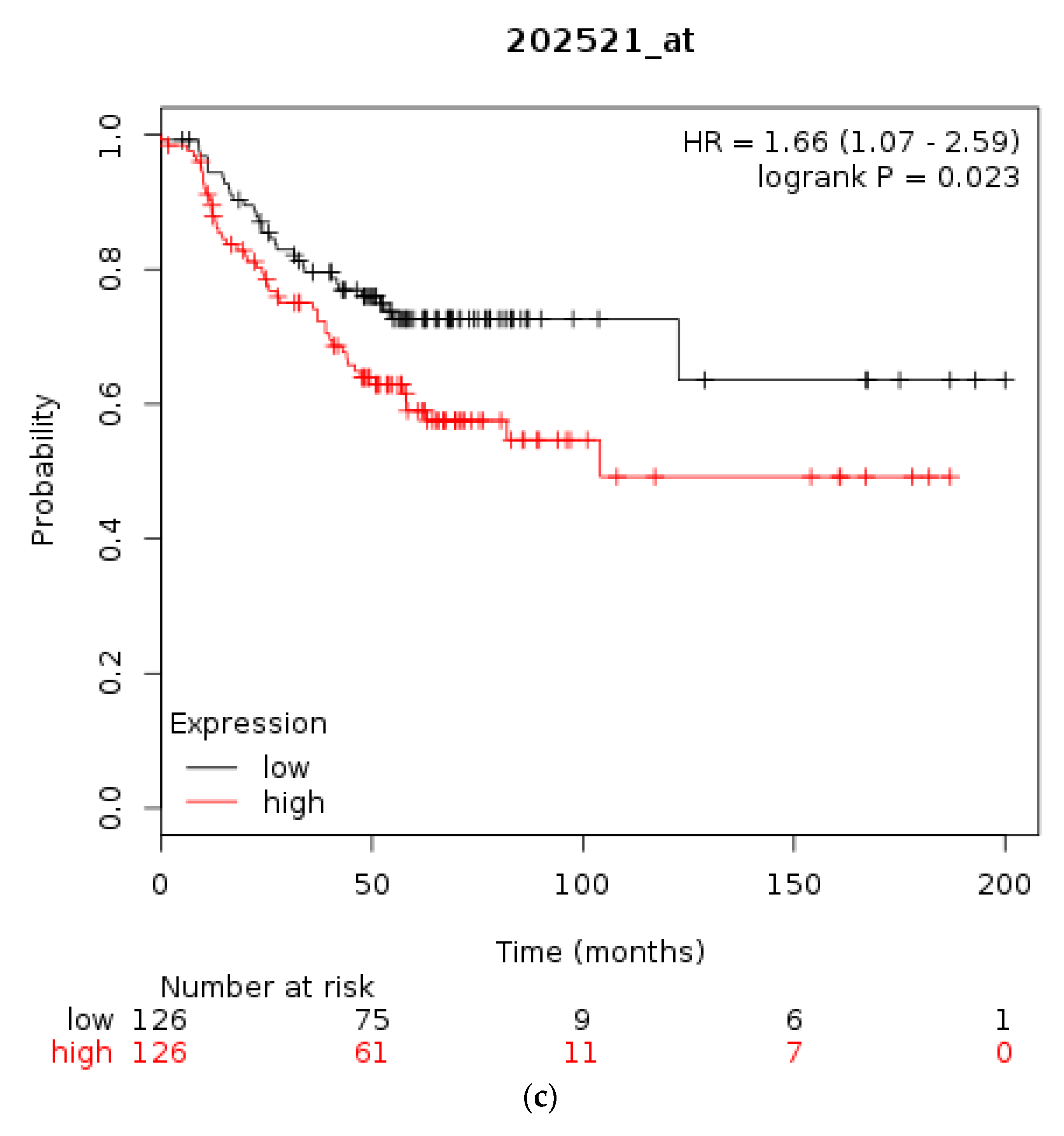
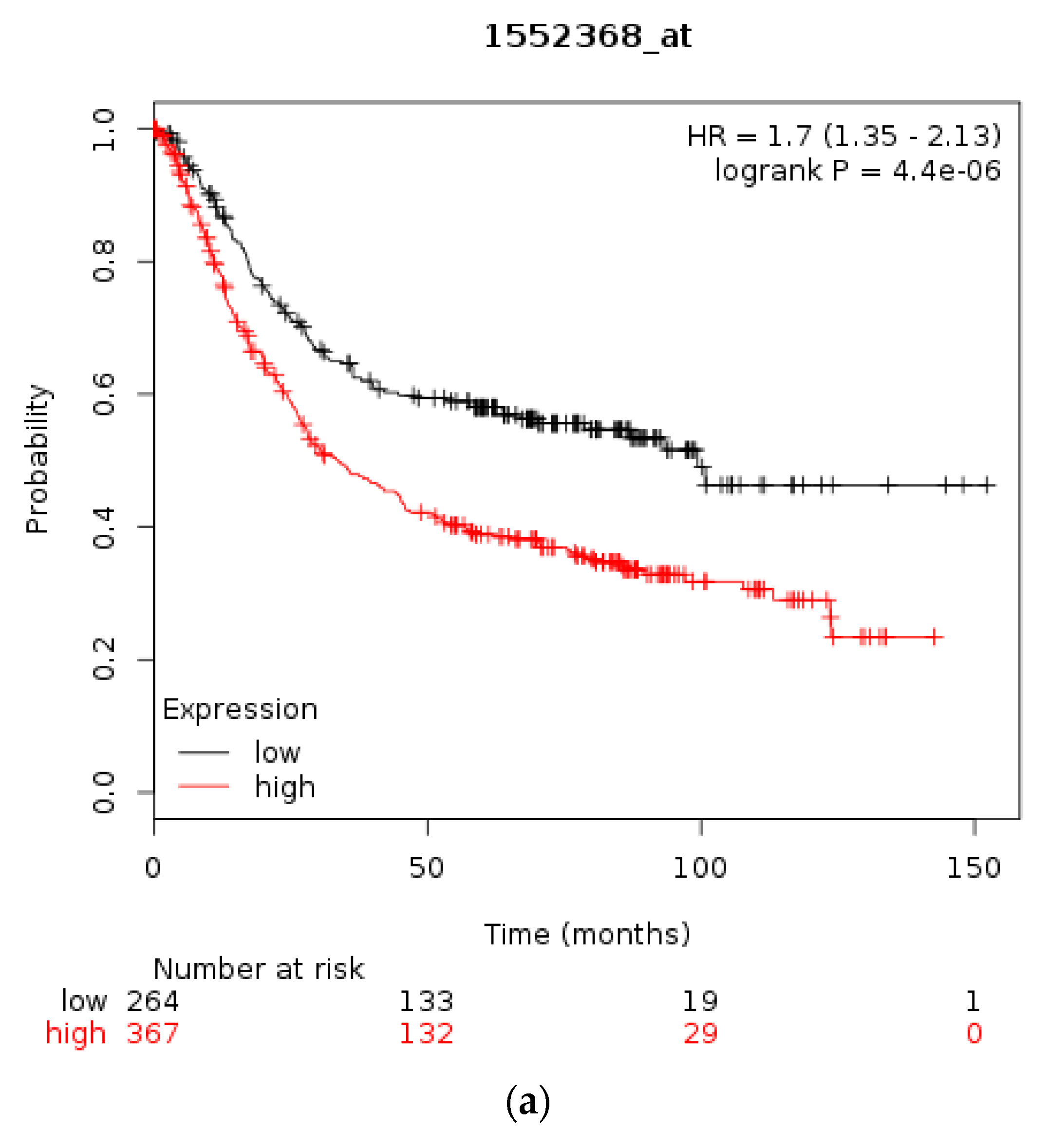
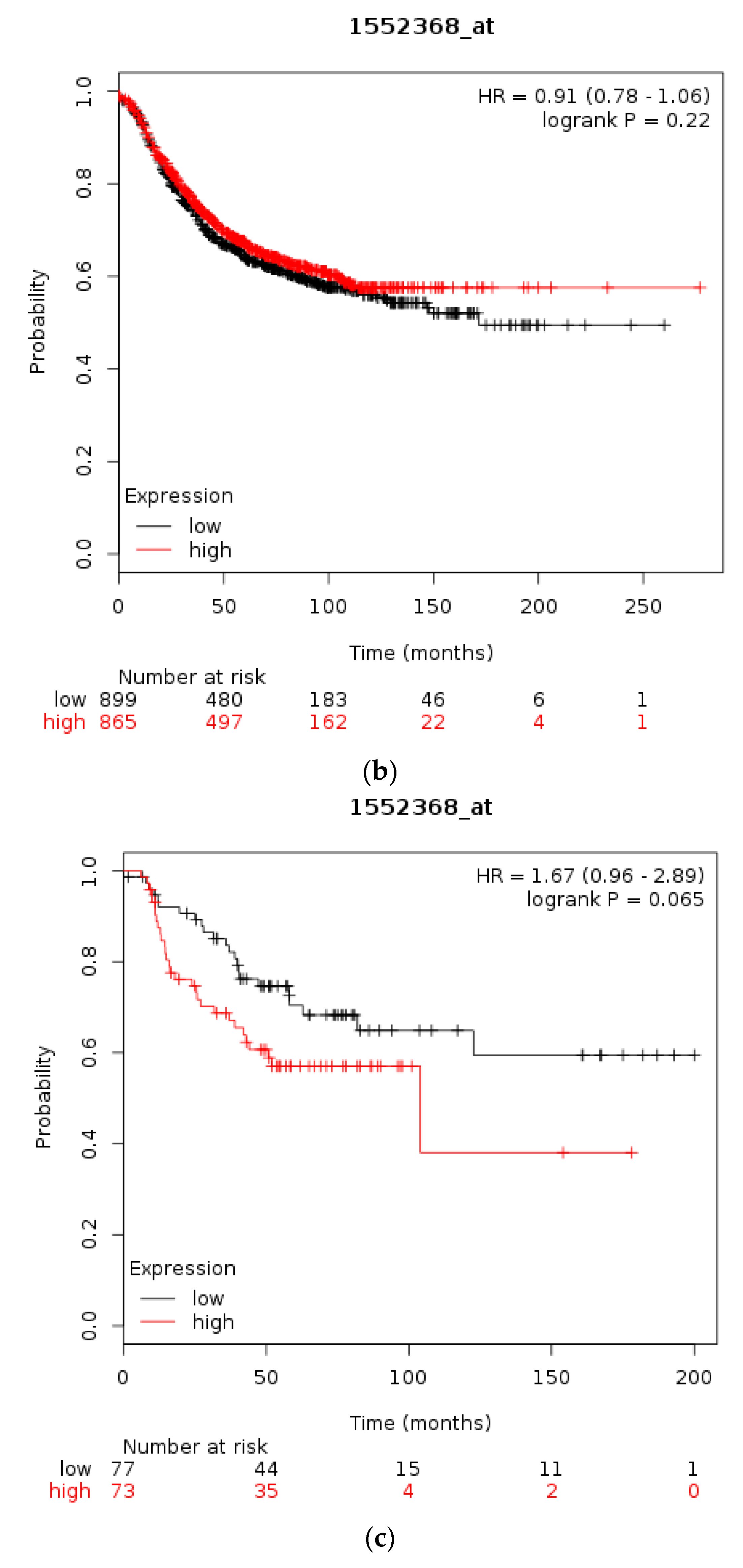
3.1. Molecular Lesions of CTCF in Cancer
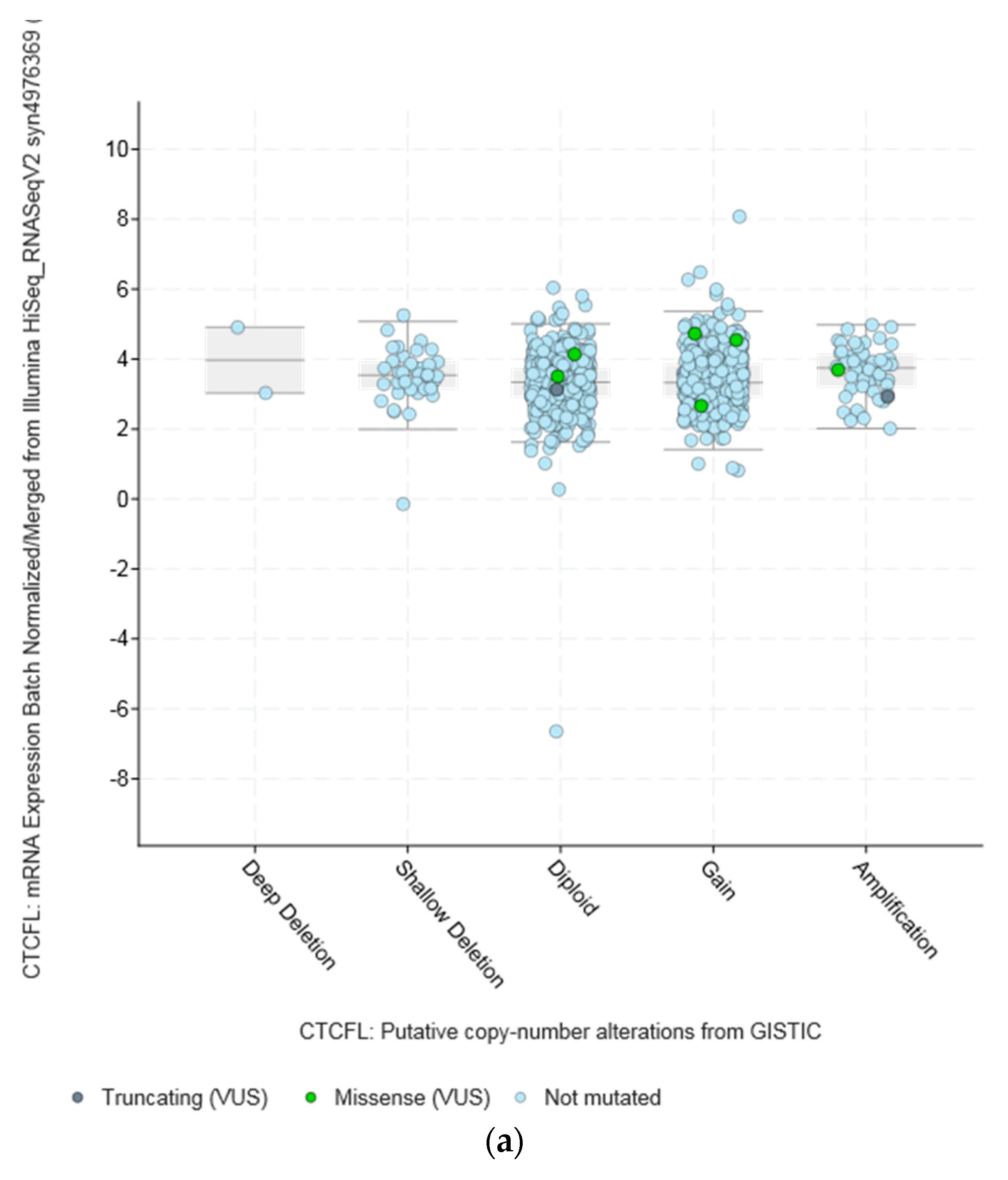
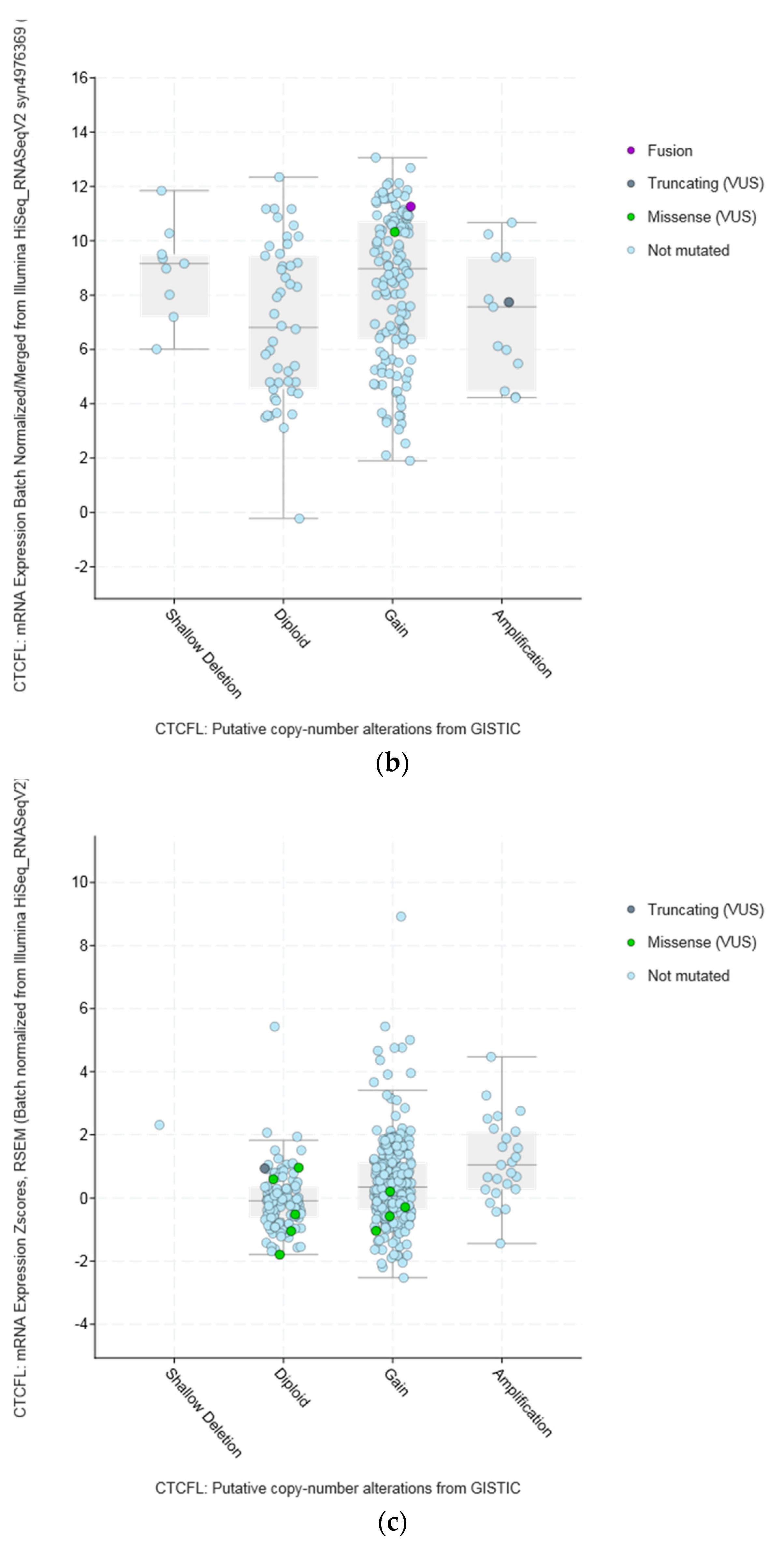
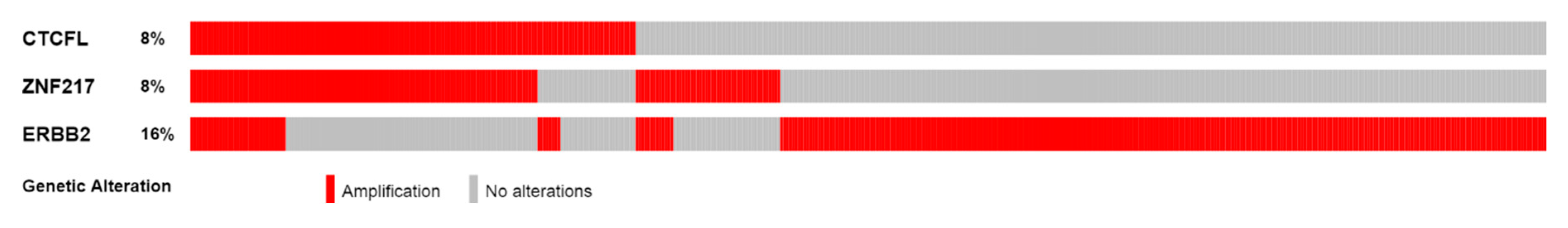
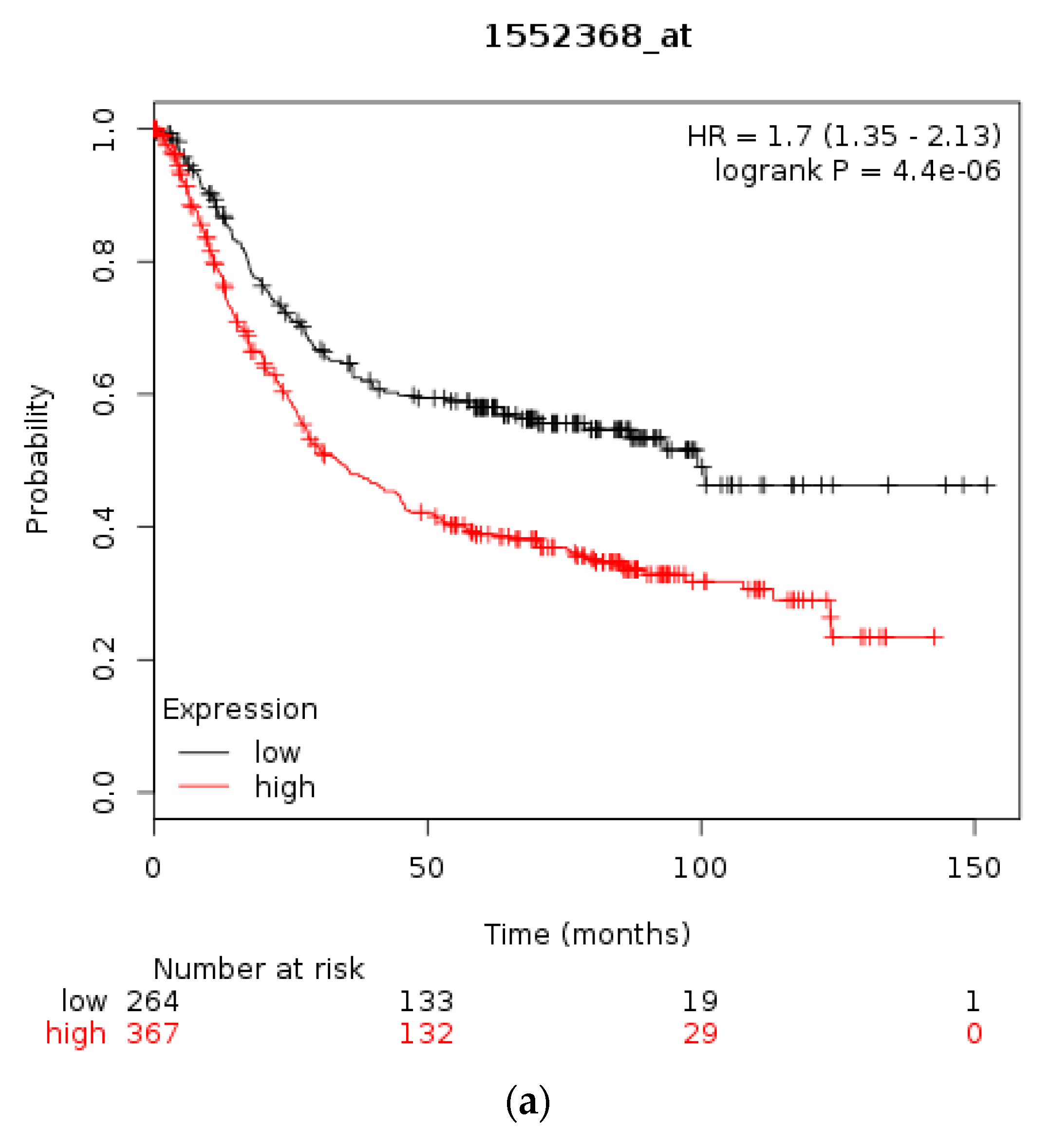
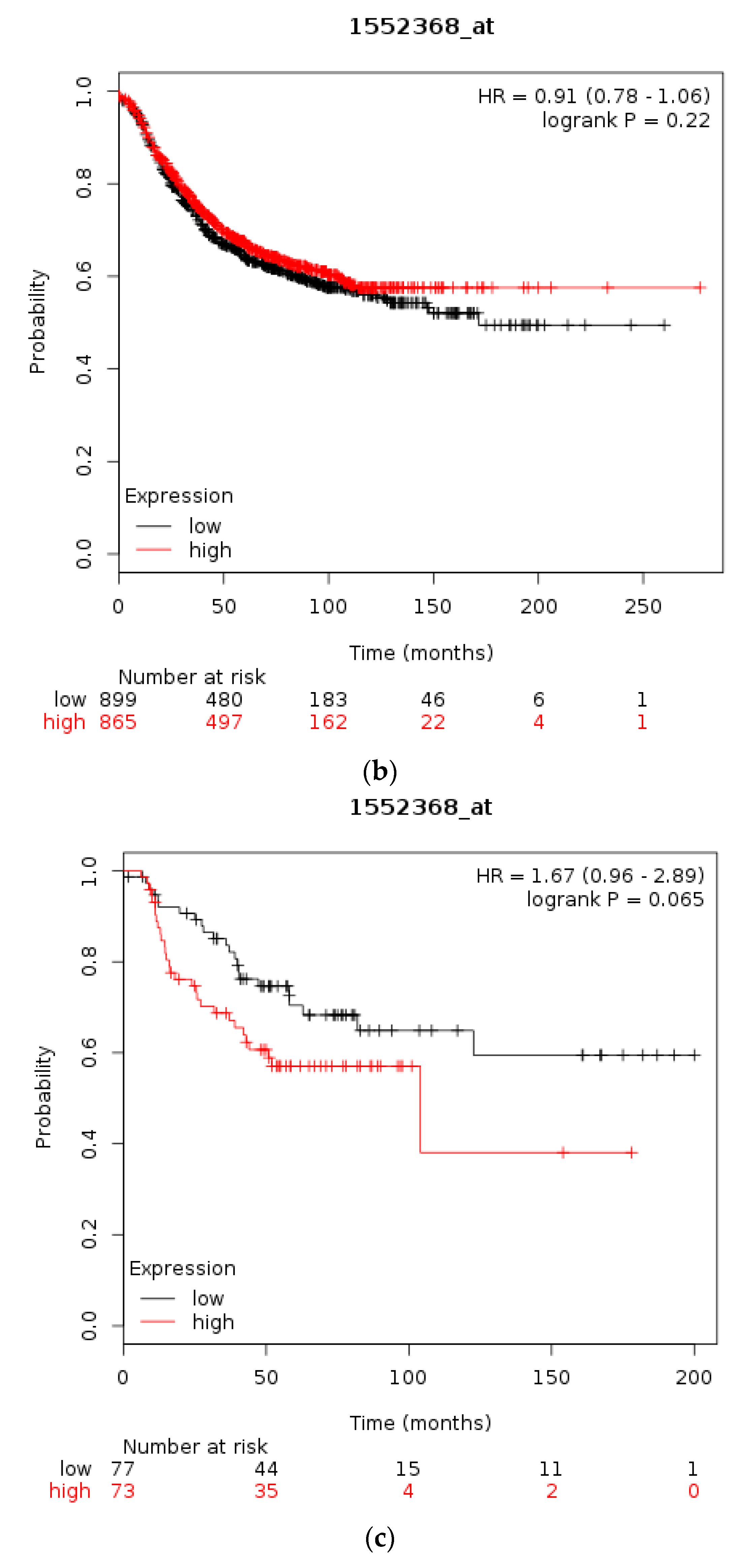
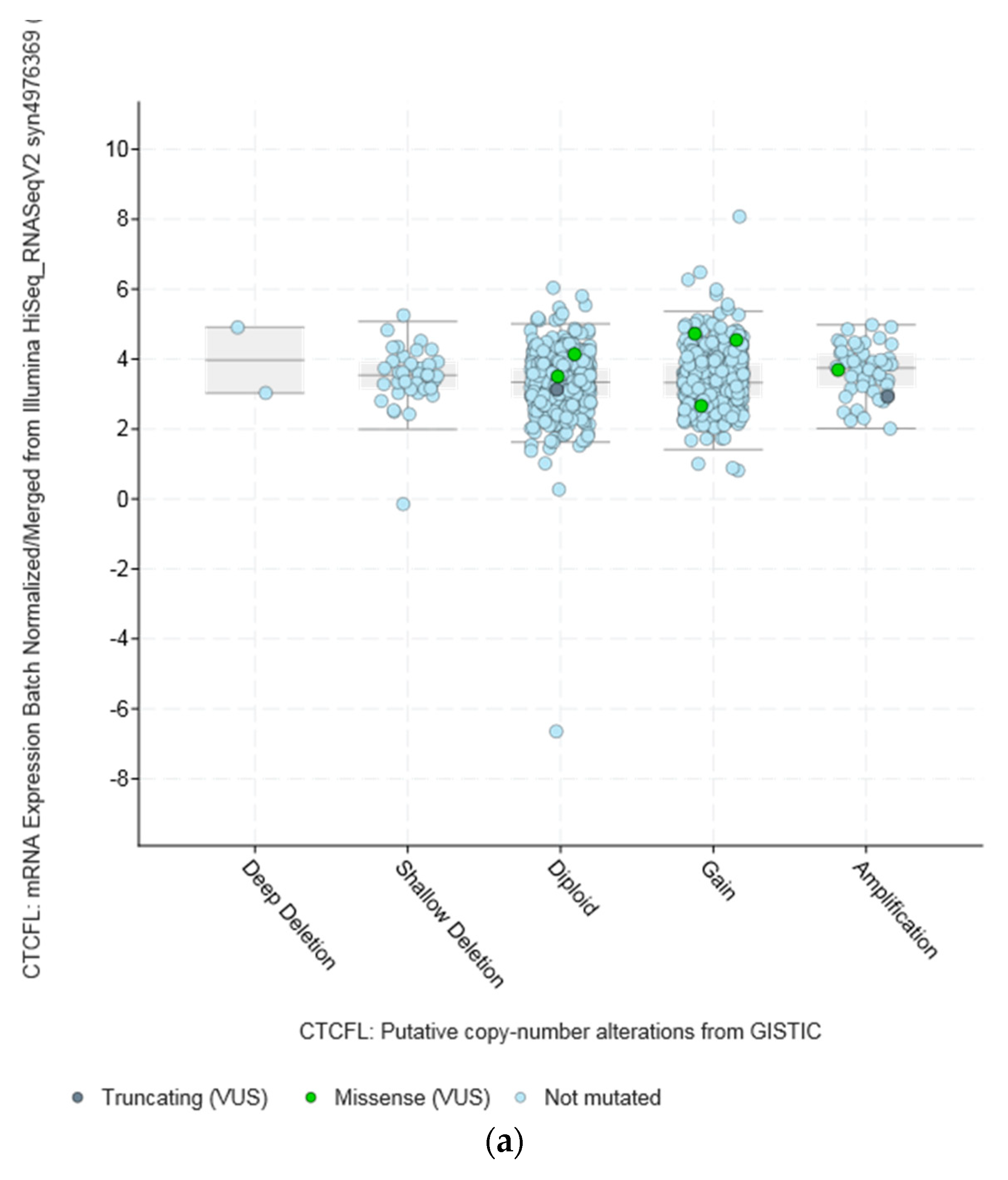
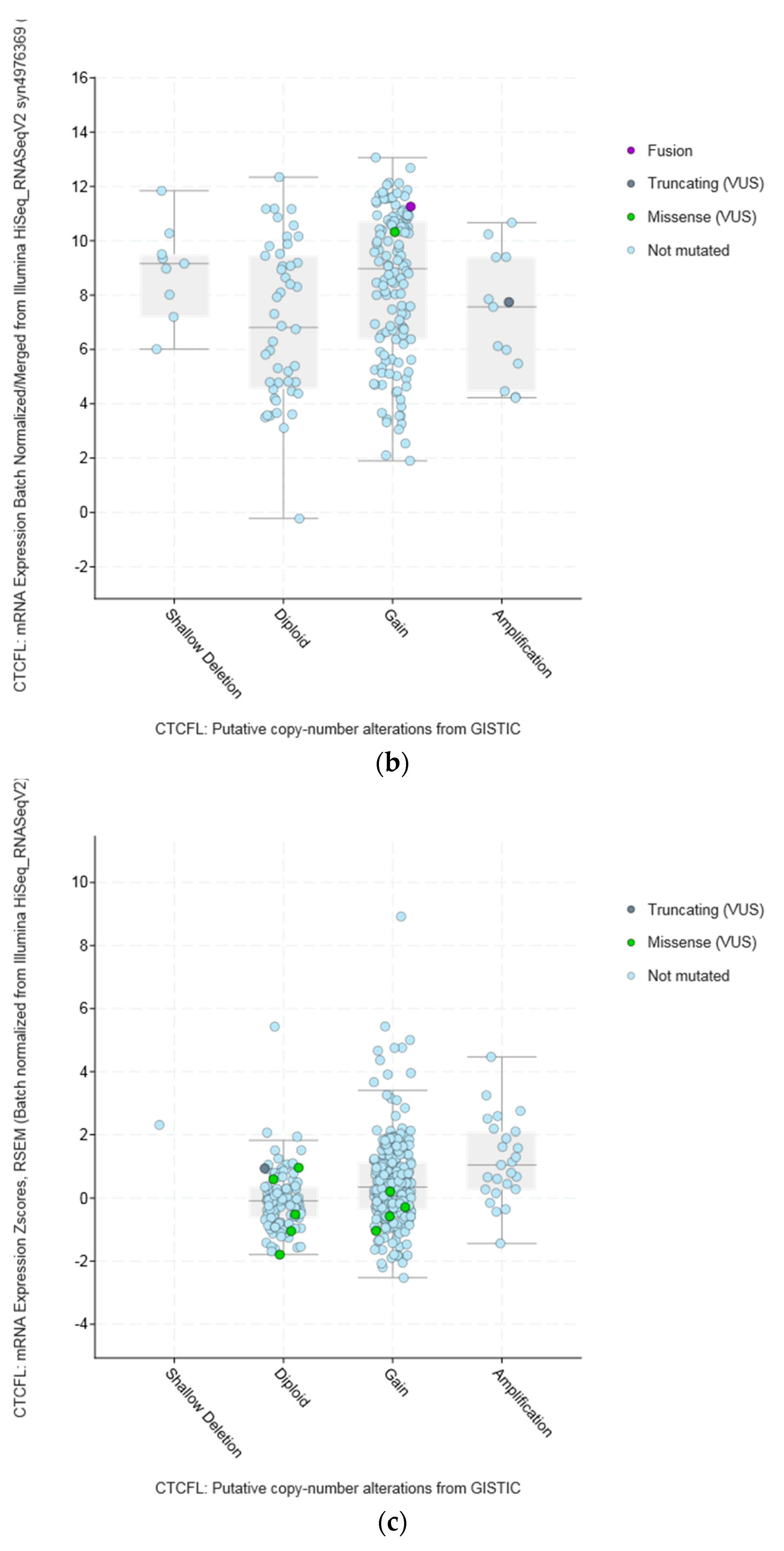
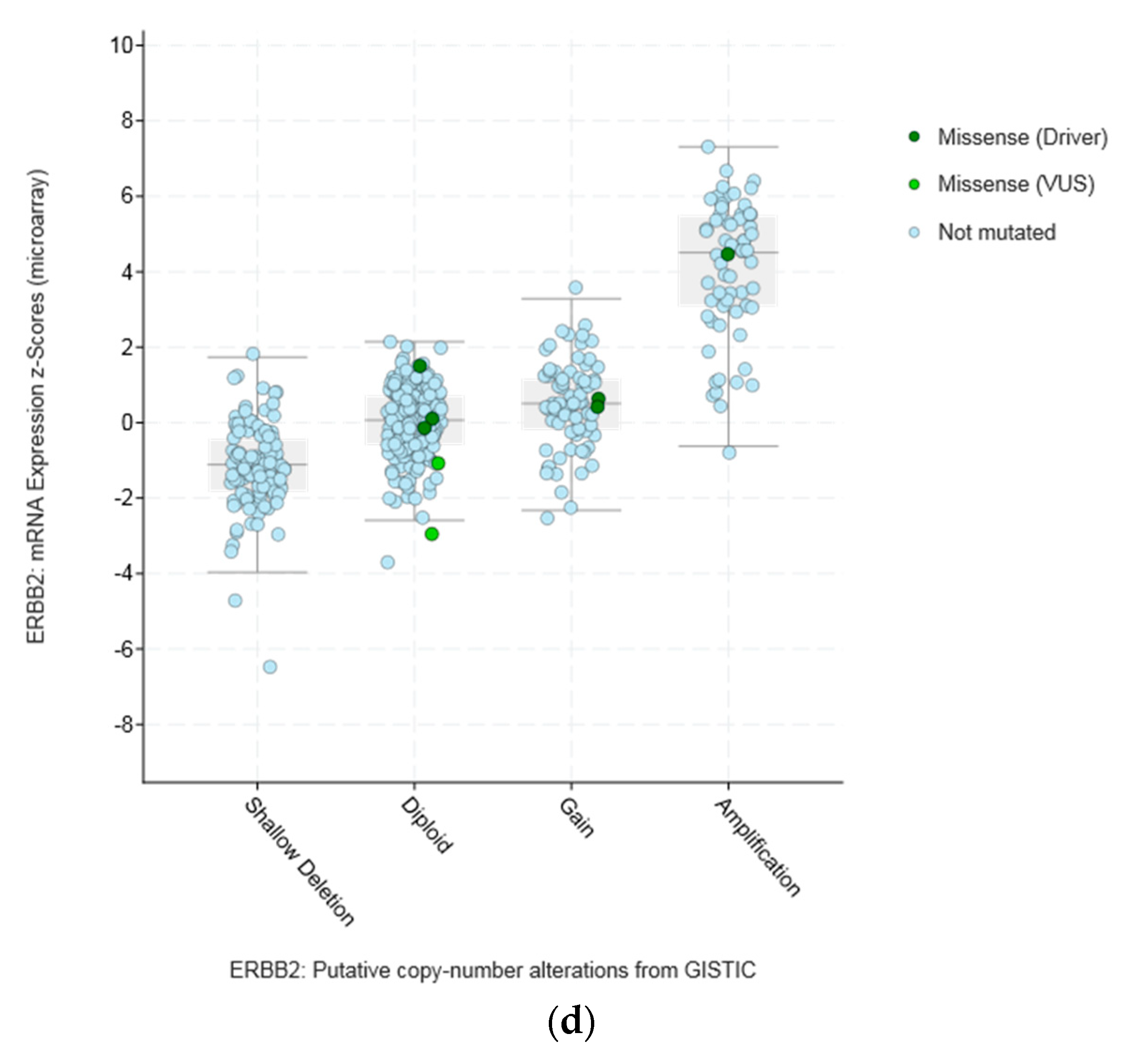
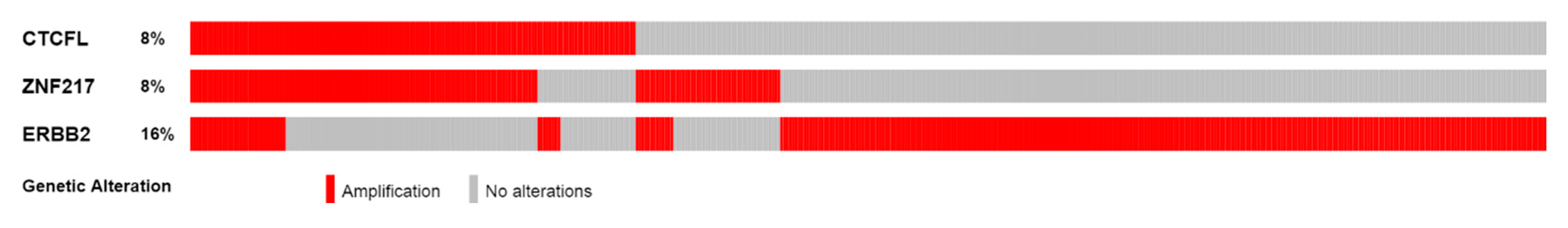
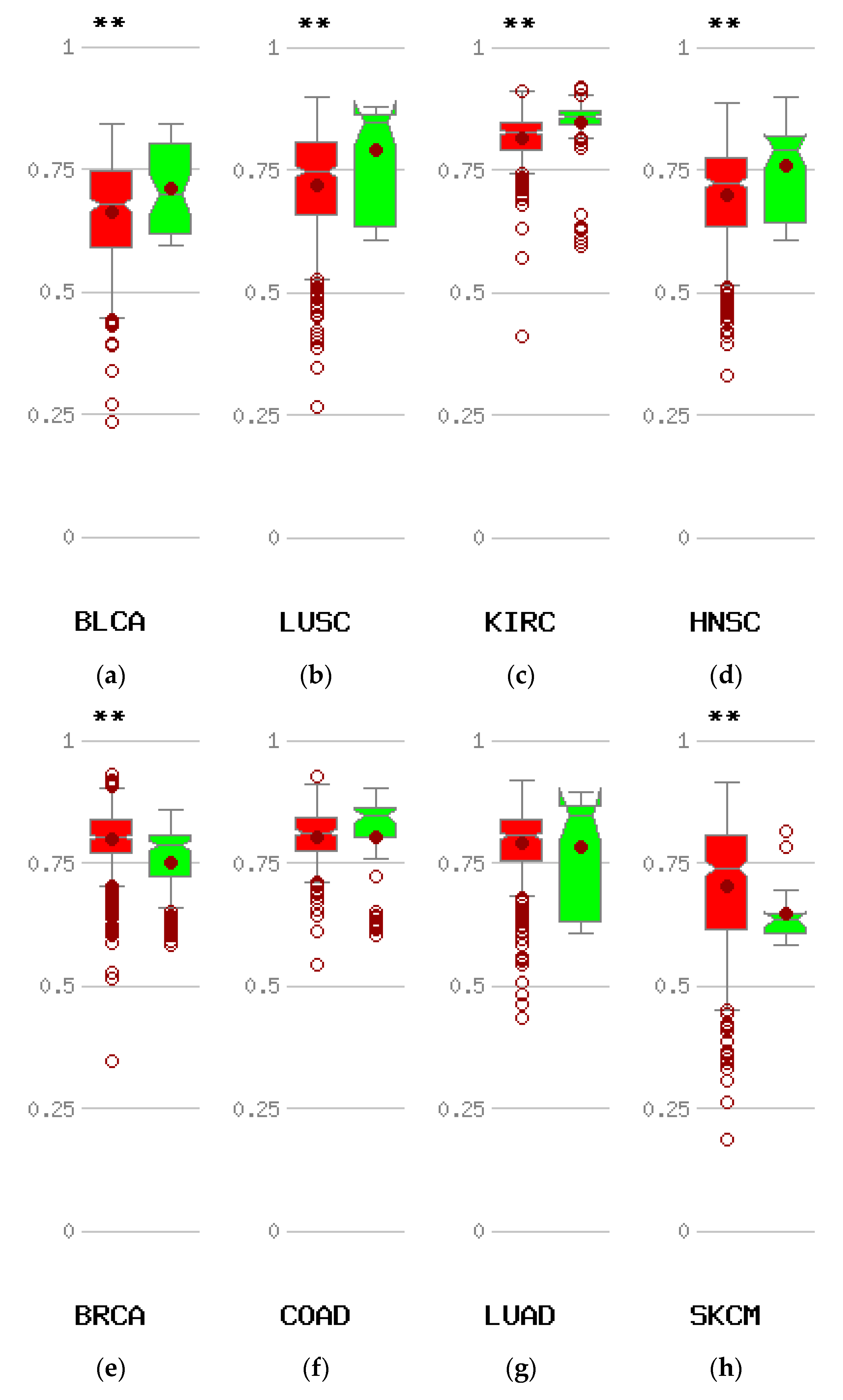
3.2. Lesions of CTCFL in Cancer
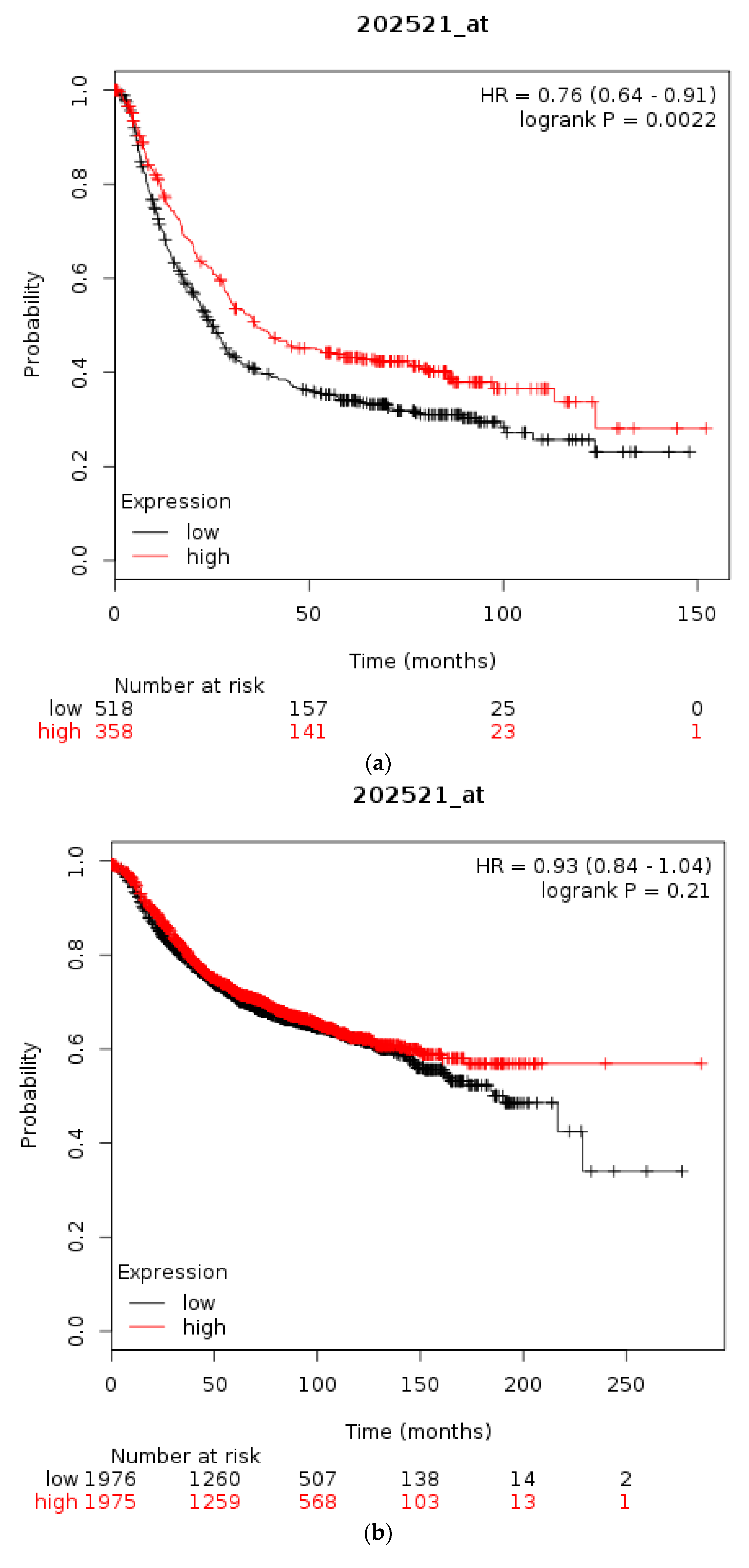
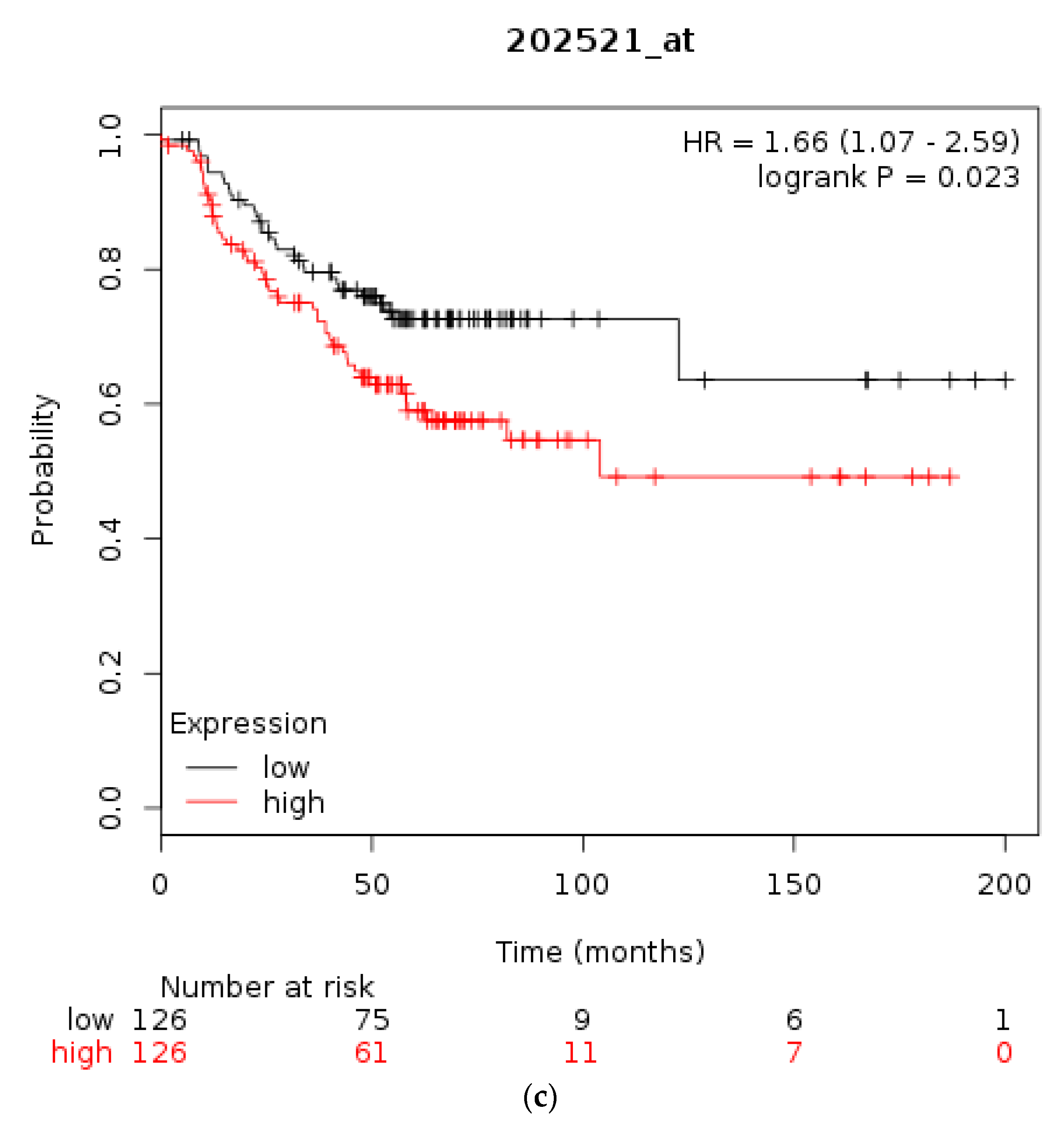
4. Discussion
Supplementary Materials
Funding
Conflicts of Interest
References
- Kaiser, V.B.; Semple, C.A. When TADs go bad: Chromatin structure and nuclear organisation in human disease. F1000 Res. 2017, 6, 314. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Day, D.S.; Young, R.A. Insulated neighborhoods: Structural and functional units of mammalian gene control. Cell 2016, 167, 1188–1200. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Gorkin, D.U.; Ren, B. Chromatin domains: The unit of chromosome organisation. Mol. Cell 2016, 62, 668–680. [Google Scholar] [CrossRef] [PubMed]
- Eukaryotic Promoter Database. Available online: https://epd.vital-it.ch (accessed on 7 July 2018).
- Wang, D.C.; Wang, W.; Zhang, L.; Wang, X. A tour of 3D genome with a focus on CTCF. Semin Cell Dev. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Tian, Y.; Shu, W.; Bo, X.; Wang, S. Comprehensive identification and annotation of cell type-specific and ubiquitous CTCF-binding sites in the human genome. PLoS ONE 2012, 7, e41374. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Abdullaev, Z.K.; Smith, A.D.; Ching, K.A.; Loukinov, D.I.; Green, R.D.; Zhang, M.Q.; Lobanenkov, V.V.; Ren, B. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell 2007, 128, 1231–1245. [Google Scholar] [CrossRef] [PubMed]
- Holwerda, S.J.B.; de Laat, W. CTCF: The protein, the binding partners, the binding sites and their chromatin loops. Phil. Trans. R. Soc. B 2013, 368, 20120369. [Google Scholar] [CrossRef] [PubMed]
- Martin-Kleiner, I. BORIS in human cancers—A review. Eur. J. Cancer 2012, 48, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.D.; Bailey, C.G.; Rasko, J.E.J. CTCF and BORIS in genome regulation and cancer. Curr. Opin. Genet. Dev. 2014, 24, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.; Cui, H.; Bisht, K.S.; Sun, L.; Patel, K.; Lee, R.S.; Kugoh, H.; Oshimura, M.; Feinberg, A.P.; Gius, D. CTCFL/BORIS is a methylation-independent DNA-binding protein that preferentially binds to the paternal H19 differentially methylated region. Cancer Res. 2008, 68, 5546–5551. [Google Scholar] [CrossRef] [PubMed]
- Maurano, M.T.; Wang, H.; John, S.; Shafer, A.; Canfield, T.; Lee, K.; Stamatoyannopoulos, J.A. Role of DNA methylation in modulating transcription factor occupancy. Cell Rep. 2015, 12, 1184–1195. [Google Scholar] [CrossRef] [PubMed]
- Van Tongelen, A.; Loriot, A.; De Smet, C. Oncogenic roles of DNA hypomethylation through the activation of cancer-germline genes. Cancer Lett. 2017, 396, 130–137. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, K.; Heger, P.; Sharma, R.; Wiehe, T. The diverging routes of BORIS and CTCF: An interactomic and phylogenomic analysis. Life 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Pugacheva, E.M.; Rivero-Hinojosa, S.; Espinoza, C.A.; Méndez-Catalá, F.; Kang, S.; Suzuki, T.; Kosaka-Suzuki, N.; Robinson, S.; Nagarajan, V.; Ye, Z.; et al. Comparative analyses of CTCF and BORIS occupancies uncover two distinct classes of CTCF binding genomic regions. Genome. Biol. 2015, 16, 161. [Google Scholar] [CrossRef] [PubMed]
- Bioportal for Cancer Genomics. Available online: http://www.cbioportal.org (accessed on 7 July 2018).
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio Cancer Genomics Portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell 2017, 171, 540–556. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014, 513, 202–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Network; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [PubMed] [Green Version]
- Curtis, C.; Shah, S.P.; Chin, S.-F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modeling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [PubMed]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012, 489, 519–525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013, 499, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed]
- Mutation Assessor. Available online: www.mutationassessor.org (accessed on 29 July 2018).
- Reva, B.; Antipin, Y.; Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 2011, 39, e118. [Google Scholar] [CrossRef] [PubMed]
- Kaplan Meier Plotter. Available online: www.kmplot.com (accessed on 14 July 2018).
- Szász, A.M.; Lánczky, A.; Nagy, Á.; Föster, S.; Hark, K.; Green, J.E.; Boussioutas, A.; Busuttil, R.; Szabó, A.; Győrffy, B. Cross-validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1065 patients. Oncotarget 2016, 7, 49322–49333. [Google Scholar] [CrossRef] [PubMed]
- MethHC A database of DNA Methylation and Gene Expression in Human Cancer. Available online: www.methhc.mbc.nctu.edu.tw (accessed on 3 July 2018).
- Huang, W.Y.; Hsu, S.D.; Huang, H.Y.; Sun, Y.M.; Chou, C.H.; Weng, S.L.; Huang, H.D. MethHC: A database of DNA methylation and gene expression in human cancer. Nucleic Acids Res. 2015, 43, D856–D861. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Matulonis, U.A. POLE mutations as an alternative pathway for Microsatellite Instability in endometrial cancer: Implications for Lynch syndrome testing. Cancer 2015, 121, 331–334. [Google Scholar] [CrossRef] [PubMed]
- Rebhandl, S.; Huemer, M.; Greil, R.; Geisberger, R. AID/APOBEC deaminases and cancer. Oncoscience 2015, 2, 320–333. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, S.; Rosenthal, R.; Kanu, N.; McGranahan, N.; Bartek, J.; Quezada, S.A.; Hare, J.; Harris, R.S.; Swanton, C. Perspective: APOBEC mutagenesis in drug resistance and immune escape in HIV and cancer evolution. Ann. Oncol. 2018, 29, 563–572. [Google Scholar] [CrossRef] [PubMed]
- Human Protein Atlas. Available online: www.proteinatlas.org (accessed on 10 July 2018).
- Lai, A.Y.; Fatemi, M.; Dhasarathy, A.; Malone, C.; Sobol, S.E.; Geigerman, C.; Jaye, D.L.; May, D.; Shah, R.; Li, L.; et al. DNA methylation prevents CTCF-mediated silencing of the oncogene BCL6 in B cell lymphomas. J. Exp. Med. 2010, 207, 1939–1950. [Google Scholar] [CrossRef] [PubMed]
- Witcher, M.; Emerson, B.M. Epigenetic silencing of the p16(INK4a) tumour suppressor is associated with loss of CTCF binding and a chromatin boundary. Mol. Cell 2009, 34, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Aitken, S.J.; Ibarra-Soria, X.; Kentepozidou, E.; Flicek, P.; Feig, C.; Marioni, J.C.; Odom, D.T. CTCF maintains regulatory homeostasis of cancer pathways. Genome. Biol. 2018, 19, 106. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.D.; Bailey, C.G.; Champ, K.; Vellozzi, M.; O’Young, P.; Metierre, C.; Feng, Y.; Thoeng, A.; Richards, M.; Schmitz, U.; et al. CTCF genetic alterations in endometrial carcinoma are pro-tumourigenic. Oncogene 2017, 36, 4100–4110. [Google Scholar] [CrossRef] [PubMed]
- Zighelboim, I.; Mutch, D.G.; Knapp, A.; Ding, L.; Xie, M.; Cohn, D.E.; Goodfellow, P.J. High frequency strand slippage mutations in CTCF in MSI-positive endometrial cancers. Human Mutat. 2014, 35, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Voutsadakis, I.A. Polymerase epsilon mutations and concomitant β2-microglobulin mutations in cancer. Gene 2018, 647, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nature Genet. 2012, 45, 136–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, F.; Li, X.; Zheng, W.; Li, Z.; Lu, D.; Chen, G.; Gong, D.; Yang, L.; Fu, J.; Shi, P.; et al. CTCF prevents genomic instability by promoting homologous recombination-directed DNA double-strand break repair. Proc. Natl. Acad. Sci. USA 2017, 114, 10912–10917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilmi, K.; Jangal, M.; Marques, M.; Zhao, T.; Saad, A.; Zhang, C.; Luo, V.M.; Syme, A.; Rejon, C.; Yu, Z.; et al. CTCF facilitates DNA double-strand break repair by enhancing homologous recombination repair. Sci. Adv. 2017, 3, e1601898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.A.; Chang, M.M.; Huang, W.; Ooi, W.F.; Xing, M.; Tan, P.; Skanderup, A.J. Mutation hotspots at CTCF binding sites coupled to chromosomal instability in gastrointestinal cancers. Nat. Commun. 2018, 9, 1520. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.M.; Glazer, C.A.; Mithani, S.K.; Ochs, M.F.; Sun, W.; Bhan, S.; Vostrov, A.; Abdullaev, Z.; Lobanenkov, V.; Gray, A.; et al. Coordinated activation of candidate proto-oncogenes and cancer testes antigens via promoter demethylation in head and neck cancer and lung cancer. PLoS ONE 2009, 4, e4961. [Google Scholar] [CrossRef] [PubMed]
- Hines, W.C.; Bazarov, A.V.; Mukhopadhyay, R.; Yaswen, R. BORIS (CTCFL) is not expressed in most human breast cell lines and high grade breast carcinomas. PLoS ONE 2010, 5, e9738. [Google Scholar] [CrossRef] [PubMed]
- D’Arcy, V.; Pore, N.; Docquier, F.; Abdullaev, Z.K.; Chernukhin, I.; Kita, G.X.; Rai, S.; Smart, M.; Farrar, D.; Pack, S.; et al. BORIS, a paralogue of the transcription factor, CTCF, is aberrantly expressed in breast tumours. Br. J. Cancer 2008, 98, 571–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Cancer (Reference) | All Lesions | Amplifications | Deep Deletions | mRNA Upregulation | mRNA Downregulation | Mutations | Multiple Lesions |
|---|---|---|---|---|---|---|---|
| Cell line encyclopedia [23] | 57/877 (6.5%) | 13 (1.48%) | 6 (0.68%) | 24 (2.74%) | 14 (1.6%) | - | - |
| Uterine endometrial TCGA [23] | 38/102 (37.25%) | - | 1 (0.98%) | 2 (1.96%) | 3 (2.94%) | 28 (27.45%) | 4 (3.92%) |
| Ovarian serous TCGA [31] | 33/200 (16.5%) | - | 2 (1%) | 3 (1.5%) | 23 (11.5%) | 1 (0.5%) | 4 (2%), two fusions |
| Bladder TCGA [20] | 55/404 (13.61%) | 3 (0.74%) | 1 (0.25%) | 25 (6.19%) | 8 (1.98%) | 13 (3.22%) | 5 (1.24%) |
| Colorectal TCGA [21] | 30/267 (11.24%) | - | - | 15 (5.62%) | 2 (0.75%) | 13 (4.87%) | - |
| Prostate TCGA [32] | 53/491 (10.79%) | - | 14 (2.85%) | 12 (2.44%) | 22 (4.48%) | 3 (0.61%) | 2 (0.41%) |
| Melanoma TCGA PanCancer (Provis.) | 36/363 (9.92%) | - | - | 14 (3.86) | 11 (3.03%) | 7 (1.93%) | 4 (1.1%) |
| Lung adenocarcinoma TCGA [28] | 40/503 (7.95%) | - | 1 (0.2%) | 21 (4.17%) | 12 (2.39%) | 5 (0.99%) | 1 (0.2%) |
| Lung squamous TCGA [29] | 34/466 (7.3) | - | - | 19 (4.08%) | 9 (1.93%) | 4 (0.86%) | 2 (0.42%), one fusion |
| Pancreatic TCGA PanCancer (Provis.) | 12/168 (7.14%) | - | - | 6 (3.57%) | 5 (2.98%) | 1 (0.6%) | - |
| TGCTs TCGA PanCancer (Provis.) | 10/144 (6.94%) | - | - | 6 (4.17%) | 3 (2.08%) | - | 1 (0.69%) |
| HCC TCGA PanCancer (Provis.) | 20/345 (5.8%) | 1 (0.29%) | - | 6 (1.74%) | 9 (2.61%) | 4 (1.16%) | - |
| RCC TCGA [30] | 20/352 (5.68%) | - | - | 6 (1.7%) | 13 (3.69%) | 1 (0.28%) | - |
| Uterine serous TCGA [23] | 3/53 (5.66%) | - | - | 2 (3.77%) | 1 (1.89%) | - | - |
| GBM TCGA [27] | 5/142 (3.52%) | - | - | 3 (2.11%) | 2 (1.41%) | - | - |
| Breast TCGA [26] | 26/816 (3.19%) | 3 (0.37%) | 5 (0.61%) | - | - | 18 (2.21%) | - |
| Gastric adenocarcinoma TCGA [22] | 4/188 (2.13%) | 1 (0.53%) | 1 (0.53%) | NA | NA | 2 (1.06%) | - |
| Oesophageal adenocarcinoma TCGA [22] | 1/77 (1.3%) | 1 (1.3%) | - | NA | NA | - | - |
| All (Not cell lines) | 420/5081 (8.27%) | 9/5081 (0.18%) | 25/5081 (0.49%) | 140/4816 (2.9%) | 123/4816 (2.55%) | 100/5081 (1.97%) | 22/5081 (0.43%) |
| Number of Sample | Protein Change | Domain | Mutation Type | Allele Frequency | Number of Mutations in Sample | Mutations in MSI-Associated Genes | Mutations in POLE/POLD1 Genes |
|---|---|---|---|---|---|---|---|
| 1 | G48Vfs*14 | N | FS del | 0.37 | 774 | No | No |
| 2 | G48Vfs*14 | N | FS del | 0.28 | 716 | MSH6 | No |
| 3 | A88V | N | Missense | 0.17 | 5737 | MSH2, MSH6, MLH1, PMS2 | POLE, POLD1 |
| 4 | G173 * | N | Nonsense | 0.25 | 7390 | MSH2, MSH6, PMS2 | POLE |
| 5 | Q180 * | N | Nonsense | 0.4 | 594 | MSH6 | No |
| 6 | E182Gfs*9 | N | FS ins | 0.33 | 9662 | MSH2, PMS2 | POLE |
| 7 | T204Qfs*18 | N | FS del | 0.19 | 597 | MLH1 | No |
| 8 | T204Nfs*26 | N | FS ins | 0.26 | 538 | MSH6, PMS2 | No |
| 9 | T204Nfs*26 | N | FS ins | 0.35 | 323 | No | No |
| 10 | T204Nfs*26 | N | FS ins | 0.5 | 864 | PMS2 | POLE |
| 11 | T204Nfs*26 | N | FS ins | 0.31 | 451 | No | No |
| 12 | T204Nfs*26 | N | FS ins | 0.25 | 56 | No | No |
| 13 | R213C | N | Missense | 0.33 | 12218 | MSH6, MLH1, PMS2 | POLE, POLD1 |
| 14 | D222Mfs*28 | N | FS del | 0.32 | 218 | No | No |
| 15 | G261C | N | Missense | 0.29 | 451 | No | No |
| 16 | Q267P | ZF | Missense | 0.32 | 13840 | MSH2, PMS2, PMS1 | POLE, POLD1 |
| 17 | H312N | ZF | Missense | 0.39 | 3190 | MSH6, PMS2 | POLE |
| 18 | T317Rfs*91 | ZF | FS del | 0.5 | 669 | PMS2 | No |
| 19 | G318Qfs*16 | ZF | FS ins | 0.22 | 562 | No | No |
| 20 | C324 * | ZF | Nonsense | 0.32 | 611 | No | POLE |
| 21 | R341H | ZF | Missense | 0.45 | 9662 | MSH2, PMS2 | POLE |
| 22 | R342C | ZF | Missense | 0.4 | 9440 | MSH2, MSH6, PMS1 | POLE |
| 23 | Y343C | ZF | Missense | 0.34 | 7644 | MSH6, MLH1 | POLE |
| 24 | H369R | ZF | Missense | 0.17 | 1326 | No | POLE |
| 25 | R377C | ZF | Missense | 0.34 | 1307 | MSH6 | POLE, POLD1 |
| 26 | P378L | ZF | Missense | 0.2 | 13840 | MSH2, PMS2 | POLE, POLD1 |
| 27 | L394del | ZF | IF del | 0.09 | 716 | MSH6 | No |
| 28 | G420C | ZF | Missense | 0.26 | 1307 | MSH6 | POLE, POLD1 |
| 29 | E432Gfs*10 | ZF | FS del | 0.38 | 41 | No | No |
| 30 | A447Vfs*61 | ZF | FS del | 0.45 | 4346 | PMS1, MLH1 | POLE, POLD1 |
| 31 | R448 * | ZF | Nonsense | 0.46 | 81 | No | No |
| 32 | K449T | ZF | Missense | 0.34 | 8511 | MSH2, MLH1, PMS1 | POLE |
| 33 | S450Kfs*2 | ZF | FS ins | 0.26 | 4096 | MSH2, MSH6 | POLE |
| 34 | X453_splice | ZF | Splice | 0.28 | 58 | No | No |
| 35 | R457 * | ZF | Nonsense | 0.37 | 10061 | MSH6, MSH2, PMS2 | POLE, POLD1 |
| 36 | Q499 * | ZF | Nonsense | 0.35 | 3925 | MSH6 | POLE |
| 37 | R533H | ZF | Missense | 0.47 | 611 | No | POLE |
| 38 | R566H | ZF | Missense | 0.2 | 884 | MSH2, MSH6 | POLD1 |
| 39 | E631 * | C | Nonsense | 0.41 | 3387 | MSH6, MLH1 | POLE |
| Number | Protein Change | Domain | Mutation Type | Allele Frequency | Number of Mutations in Sample | Mutations MSI | POLE/POLD1 Mutations | APOBEC Mutations |
|---|---|---|---|---|---|---|---|---|
| 1 | R29W | N | Missense | 0.44 | 170 | No | No | No |
| 2 | E182Gfs*9 | N | FS ins | 0.29 | 1858 | No | POLD1 | APOBEC2 |
| 3 | D194Rfs*36 | N | FS ins | 0.25 | 814 | No | No | APOBEC4 |
| 4 | T204Qfs*18 | N | FS del | 0.1 | 530 | MLH1, PMS1 | No | APOBEC4 |
| 5 | T204Nfs*26, D194_A201delinsTQTIS | N | FS ins, Missense | 0.11, 0.17 | 1917 | PMS1 | POLE | APOBEC1 |
| 6 | K260 * | N | Nonsense | 0.25 | 181 | No | No | APOBEC3B |
| 7 | R278C | ZF | Missense | 0.24 | 1002 | MSH2 | POLE, POLD1 | APOBEC3C |
| 8 | R368C | ZF | Missense | 0.58 | 171 | No | No | No |
| 9 | R377C | ZF | Missense | 0.41 | 2139 | MSH6, PMS2 | POLE, POLD1 | APOBEC4, APOBEC3C |
| 10 | E464K | ZF | Missense | 0.28 | 1107 | No | POLD1 | APOBEC3A |
| 11 | G519R | ZF | Missense | 0.18 | 662 | No | POLD1 | No |
| 12 | A524T | ZF | Missense | 0.29 | 4195 | MSH6 | POLE, POLD1 | APOBEC3G |
| 13 | E691Sfs*30 | C | FS del | 0.33 | 1057 | No | POLD1 | APOBEC3C |
| Number | Protein Change | Domain | Mutation Type | Number of Mutations in Sample | Mutations in MSI | Mutations in POLE/POLD1 |
|---|---|---|---|---|---|---|
| 1 | E104 *, E145Q, K264N | N | Nonsense | 580 | No | No |
| 2 | Q117 * | N | Nonsense | 58 | No | No |
| 3 | D290N | ZF | Missense | 291 | No | No |
| 4 | H322Y | ZF | Missense | 588 | No | No |
| 5 | T346N | ZF | Missense | 881 | No | No |
| 6 | F351L | ZF | Missense | 508 | No | No |
| 7 | S354F | ZF | Missense | 182 | No | No |
| 8 | S354Y | ZF | Missense | 124 | No | No |
| 9 | G375A | ZF | Missense | 857 | MSH2 | No |
| 10 | E376 * | ZF | Nonsense | 133 | No | No |
| 11 | S388N | ZF | Missense | 283 | No | No |
| 12 | E631 * | C | Nonsense | 3545 | MSH2, MLH1 | POLE |
| 13 | E687 * | C | Nonsense | 766 | MSH6 | No |
| Type of Cancer (Reference) | All Lesions | Amplifications | Deep Deletions | mRNA Upregulation | mRNA Downregulation | Mutations | Multiple Lesions |
|---|---|---|---|---|---|---|---|
| Cell line encyclopedia [25] | 100/877 (11.4%) | 79 (9.01%) | - | 19 (2.17%) | - | - | 2 (0.23%) |
| Uterine serous TCGA [23] | 17/53 (32.08%) | 4 (7.55%) | - | 13 (24.53%) | - | - | - |
| Ovarian serous TCGA [31] | 37/200 (18.5%) | 12 (6%) | - | 22 (11%) | - | 1 (0.5%) | 2 (1%) 1 fusion |
| Gastric adenocarcinoma TCGA [22] | 26/188 (13.83%) | 24 (12.77%) | - | NA | NA | 2 (1.06%) | - |
| Melanoma TCGA PanCancer (Provisional) | 49/363 (13.5%) | 4 (1.1%) | - | 17 (4.68%) | - | 27 (7.44%) | 1 (0.28%) |
| Colorectal TCGA [21] | 33/267 (12.36%) | 9 (3.37%) | - | 13 (4.87%) | - | 5 (1.87%) | 6 (2.25%) |
| Uterine endometrial [23] | 12/102 (11.76%) | - | - | 3 (2.94%) | - | 7 (6.86%) | 2 (1.96%) |
| Lung adenocarcinoma TCGA [28] | 54/503 (10.74%) | 19 (3.78%) | 1 (0.2%) | 13 (2.58%) | - | 19 (3.78%) | 2 (0.4%) |
| Esophageal adenocarcinoma TCGA [22] | 7/77 (9.09%) | 7/77 (9.09%) | - | NA | NA | - | - |
| Breast TCGA [26] | 62/816 (7.6%) | 56 (6.86%) | 2 (0.25%) | - | - | 3 (0.37%) | - |
| Lung squamous TCGA [29] | 33/466 (7.08%) | 1 (0.21%) | 1 (0.2%) | 22 (4.72%) | - | 8 (1.72%) | 1 (0.2%) |
| Prostate TCGA [32] | 32/491 (6.52%) | 4 (0.81%) | 1 (0.2%) | 13 (2.65%) | 12 (2.44%) | 2 (0.41%) | - |
| TGCTs TCGA PanCancer [Provisional] | 9/144 (6.25%) | - | - | 9 (6.25%) | - | - | - |
| Pancreatic TCGA PanCancer (Provisional) | 10/168 (5.95%) | 1 (0.6%) | - | 6 (3.57%) | 1 (0.6%) | - | 2 (1.19%) |
| HCC TCGA PanCancer (Provisional) | 19/345 (5.51%) | 5 (1.45%) | - | 12 (3.48%) | - | 2 (0.58%) | - |
| GBM TCGA [27] | 6/142 (4.23%) | - | - | 6/142 (4.23%) | - | - | - |
| RCC TCGA [30] | 10/352 (2.84%) | - | - | 8 (2.27%) | 1 (0.28%) | 1 (0.28%) | - |
| Bladder TCGA [20] | 10/404 (2.48%) | 4 (0.99%) | - | 3 (0.74%) | - | 3 (0.74%) | - |
| All (Not lines) | 426/5081 (8.38%) | 150/5081 (2.95%) | 5/5081 (0.1%) | 160/4816 (3.3%) | 14/4816 (0.29%) | 80/5081 (1.57%) | 16/5081 (0.31%) |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voutsadakis, I.A. Molecular Lesions of Insulator CTCF and Its Paralogue CTCFL (BORIS) in Cancer: An Analysis from Published Genomic Studies. High-Throughput 2018, 7, 30. https://doi.org/10.3390/ht7040030
Voutsadakis IA. Molecular Lesions of Insulator CTCF and Its Paralogue CTCFL (BORIS) in Cancer: An Analysis from Published Genomic Studies. High-Throughput. 2018; 7(4):30. https://doi.org/10.3390/ht7040030
Chicago/Turabian StyleVoutsadakis, Ioannis A. 2018. "Molecular Lesions of Insulator CTCF and Its Paralogue CTCFL (BORIS) in Cancer: An Analysis from Published Genomic Studies" High-Throughput 7, no. 4: 30. https://doi.org/10.3390/ht7040030
APA StyleVoutsadakis, I. A. (2018). Molecular Lesions of Insulator CTCF and Its Paralogue CTCFL (BORIS) in Cancer: An Analysis from Published Genomic Studies. High-Throughput, 7(4), 30. https://doi.org/10.3390/ht7040030