Current High-Throughput Approaches of Screening Modulatory Effects of Xenobiotics on Cytochrome P450 (CYP) Enzymes

Abstract
:1. Introduction
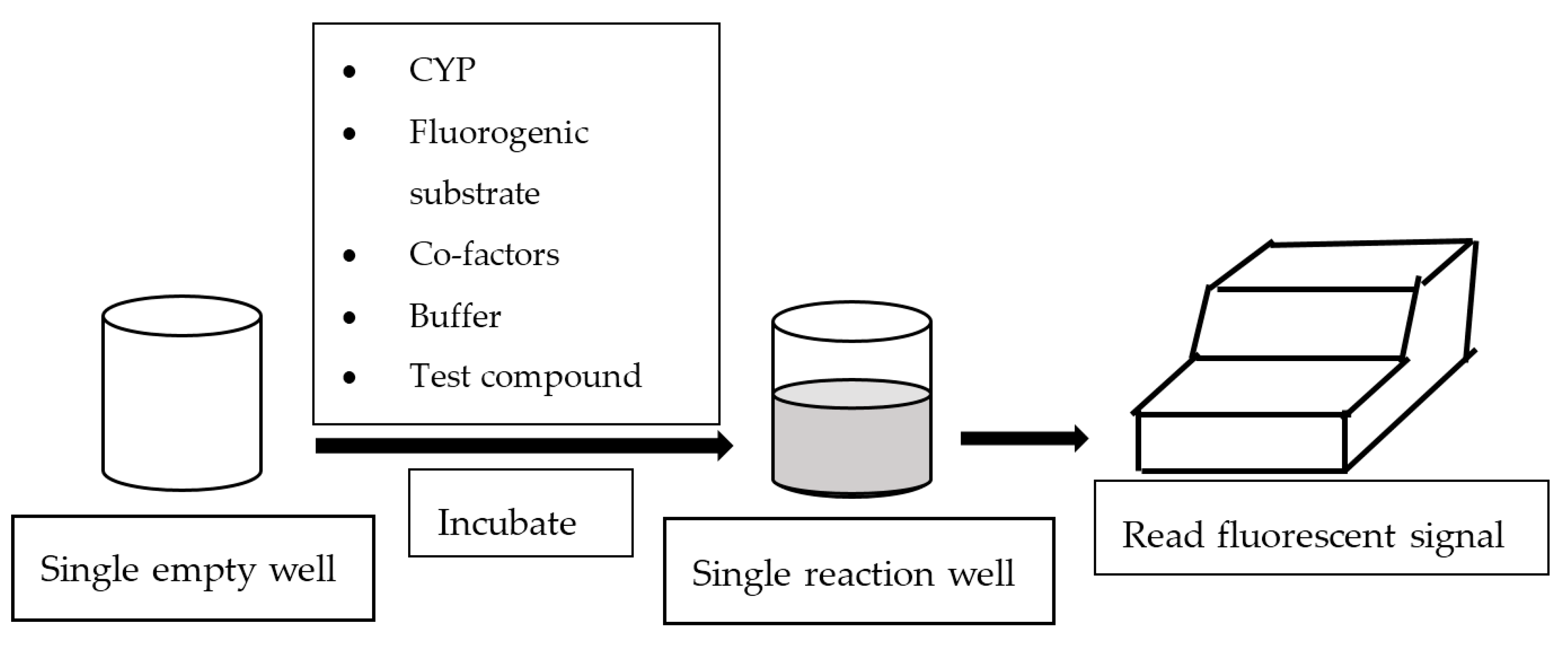
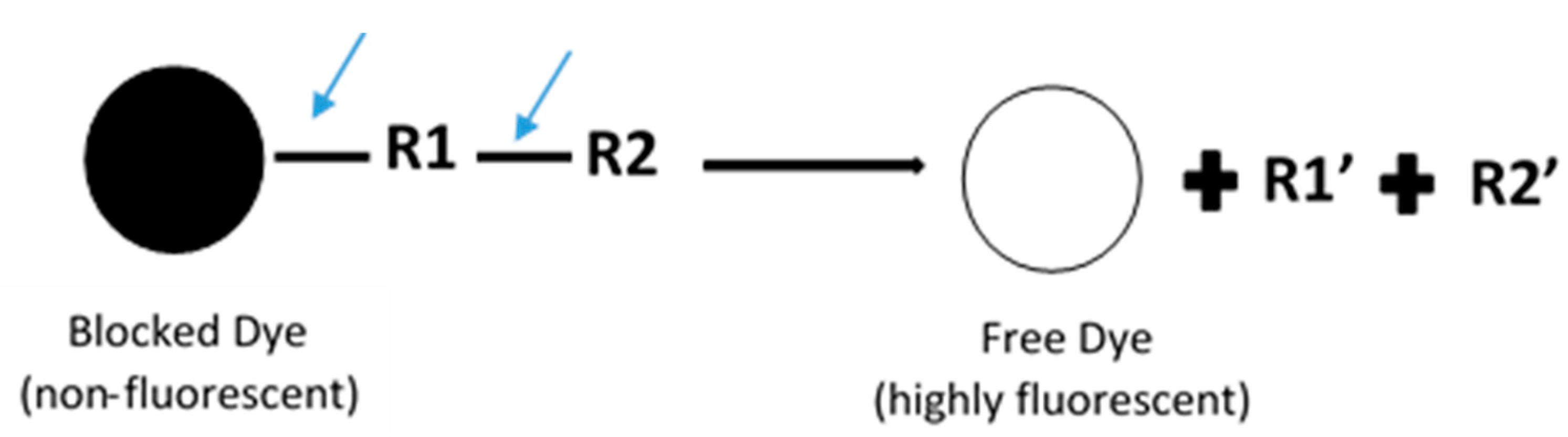
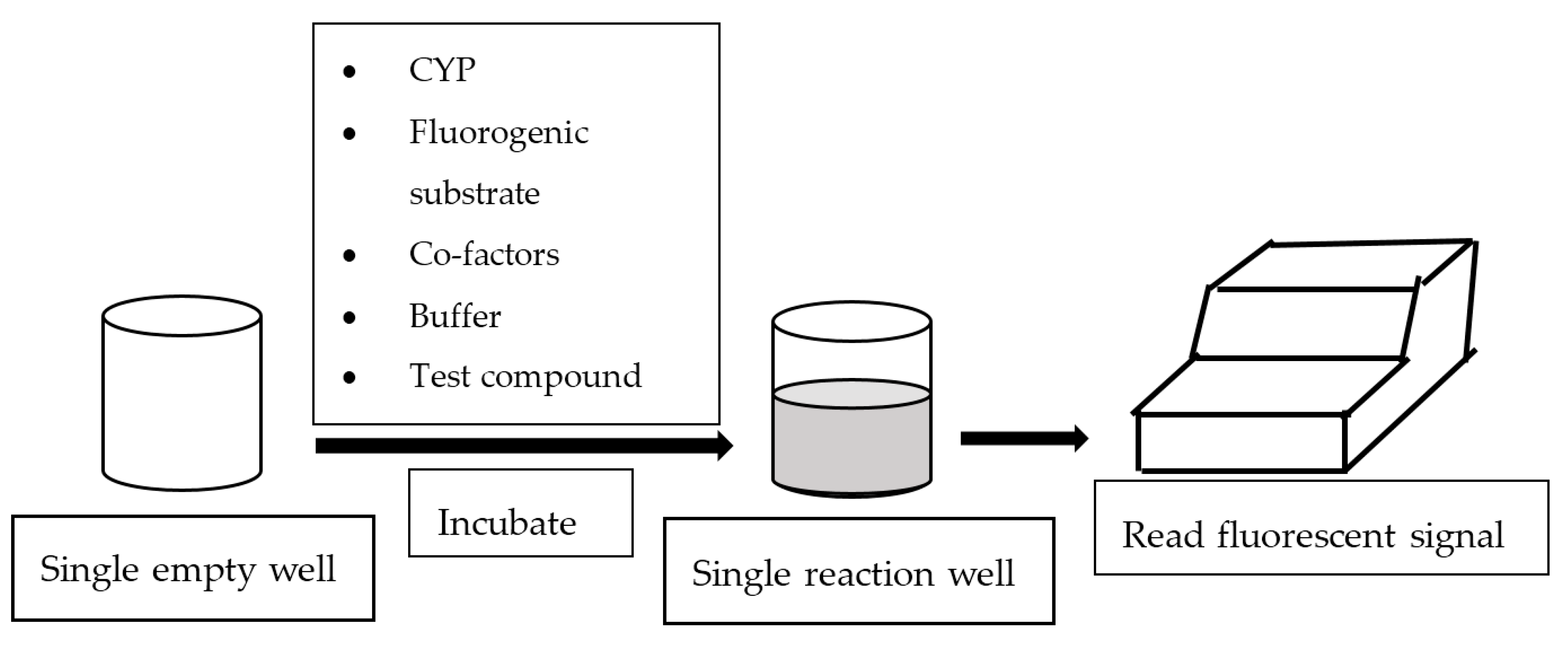
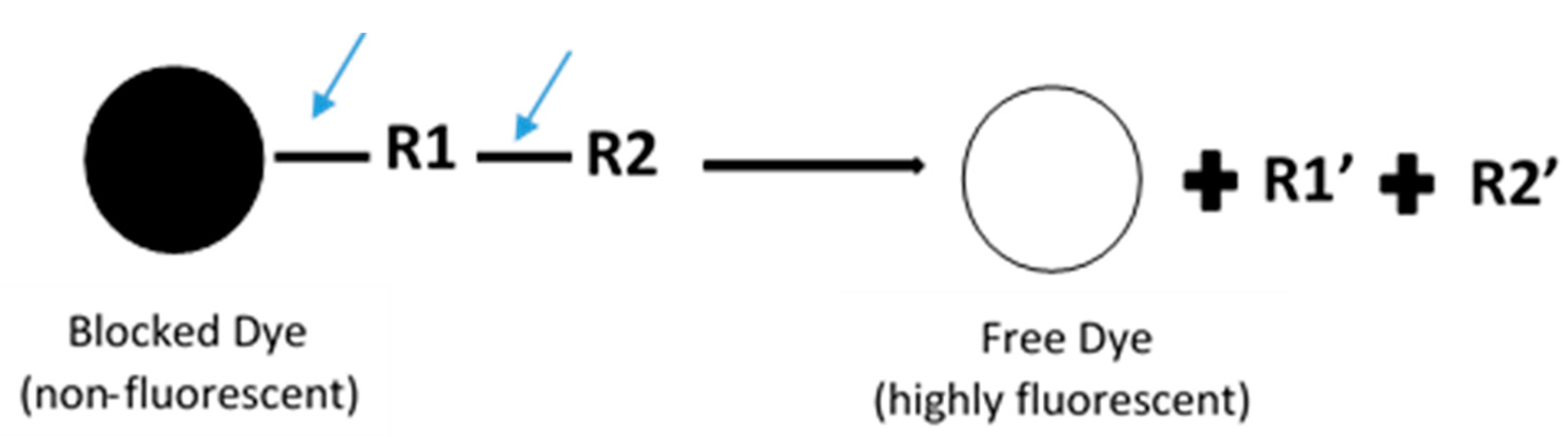
2. Fluorescence-Based Assay
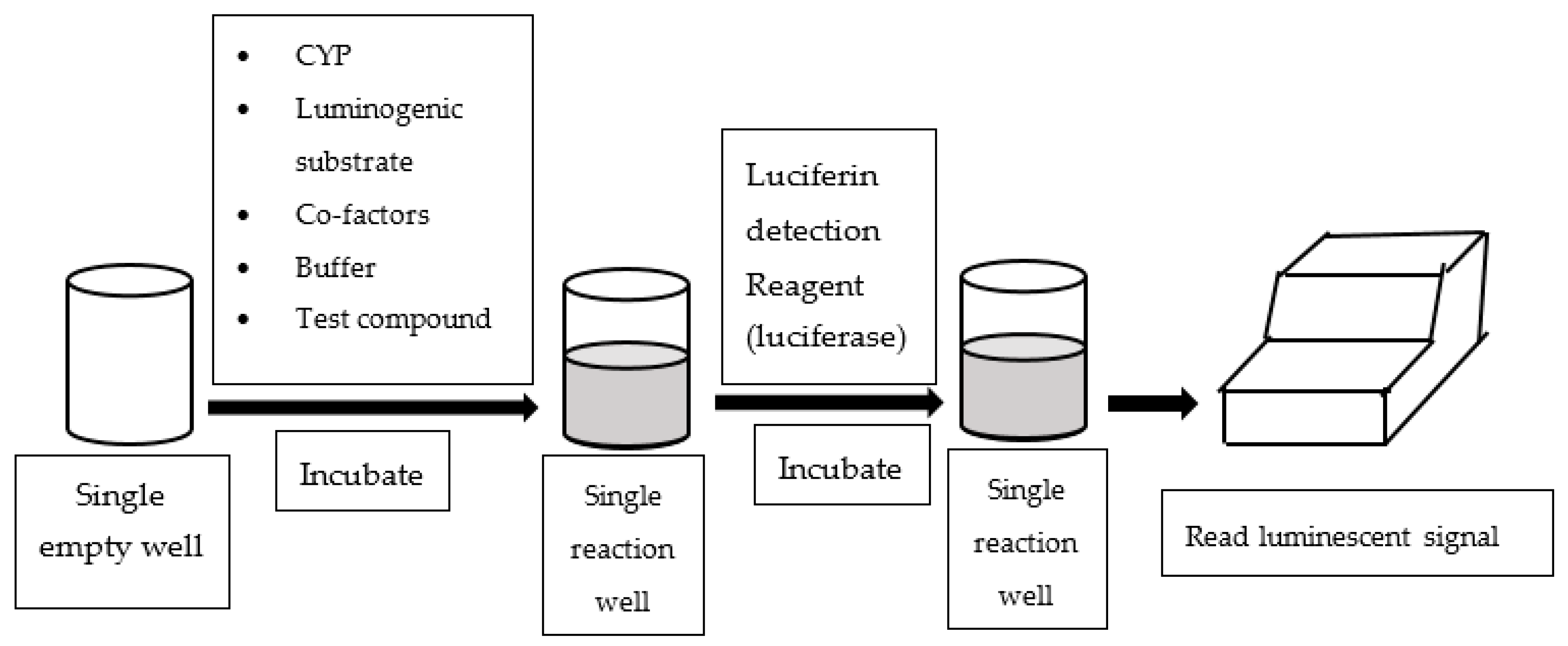
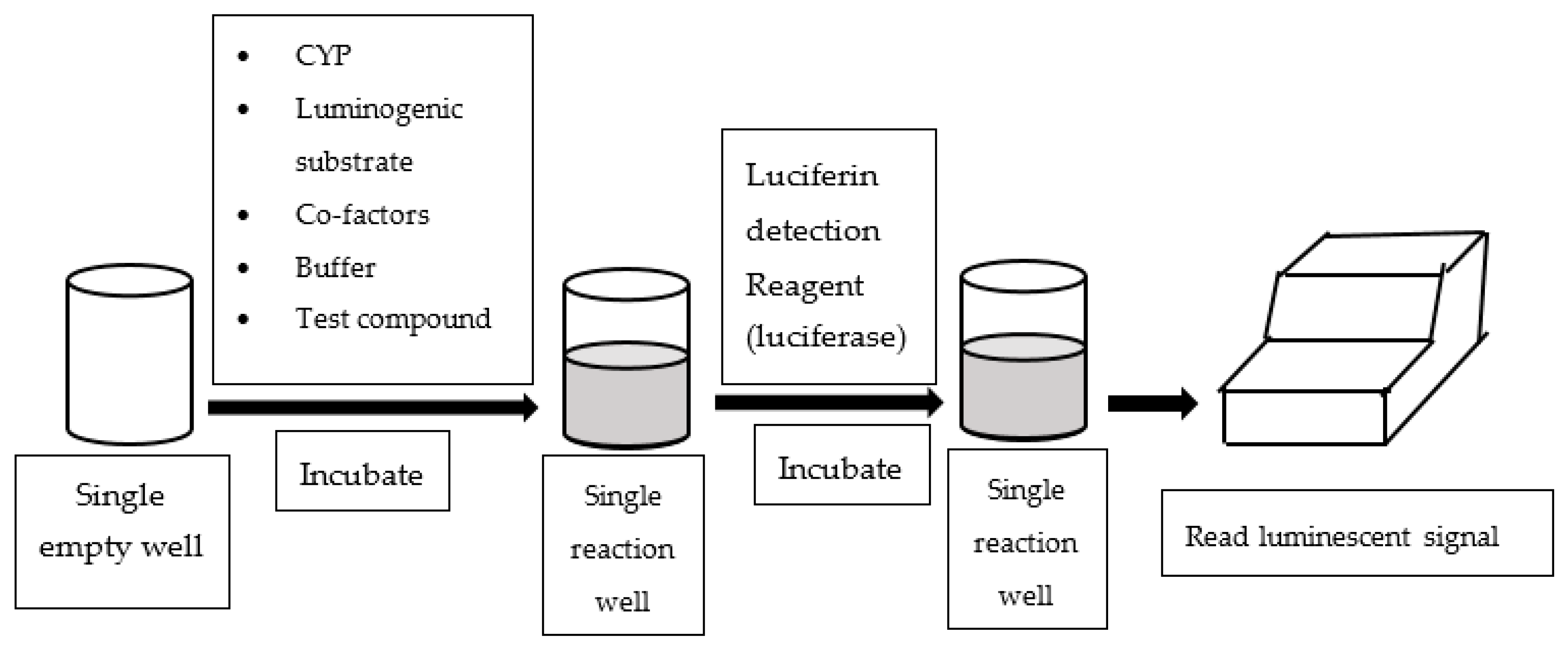
3. Luminescence-Based Assay
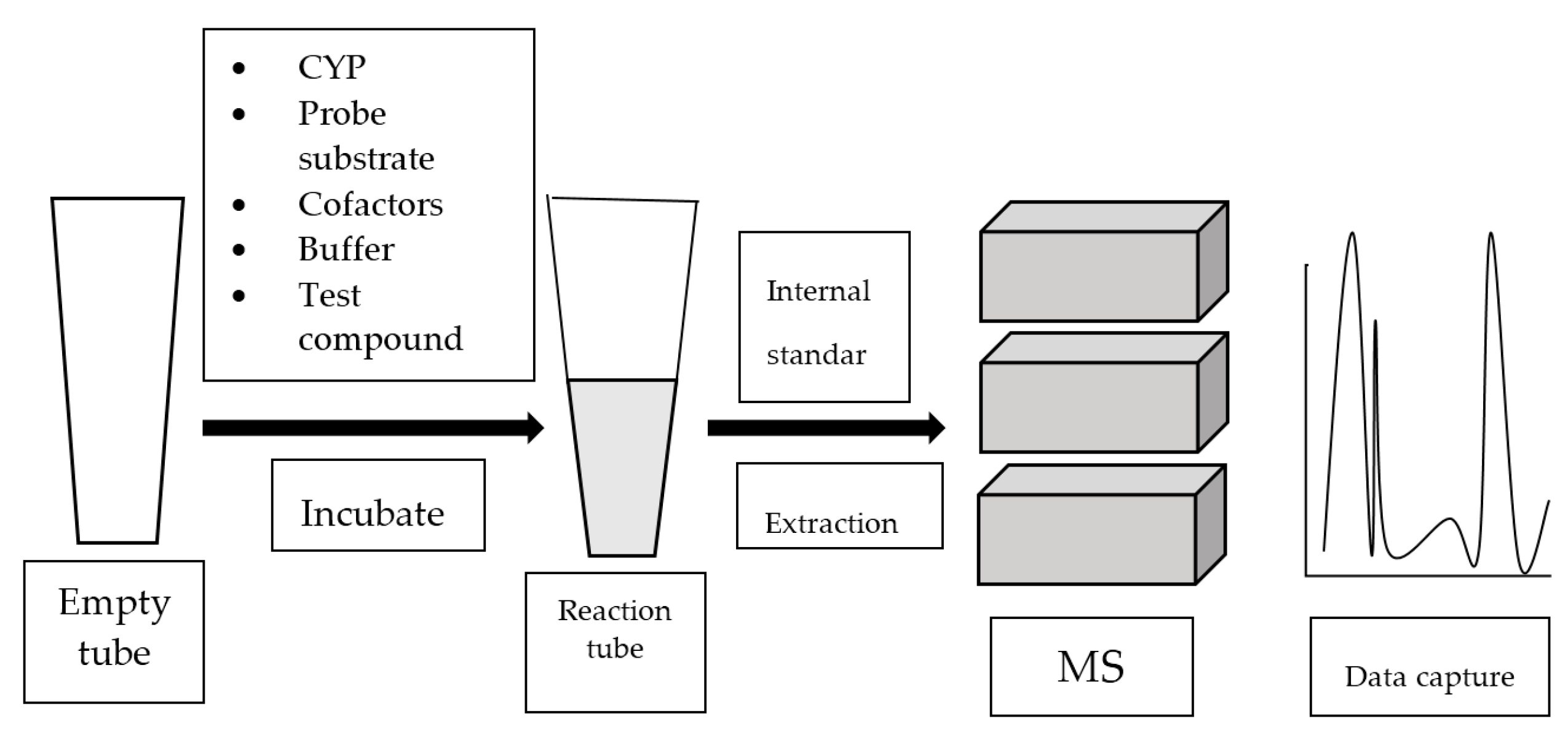
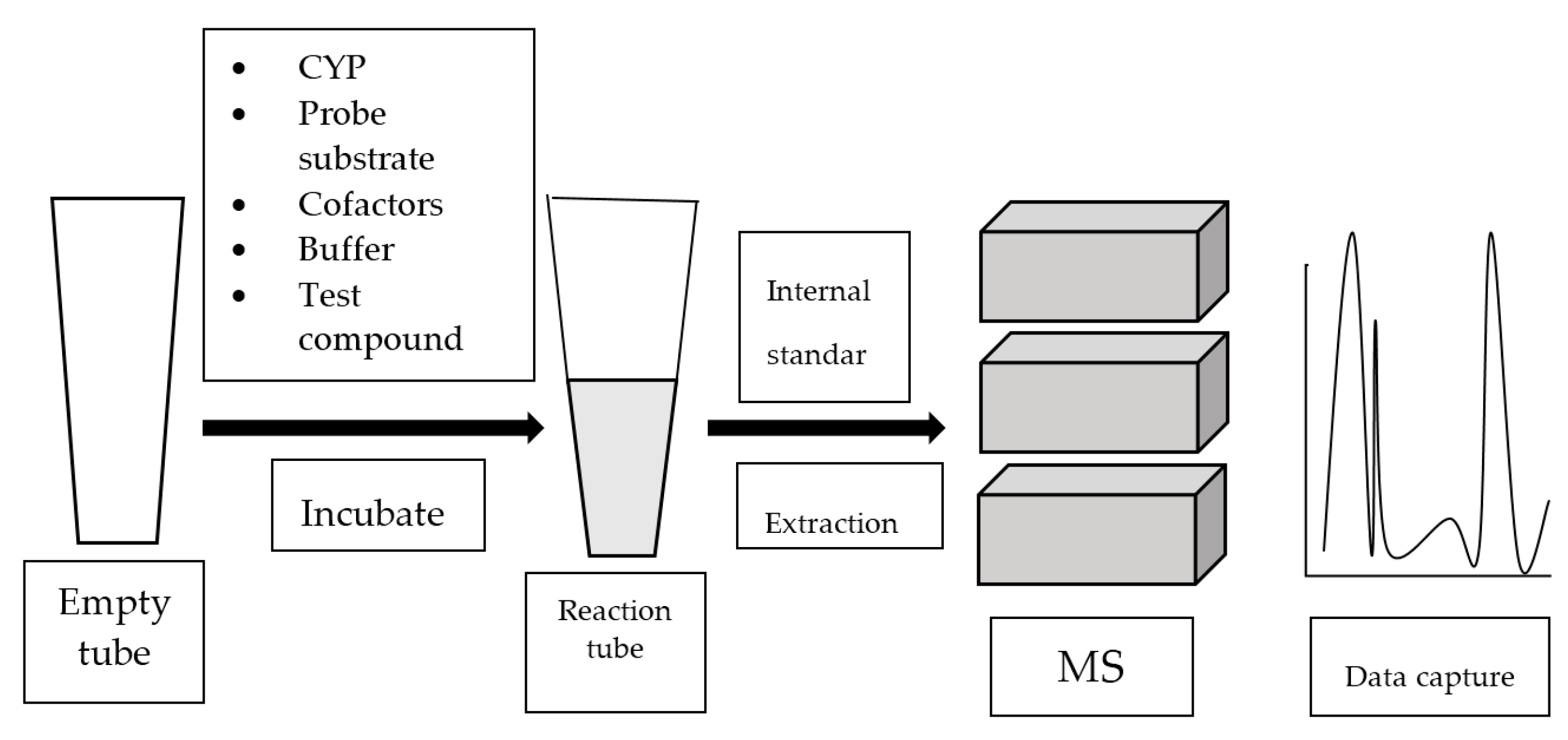
4. Mass Spectrometry-Based Assay
4.1. GC-MS-Based Assay
4.2. LC-MS or LC-MS/MS-Based Assay
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Anzenbacher, P.; Anzenbacherová, E. Cytochromes P450 and metabolism of xenobiotics. Cell. Mol. Life Sci. 2001, 58, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Doogue, M.P.; Polasek, T.M. The ABCD of clinical pharmacokinetics. Ther. Adv. Drug Saf. 2013, 4, 5–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Testa, B.; Pedretti, A.; Vistoli, G. Reactions and enzymes in the metabolism of drugs and other xenobiotics. Drug Discov. Today 2012, 17, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Edwards, I.R.; Aronson, J.K. Adverse drug reactions: Definitions, diagnosis, and management. Lancet 2000, 356, 1255–1259. [Google Scholar] [CrossRef]
- Danton, A.C.; Montastruc, F.; Sommet, A.; Durrieu, G.; Bagheri, H.; Bondon-Guitton, E.; Lapeyre-Mestre, M.; Montastruc, J.L. Importance of cytochrome P450 (CYP450) in adverse drug reactions due to drug–drug interactions: A PharmacoVigilance study in France. Eur. J. Clin. Pharmacol. 2013, 69, 885–888. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.J.; Pontefract, S.K. Adverse drug reactions. Clin. Med. 2016, 16, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Rautio, J.; Kumpulainen, H.; Heimbach, T.; Oliyai, R.; Oh, D.; Järvinen, T.; Savolainen, J. Prodrugs: Design and clinical applications. Nat. Rev. Drug Discov. 2008, 7, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Mannheimer, B.; Eliasson, E. Drug-drug interactions that reduce the formation of pharmacologically active metabolites: A poorly understood problem in clinical practice. J. Intern. Med. 2000, 268, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.K.; Ponnusankar, S.; Pandit, S.; Hazam, P.K.; Ahmmed, M.; Mukherjee, K. Botanicals as medicinal food and their effects on drug metabolizing enzymes. Food Chem. Toxicol. 2011, 49, 3142–3153. [Google Scholar] [CrossRef] [PubMed]
- Awortwe, C.; Makiwane, M.; Reuter, H.; Muller, C.; Louw, J.; Rosenkranz, B. Critical evaluation of causality assessment of herb-drug interactions in patients. Br. J. Clin. Pharmacol. 2018, 84, 679–693. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.H.; Ahemad, N.; Pan, Y.; Palanisamy, U.D.; Othman, I.; Yiap, B.C.; Ong, C.E. Cytochrome P450 2C9-natural antiarthritic interactions: Evaluation of inhibition magnitude and prediction from in vitro data. Biopharm. Drug Dispos. 2018, 39, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Mak, J.W.; Ong, C.E. Heterologous expression of human cytochrome P450 (CYP) 2C19 in Escherichia coli and establishment of RP-HPLC method to serve as activity marker. Biomed. Chromatogr. 2013, 27, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Abd-Rashid, B.A.; Ismail, Z.; Ismail, R.; Mak, J.W.; Pook, P.C.K.; Er, H.M.; Ong, C.E. In vitro determination of the effect of Andrographis paniculata extracts and andrographolide on human hepatic cytochrome P450 activities. J. Nat. Med. 2011, 65, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Abd-Rashid, B.A.; Ismail, Z.; Ismail, R.; Mak, J.W.; Pook, P.C.K.; Er, H.M.; Ong, C.E. In vitro effects of active constituents and extracts of Orthosiphon stamineus on the activities of three major human cDNA-expressed cytochrome P450 enzymes. Chem. Biol. Interact. 2011, 190, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Abd-Rashid, B.A.; Ismail, Z.; Ismail, R.; Mak, J.W.; Pook, P.C.K.; Er, H.M.; Ong, C.E. In vitro modulatory effects of Andrographis paniculata, Centella asiatica and Orthosiphon stamineus on cytochrome P450 2C19 (CYP2C19). J. Ethnopharmacol. 2011, 133, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, A.D.; Lin, J.H. Screening of drug candidates for their drug–drug interaction potential. Curr. Opin. Chem. Biol. 2001, 5, 396–401. [Google Scholar] [CrossRef]
- Burke, M.D.; Thompson, S.; Elcombe, C.R.; Halpert, J.; Haaparanta, T.; Mayer, R.T. Ethoxy-, pentoxy- and benzyloxyphenoxazones and homologues: A series of substrates to distinguish between different induced cytochromes P-450. Biochem. Pharmacol. 1985, 34, 3337–3345. [Google Scholar] [CrossRef]
- Stresser, D.M.; Blanchard, A.P.; Turner, S.D.; Erve, J.C.; Dandeneau, A.A.; Miller, V.P.; Crespi, C.L. Substrate-dependent modulation of CYP3A4 catalytic activity: Analysis of 27 test compounds with four fluorometric substrates. Drug Metab. Dispos. 2000, 28, 1440–1448. [Google Scholar] [PubMed]
- White, I.N. A continuous fluorometric assay for cytochrome P-450-dependent mixed function oxidases using 3-cyano-7-ethoxycoumarin. Anal. Biochem. 1988, 172, 304–310. [Google Scholar] [CrossRef]
- Donato, M.T.; Jiménez, N.; Castell, J.V.; Gómez-Lechón, M.J. Fluorescence-based assays for screening nine cytochrome P450 (P450) activities in intact cells expressing individual human P450 enzymes. Drug Metab. Dispos. 2004, 32, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Chauret, N.; Tremblay, N.; Lackman, R.L.; Gauthier, J.-Y.; Silva, J.M.; Marois, J.; Yergey, J.A.; Nicoll-Griffith, D.A. Description of a 96-well plate assay to measure cytochrome P4503A inhibition in human liver microsomes using a selective fluorescent probe. Anal. Biochem. 1999, 276, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Pérez, J.; Díaz, C.; Asensio, F.; Palafox, A.; Genilloud, O.; Vicente, F. A novel in vitro approach for simultaneous evaluation of CYP3A4 inhibition and kinetic aqueous solubility. J. Biomol. Screen. 2015, 20, 254–264. [Google Scholar] [CrossRef] [PubMed]
- Raymond, L.; Rayani, N.; Polson, G.; Sikorski, K.; Lian, A.; VanAlstine-Parris, M.A. Determining the IC 50 values for vorozole and letrozole, on a series of human liver cytochrome P450s, to help determine the binding site of vorozole in the liver. Enzyme Res. 2015, 2015, 321820. [Google Scholar] [CrossRef] [PubMed]
- Bapiro, T.E.; Egnell, A.C.; Hasler, J.A.; Masimirembwa, C.M. Application of higher throughput screening (HTS) inhibition assays to evaluate the interaction of antiparasitic drugs with cytochrome P450s. Drug Metab. Dispos. 2001, 29, 30–35. [Google Scholar] [PubMed]
- Kenaan, C.; Zhang, H.; Hollenberg, P.F. A quantitative high-throughput 96-well plate fluorescence assay for mechanism-based inactivators of cytochromes P450 exemplified using CYP2B6. Nat. Protoc. 2010, 5, 1652–1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahadur, S.; Mukherjee, P.; Milan Ahmmed, S.; Kar, A.; Harwansh, R.; Pandit, S. Metabolism-mediated interaction potential of standardized extract of Tinospora cordifolia through rat and human liver microsomes. Indian J. Pharmacol. 2016, 48, 576–581. [Google Scholar] [CrossRef] [PubMed]
- Ahmmed, S.; Mukherjee, P.; Bahadur, S.; Kar, A.; Mukherjee, K.; Karmakar, S.; Bandyopadhyay, A. Interaction potential of Trigonella foenum graceum through cytochrome P450 mediated inhibition. Indian J. Pharmacol. 2015, 47, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Siqueira-Neto, J.L.; Song, O.-R.; Oh, H.; Sohn, J.-H.; Yang, G.; Nam, J.; Jang, J.; Cechetto, J.; Lee, C.B.; Moon, S.; et al. Antileishmanial high-throughput drug screening reveals drug candidates with new scaffolds. PLoS Negl. Trop. Dis. 2010, 4, e675. [Google Scholar] [CrossRef] [PubMed]
- Awortwe, C.; Bouic, P.J.; Masimirembwa, C.M.; Rosenkranz, B. Inhibition of major drug metabolizing CYPs by common herbal medicines used by HIV/AIDS patients in Africa—Implications for herb-drug interactions. Drug Metab. Lett. 2014, 7, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Zhang, B.; He, M.L.; Chang, V.S.C.; Kung, H.F. Screening of active ingredients of herbal medicine for interaction with CYP450 3A4. Phytother. Res. 2007, 21, 1096–1099. [Google Scholar] [CrossRef] [PubMed]
- Sukumaran, S.M.; Potsaid, B.; Lee, M.-Y.; Clark, D.S.; Dordick, J.S. Development of a fluorescence-based, ultra high-throughput screening platform for nanoliter-scale cytochrome p450 microarrays. J. Biomol. Screen. 2009, 14, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Auld, D.S.; Veith, H.; Cali, J.J. Bioluminescent Assays for Cytochrome P450 Enzymes. Methods Mol. Biol. 2013, 987, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Bae, Y.J.; Kim, H.S.; Cha, H.J.; Yun, J.S.; Shin, J.S.; Seong, W.K.; Lee, Y.M.; Han, K.M. Measurement of Human Cytochrome P450 Enzyme Induction Based on Mesalazine and Mosapride Citrate Treatments Using a Luminescent Assay. Biomol. Ther. 2015, 23, 486–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Lu, J.; He, B.; Tang, L.; Liu, X.; Zhu, D.; Cao, H.; Wang, Y.; Li, L. A tryptophan derivative, ITE, enhances liver cell metabolic functions in vitro. Int. J. Mol. Med. 2017, 39, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Satoh, D.; Iwado, S.; Abe, S.; Kazuki, K.; Wakuri, S.; Oshimura, M.; Kazuki, Y. Establishment of a novel hepatocyte model that expresses four cytochrome P450 genes stably via mammalian-derived artificial chromosome for pharmacokinetics and toxicity studies. PLoS ONE 2017, 12, e0187072. [Google Scholar] [CrossRef] [PubMed]
- Sa-ngiamsuntorn, K.; Wongkajornsilp, A.; Kasetsinsombat, K.; Duangsa-ard, S.; Nuntakarn, L.; Borwornpinyo, S.; Akarasereenont, P.; Limsrichamrern, S.; Hongeng, S. Upregulation of CYP 450s expression of immortalized hepatocyte-like cells derived from mesenchymal stem cells by enzyme inducers. BMC Biotechnol. 2011, 11, 89. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.; Tseng, H.; Souza, G. Assembly of hepatocyte spheroids using magnetic 3D cell culture for CYP450 inhibition/induction. Int. J. Mol. Sci. 2017, 18, 1085. [Google Scholar] [CrossRef] [PubMed]
- Kostiainen, R.; Kotiaho, T.; Kuuranne, T.; Auriola, S. Liquid chromatography/atmospheric pressure ionization-mass spectrometry in drug metabolism studies. J. Mass Spectrom. 2003, 38, 357–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, M.R.; Vollmar, C.; Schwaninger, A.E.; Wolf, E.U.; Maurer, H.H. New cathinone-derived designer drugs 3-bromomethcathinone and 3-fluoromethcathinone: Studies on their metabolism in rat urine and human liver microsomes using GC-MS and LC-high-resolution MS and their detectability in urine. J. Mass Spectrom. 2012, 47, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.-A.; Lee, H.; Kim, D.; Jung, B.H. Development of GC-MS based cytochrome P450 assay for the investigation of multi-herb interaction. Anal. Biochem. 2017, 519, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Nelson, L.J.; Morgan, K.; Treskes, P.; Samuel, K.; Henderson, C.J.; LeBled, C.; Homer, N.; Grant, M.H.; Hayes, P.C.; Plevris, J.N. Human hepatic HepaRG cells maintain an organotypic phenotype with high intrinsic CYP450 activity/metabolism and significantly outperform standard HepG2/C3A cells for pharmaceutical and therapeutic applications. Basic Clin. Pharmacol. Toxicol. 2017, 120, 30–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szultka-Mlynska, M.; Buszewski, B. Study of in-vitro metabolism of selected antibiotic drugs in human liver microsomes by liquid chromatography coupled with tandem mass spectrometry. Anal. Bioanal. Chem. 2016, 408, 8273–8287. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Ji, H.; Yang, B.; Ma, L.; Bei, Z.; Li, X.; Dang, H.; Yang, X.; Liu, C.; Wu, X.; et al. Impact of chrysosplenetin on the pharmacokinetics and anti-malarial efficacy of artemisinin against Plasmodium berghei as well as in vitro CYP450 enzymatic activities in rat liver microsome. Malar. J. 2015, 14, 432. [Google Scholar] [CrossRef] [PubMed]
- Urban, P.; Truan, G.; Pompon, D. High-throughput functional screening of steroid substrates with wild-type and chimeric P450 enzymes. BioMed Res. Int. 2014, 2014, 764102. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Zhang, Q.; Lin, K.; Hu, L.; Zheng, Y. The effect of MGCD0103 on CYP450 isoforms activity of rats by cocktail method. BioMed Res. Int. 2015, 2015, 517295. [Google Scholar] [CrossRef] [PubMed]
- Dierks, E.A.; Stams, K.R.; Lim, H.K.; Cornelius, G.; Zhang, H.; Ball, S.E. A method for the simultaneous evaluation of the activities of seven major human drug-metabolizing cytochrome P450s using an in vitro cocktail of probe substrates and fast gradient liquid chromatography tandem mass spectrometry. Drug Metab. Dispos. 2001, 29, 23–29. [Google Scholar] [PubMed]
- Qin, C.Z.; Ren, X.; Tan, Z.R.; Chen, Y.; Yin, J.Y.; Yu, J.; Qu, J.; Zhou, H.H.; Liu, Z.Q. A high-throughput inhibition screening of major human cytochrome P450 enzymes using an in vitro cocktail and liquid chromatography-tandem mass spectrometry. Biomed. Chromatogr. 2014, 28, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Guo, J.J.; Zhan, J.; Bu, H.Z.; Lin, J.H. An in-vitro cocktail assay for assessing compound-mediated inhibition of six major cytochrome P450 enzymes. J. Pharm. Anal. 2014, 4, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Kozakai, K.; Yamada, Y.; Oshikata, M.; Kawase, T.; Suzuki, E.; Haramaki, Y.; Taniguchi, H. Cocktail-substrate approach-based high-throughput assay for evaluation of direct and time-dependent inhibition of multiple cytochrome P450 isoforms. Drug Metab. Pharmacokinet. 2014, 29, 198–207. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate | CYP | Metabolite | Ex/Em (nm) |
|---|---|---|---|
| CEC | CYP1A1/CYP1A2/CYP2C19 | CHC | 408/455 |
| Coumarin | CYP2A6 | 7-HC | 355/460 |
| BFC | CYP3A4 | HFC | 410/510 |
| EFC | CYP2B6 | HFC | 410/510 |
| MFC | CYP2C9/CYP2E1/CYP2C19 | HFC | 410/510 |
| DBF | CYP2C8/CYP3A4/CYP2C9/CYP2C19 | Fluorescein | 485/538 |
| AMMC | CYP2D6 | AHMC | 390/460 |
| MAMC | CYP2D6 | HAMC | 390/460 |
| DFB | CYP3A4 | DFH | 360/440 |
| EOMCC | CYP1A2/CYP2C19/CYP2D6 | CHC | 408/455 |
| BOMCC | CYP2C9/CYP3A4 | CHC | 408/455 |
| BOMF | CYP2C9 | Fluorescein | 485/538 |
| BQ | CYP3A4 | 7-hydroxyquinoline | 358/505 |
| BzRes | CYP3A4 | Fluorescein | 485/538 |
| DBOMF | CYP3A4 | Fluorescein | 485/538 |
| Advantages/Disadvantages | Fluorescence-Based Assay | Luminescence-Based Assay | MS-Based Assay |
|---|---|---|---|
| Advantages |
|
|
|
| Disadvantages |
|
|
|
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ung, Y.T.; Ong, C.E.; Pan, Y. Current High-Throughput Approaches of Screening Modulatory Effects of Xenobiotics on Cytochrome P450 (CYP) Enzymes. High-Throughput 2018, 7, 29. https://doi.org/10.3390/ht7040029
Ung YT, Ong CE, Pan Y. Current High-Throughput Approaches of Screening Modulatory Effects of Xenobiotics on Cytochrome P450 (CYP) Enzymes. High-Throughput. 2018; 7(4):29. https://doi.org/10.3390/ht7040029
Chicago/Turabian StyleUng, Yee Tze, Chin Eng Ong, and Yan Pan. 2018. "Current High-Throughput Approaches of Screening Modulatory Effects of Xenobiotics on Cytochrome P450 (CYP) Enzymes" High-Throughput 7, no. 4: 29. https://doi.org/10.3390/ht7040029
APA StyleUng, Y. T., Ong, C. E., & Pan, Y. (2018). Current High-Throughput Approaches of Screening Modulatory Effects of Xenobiotics on Cytochrome P450 (CYP) Enzymes. High-Throughput, 7(4), 29. https://doi.org/10.3390/ht7040029