
Deciphering Microbiota of Acute Upper Respiratory Infections: A Comparative Analysis of PCR and mNGS Methods for Lower Respiratory Trafficking Potential

 ,
,  and
and
Abstract
Highlights
- Although there was a high concordance between methodologies, a hybridization-capture-based mNGS workflow was able to detect 29 additional upper respiratory microorganisms versus PCR.
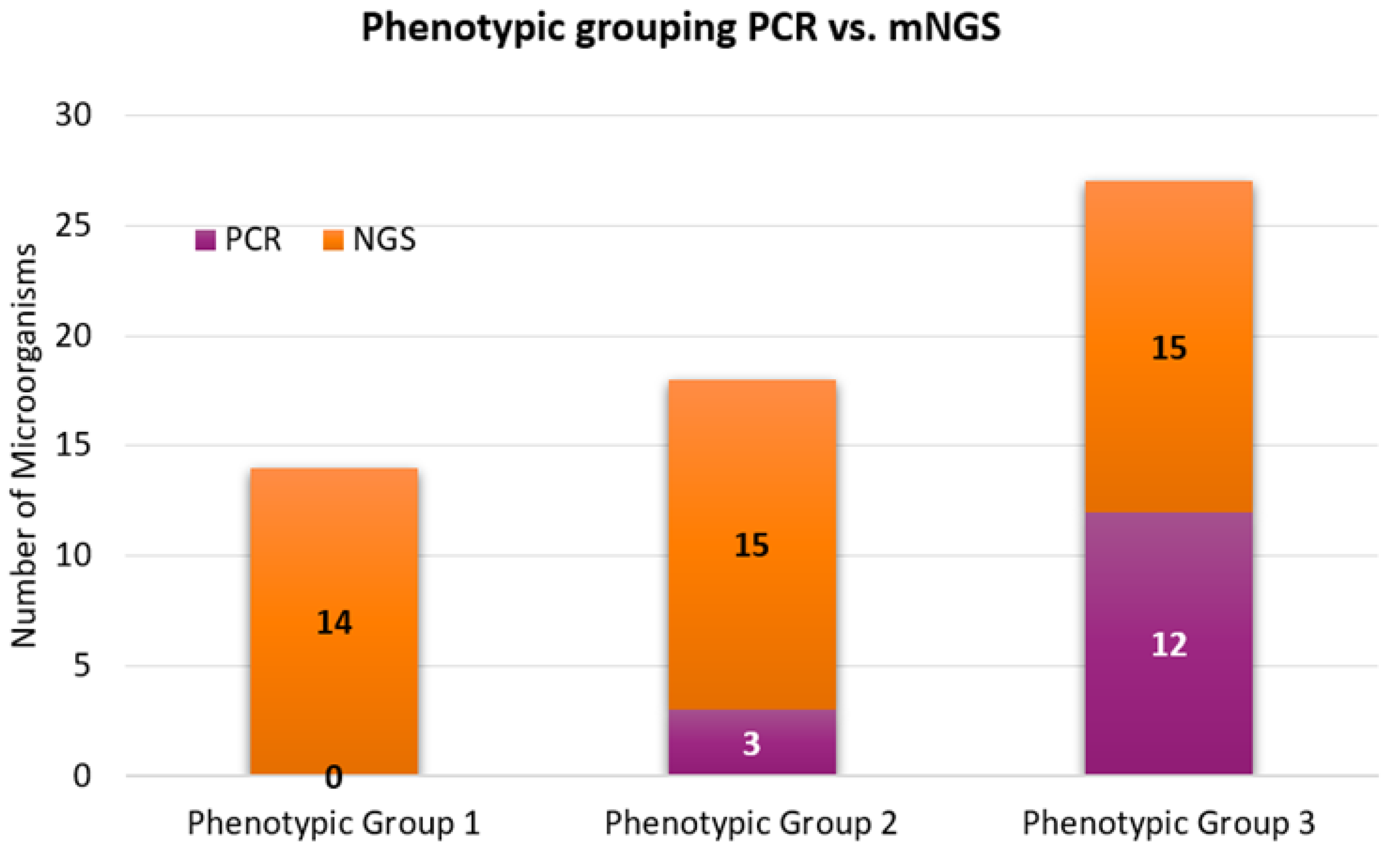
- The identified microorganisms were rapidly characterized into three phenotypic groups for infectivity and trafficking potential.
- A hybridization-capture-based mNGS workflow can provide a comprehensive yet clinically relevant microbiology profile of acute upper respiratory infection.
- Deciphering upper respiratory microbiota with phenotypic grouping has potential to provide respiratory medicine a tool to better manage immunocompromised, immunocompetent with comorbidity and complex respiratory cases.
Abstract
1. Introduction
2. Materials and Methods
2.1. Nucleic Acid Isolation
2.2. PCR Testing Using Fast Track Diagnostic® Assay
2.3. Library Preparation and Enrichment
2.4. Bioinformatic Analysis Using the Explify® Platform
3. Results
3.1. Bioinformatic Analysis Using the Explify® Platform
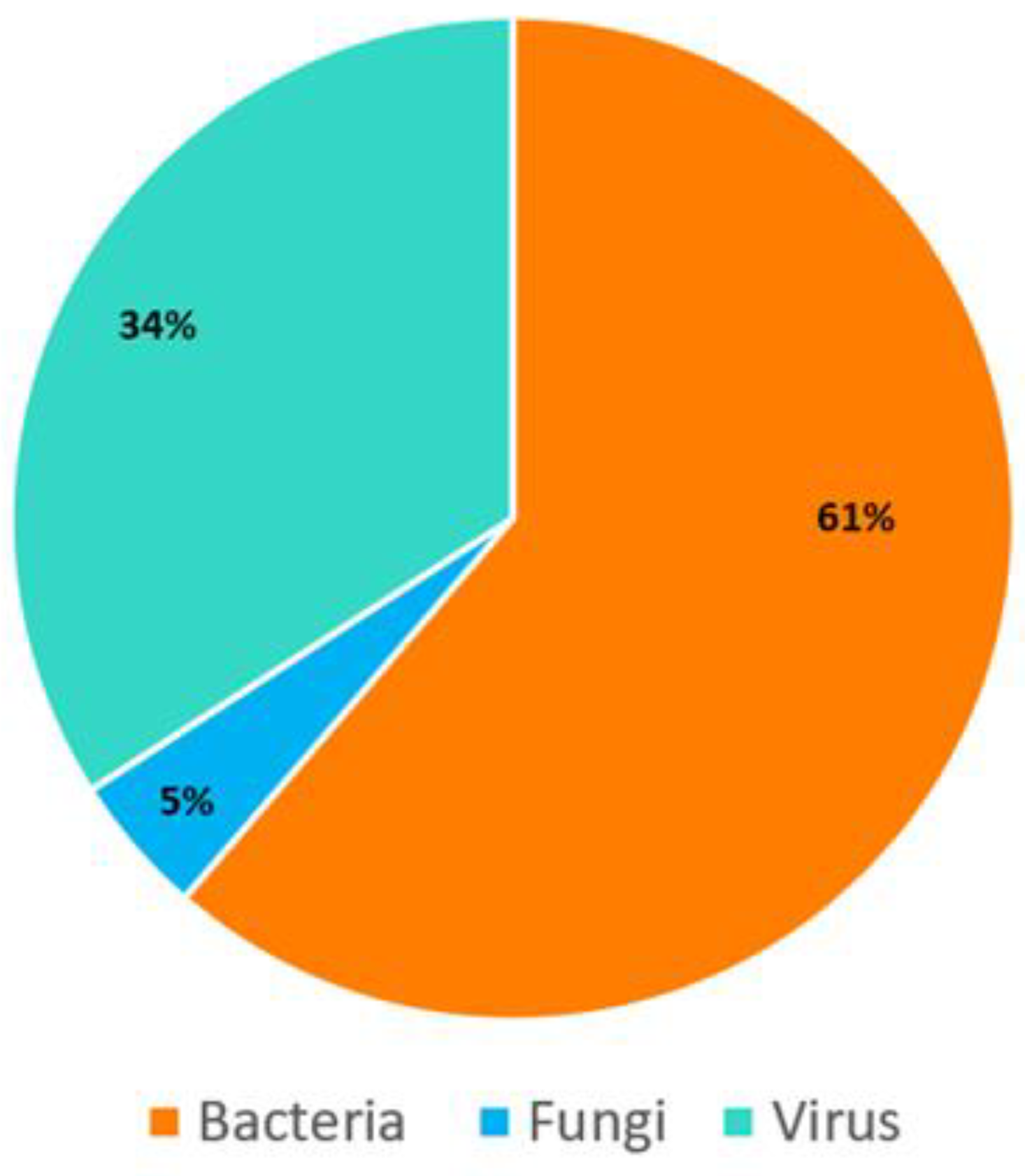
3.2. Bioinformatic Analysis Using Hybridization-Capture-Based mNGS Workflow
3.3. Comparative Analysis between PCR and mNGS Assays
4. Discussion
Implications for Respiratory Medicine
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cookson, W.O.; Cox, M.J.; Moffatt, M.F. New Opportunities for Managing Acute and Chronic Lung Infections. Nat. Rev. Microbiol. 2018, 16, 111–120. [Google Scholar] [CrossRef]
- Besen, B.A.; Park, M.; Ranzani, O.T. Noninvasive Ventilation in Critically Ill Very Old Patients with Pneumonia: A Multicenter Retrospective Cohort Study. PLoS ONE 2021, 16, e0246072. [Google Scholar] [CrossRef]
- 3. Hassan, A.; Blanchard, N. Microbial (co) Infections: Powerful Immune Influencers. PLoS Pathog. 2022, 18, e1010212. [Google Scholar] [CrossRef]
- Theilacker, C.; Sprenger, R.; Leverkus, F.; Walker, J.; Häckl, D.; Von Eiff, C.; Schiffner-Rohe, J. Population-Based Incidence and Mortality of Community-Acquired Pneumonia in Germany. PLoS ONE 2021, 16, e0253118. [Google Scholar] [CrossRef]
- Valley, T.S.; Sjoding, M.W.; Ryan, A.M.; Iwashyna, T.J.; Cooke, C.R. Association of Intensive Care Unit Admission with Mortality Among Older Patients with Pneumonia. JAMA 2015, 314, 1272–1279. [Google Scholar] [CrossRef]
- Voiriot, G.; Visseaux, B.; Cohen, J.; Nguyen, L.B.L.; Neuville, M.; Morbieu, C.; Burdet, C.; Radjou, A.; Lescure, F.X.; Smonig, R.; et al. Viral-Bacterial Coinfection Affects the Presentation and Alters the Prognosis of Severe Community-Acquired Pneumonia. Crit. Care 2016, 20, 375. [Google Scholar] [CrossRef]
- Rothberg, M.B.; Haessler, S.D.; Brown, R.B. Complications of Viral Influenza. Am. J. Med. 2008, 121, 258–264. [Google Scholar] [CrossRef]
- Silva, D.L.; Lima, C.M.; Magalhaes, V.C.R.; Baltazar, L.M.; Peres, N.T.A.; Caligiorne, R.B.; Moura, A.S.; Fereguetti, T.; Martins, J.C.; Rabelo, L.F.; et al. Fungal and Bacterial Coinfections Increase Mortality of Severely Ill COVID-19 Patients. J. Hosp. Infect. 2021, 113, 145–154. [Google Scholar] [CrossRef]
- Bradley, B.T.; Bryan, A. Emerging Respiratory Infections: The Infectious Disease Pathology of SARS, MERS, Pandemic Influenza, and Legionella. Semin. Diagn. Path 2019, 36, 152–159. [Google Scholar] [CrossRef]
- Troeger, C.; Blacker, B.; Khalil, I.A.; Rao, P.C.; Cao, J.; Zimsen, S.R.; Albertson, S.B.; Deshpande, A.; Farag, T.; Abebe, Z.; et al. Estimates of the Global, Regional, And National Morbidity, Mortality, And Aetiologies of Lower Respiratory Infections In 195 Countries, 1990–2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Infect. Dis. 2018, 18, 1191–1210. [Google Scholar] [CrossRef]
- Havelka, A.; Sejersen, K.; Venge, P.; Pauksens, K.; Larsson, A. Calprotectin, a New Biomarker for Diagnosis of Acute Respiratory Infections. Sci. Rep. 2020, 10, 4208. [Google Scholar] [CrossRef]
- Versporten, A.; Zarb, P.; Caniaux, I.; Gros, M.F.; Drapier, N.; Miller, M.; Jarlier, V.; Nathwani, D.; Goossens, H.; Koraqi, A.; et al. Antimicrobial Consumption and Resistance in Adult Hospital Inpatients In 53 Countries: Results of an Internet-Based Global Point Prevalence Survey. Lancet Glob. Health 2018, 6, e619–e629. [Google Scholar] [CrossRef]
- Schreckenberger, P.C.; McAdam, A.J. Point-Counterpoint: Large Multiplex PCR Panels Should Be First-Line Tests for Detection of Respiratory and Intestinal Pathogens. J. Clin. Microbiol. 2015, 53, 3110–3115. [Google Scholar] [CrossRef]
- Graf, E.H.; Simmon, K.E.; Tardif, K.D.; Hymas, W.; Flygare, S.; Eilbeck, K.; Yandell, M.; Schlaberg, R. Unbiased Detection of Respiratory Viruses by Use of RNA Sequencing-Based Metagenomics: A Systematic Comparison to a Commercial PCR Panel. J. Clin. Microbiol. 2016, 54, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Fan, Y.; Zhang, D.; Yang, L.; Wang, X.; Wang, S.; Xu, J.; Zhang, J. Metagenomic Next-Generation Sequencing for Accurate Diagnosis and Management of Lower Respiratory Tract Infections. Int. J. Infect. Dis. 2022, 122, 921–929. [Google Scholar] [CrossRef]
- Zhou, H.; Larkin, P.M.; Zhao, D.; Ma, Q.; Yao, Y.; Wu, X.; Wang, J.; Zhou, X.; Li, Y.; Wang, G.; et al. Clinical Impact of Metagenomic Next-Generation Sequencing of Bronchoalveolar Lavage in The Diagnosis and Management of Pneumonia: A Multicenter Prospective Observational Study. J. Mol. Diagn. 2021, 23, 1259–1268. [Google Scholar] [CrossRef]
- Xie, F.; Duan, Z.; Zeng, W.; Xie, S.; Xie, M.; Fu, H.; Xie, L. Clinical Metagenomics Assessments Improve Diagnosis and Outcomes in Community-Acquired Pneumonia. BMC Infect. Dis. 2021, 21, 352. [Google Scholar] [CrossRef]
- Mostafa, H.H.; Fissel, J.A.; Fanelli, B.; Bergman, Y.; Gniazdowski, V.; Dadlani, M.; Simner, P.J. Metagenomic Next-Generation Sequencing of Nasopharyngeal Specimens Collected from Confirmed and Suspect COVID-19 Patients. MBio 2020, 11, e01969. [Google Scholar] [CrossRef] [PubMed]
- Zinter, M.S.; Mayday, M.Y.; Ryckman, K.K.; Jelliffe-Pawlowski, L.L.; DeRisi, J.L. Towards Precision Quantification of Contamination in Metagenomic Sequencing Experiments. Microbiome 2019, 7, 62. [Google Scholar] [CrossRef]
- Afshinnekoo, E.; Chou, C.; Alexander, N.; Ahsanuddin, S.; Schuetz, A.N.; Mason, C.E. Precision Metagenomics: Rapid Metagenomic Analyses for Infectious Disease Diagnostics and Public Health Surveillance. J. Biomol. Tech. JBT 2017, 28, 40–45. [Google Scholar] [CrossRef]
- Gaudin, M.; Desnues, C. Hybrid Capture-Based Next Generation Sequencing and Its Application to Human Infectious Diseases. Front. Microbiol. 2018, 9, 2924. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Miller, S.; Chiu, C.Y. Clinical Metagenomic Next-Generation Sequencing for Pathogen Detection. Annu. Rev. Pathol. 2019, 14, 319–338. [Google Scholar] [CrossRef]
- Shi, Y.; Wang, G.; Lau, H.C.H.; Yu, J. Metagenomic Sequencing for Microbial DNA in Human Samples: Emerging Technological Advances. Int. J. Mol. Sci. 2022, 23, 2181. [Google Scholar] [CrossRef] [PubMed]
- Gaston, D.C.; Miller, H.B.; Fissel, J.A.; Jacobs, E.; Gough, E.; Wu, J.; Simner, P.J. Evaluation of Metagenomic and Targeted Next-Generation Sequencing Workflows for Detection of Respiratory Pathogens from Bronchoalveolar Lavage Fluid Specimens. J. Clin. Microbiol. 2022, 60, e00526-22. [Google Scholar] [CrossRef]
- Royston, L.; Tapparel, C. Rhinoviruses and Respiratory Enteroviruses: Not as Simple as ABC. Viruses 2016, 8, 16. [Google Scholar] [CrossRef]
- 26. Alon, R.; Sportiello, M.; Kozlovski, S.; Kumar, A.; Reilly, E.C.; Zarbock, A.; Garbi, N.; Topham, D.J. Leukocyte Trafficking to the Lungs and Beyond: Lessons from Influenza for COVID-19. Nat. Rev. Immunol. 2021, 1, 49–64. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Qiu, X.; Wang, T.; Zhang, J. The Diagnostic Value of Metagenomic Next–Generation Sequencing in Lower Respiratory Tract Infection. Front. Cell. Infect. Microbiol. 2021, 11, 694756. [Google Scholar] [CrossRef] [PubMed]
- Silberman, D.; Carpenter, R.E.; Cabrera, E.; Kernaleguen, J. Organizational Silofication: Implications in Grouping Experts for Organizational Performance. Dev. Learn. Org. Int. J. 2022, 36, 15–18. [Google Scholar] [CrossRef]
- Carpenter, R.E.; Tamrakar, V.; Almas, S.; Rowen, C.; Sharma, R. Optimization of the Illumina COVIDSeq™ Protocol for Decentralized, Cost-Effective Genomic Surveillance. BioRxiv 2022. [Google Scholar] [CrossRef]
- Carpenter, R.E.; Tamrakar, V.; Chahar, H.; Vine, T.; Sharma, R. Confirming Multiplex Q-PCR Use In COVID-19 With Next Generation Sequencing: Strategies for Epidemiological Advantage. Glob. Health Epidemiol. Genom. 2022, 2022, 2270965. [Google Scholar] [CrossRef]
- Adam, A.; Htet, Z.M.; Stojsavljevic, J.; Chao, C.; Antic, M.; Khan, Z.; Bachan, M. Acute Chronic Pericarditis Caused by Coxsackievirus A. Chest 2022, 162, A441–A442. [Google Scholar] [CrossRef]
- Chambers, R.; Takimoto, T. Trafficking of Sendai Virus Nucleocapsids is Mediated by Intracellular Vesicles. PLoS ONE 2010, 5, e10994. [Google Scholar] [CrossRef] [PubMed]
- Legay, F.; Lévêque, N.; Gacouin, A.; Tattevin, P.; Bouet, J.; Thomas, R.; Chomel, J.J. Fatal Coxsackievirus A-16 Pneumonitis in Adult. Emerg. Infect. Dis. 2007, 13, 1084–1086. [Google Scholar] [CrossRef] [PubMed]
- Pelton, S.I. Regulation of Bacterial Trafficking in the Nasopharynx. Paediatr. Resp. Rev. 2012, 13, 150–153. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.; Wang, X.; Briere, E.C.; Katz, L.S.; Cohn, A.C.; Clark, T.A.; Mayer, L.W. Haemophilus Haemolyticus Isolates Causing Clinical Disease. J. Clin. Microbiol. 2012, 50, 2462–2465. [Google Scholar] [CrossRef]
- Huang, L.; Ma, L.; Fan, K.; Li, Y.; Xie, L.; Xia, W.; Liu, G. Necrotizing Pneumonia and Empyema Caused by Neisseria Flavescens Infection. J. Thorac. Dis 2014, 6, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Lécuyer, H.; Audibert, J.; Bobigny, A.; Eckert, C.; Jannière-Nartey, C.; Buu-Hoï, A.; Podglajen, I. Dolosigranulum Pigrum Causing Nosocomial Pneumonia and Septicemia. J. Clin. Microbiol. 2007, 45, 3474–3475. [Google Scholar] [CrossRef]
- Ehrmann, E.; Jolivet-Gougeon, A.; Bonnaure-Mallet, M.; Fosse, T. Multidrug-Resistant Oral Capnocytophaga Gingivalis Responsible for an Acute Exacerbation of Chronic Obstructive Pulmonary Disease: Case Report and Literature Review. Anaerobe 2016, 42, 50–54. [Google Scholar] [CrossRef]
- Carmody, L.A.; Zhao, J.; Schloss, P.D.; Petrosino, J.F.; Murray, S.; Young, V.B.; LiPuma, J.J. Changes in Cystic Fibrosis Airway Microbiota at Pulmonary Exacerbation. Ann. Am. Thorac. Soc. 2013, 10, 179–187. [Google Scholar] [CrossRef]
- Ryan, M.P.; Pembroke, J.T. The Genus Ochrobactrum as Major Opportunistic Pathogens. Microorganisms 2020, 8, 1797. [Google Scholar] [CrossRef]
- Baptista, I.M.D.C.; Martinho, F.C.; Nascimento, G.G.; Da Rocha Santos, C.E.; Do Prado, R.F.; Valera, M.C. Colonization of Oropharynx and Lower Respiratory Tract in Critical Patients: Risk of Ventilator-Associated Pneumonia. Arch. Oral Biol. 2018, 85, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Rigauts, C.; Aizawa, J.; Taylor, S.L.; Rogers, G.B.; Govaerts, M.; Cos, P.; Crabbé, A. Rothia Mucilaginosa Is an Anti-Inflammatory Bacterium in the Respiratory Tract of Patients with Chronic Lung Disease. Eur. Respir. J. 2022, 59, 2101293. [Google Scholar] [CrossRef] [PubMed]
- Craig, T.J.; Maguire, F.E.; Wallace, M.R. Tracheobronchitis Due to Corynebacterium Pseudodiphtheriticum. South. Med. J. 1991, 84, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Bongaerts, G.P.A.; Schreurs, B.W.; Lunel, F.V.; Lemmens, J.A.M.; Pruszczynski, M.; Merkx, M.A.W. Was Isolation of Veillonella from Spinal Osteomyelitis Possible Due to Poor Tissue Perfusion? Med. Hypotheses 2004, 63, 659–661. [Google Scholar] [CrossRef]
- Wiest, P.M.; Wiese, K.; Jacobs, M.R.; Morrissey, A.B.; Abelson, T.I.; Witt, W.; Lederman, M.M. Alternaria Infection in A Patient with Acquired Immunodeficiency Syndrome: Case Report and Review of Invasive Alternaria Infections. Rev. Infect. Dis. 1987, 9, 799–803. [Google Scholar] [CrossRef]
- Goldstein, E.J.; Murphy, T.F.; Parameswaran, G.I. Moraxella Catarrhalis, a Human Respiratory Tract Pathogen. Clin. Infect. Dis. 2009, 49, 124–131. [Google Scholar] [CrossRef]
- Winstanley, T.G.; Spencer, R.C. Moraxella Catarrhalir: Antibiotic Susceptibility with Special Reference to Trimethoprim. J. Antimicrob. Chemother. 1986, 18, 425–426. [Google Scholar] [CrossRef]
- World Health Organization. Pneumonia in Children. Available online: https://www.who.int/news-room/fact-sheets/detail/pneumonia (accessed on 26 December 2022).
- Pillai, A.; Mitchell, J.L.; Hill, S.L.; Stockley, R.A. A Case of Haemophilus Parainfluenzaepneumonia. Thorax 2000, 55, 623–624. [Google Scholar] [CrossRef]
- Nagaoka, K.; Izumikawa, K.; Yamamoto, Y.; Yanagihara, K.; Ohkusu, K.; Kohno, S. Multiple Lung Abscesses Caused by Actinomyces Graevenitzii Mimicking Acute Pulmonary Coccidioidomycosis. J. Clin. Microbiol. 2012, 50, 3125–3128. [Google Scholar] [CrossRef]
- De Baets, F.; Schelstraete, P.; Van Daele, S.; Haerynck, F.; Vaneechoutte, M. Achromobacter Xylosoxidans in Cystic Fibrosis: Prevalence and Clinical Relevance. J. Cyst. Fibros. 2007, 6, 75–78. [Google Scholar] [CrossRef]
- Denton, M.; Kerr, K.G. Microbiological and Clinical Aspects of Infection Associated with Stenotrophomonas Maltophilia. Clin. Microbiol. Rev. 1998, 11, 57–80. [Google Scholar] [CrossRef]
- Rigvava, S.; Tchgkonia, I.; Jgenti, D.; Dvalidze, T.; Carpino, J.; Goderdzishvili, M. Comparative Analysis of The Biological and Physical Properties of Enterococcus Faecalis Bacteriophage vB_EfaS_GEC-EfS_3 and Streptococcus Mitis Bacteriophage vB_SmM_GEC-SmitisM_2. Can. J. Microbiol. 2013, 59, 18–21. [Google Scholar] [CrossRef]
- Grilli, E.; Galati, V.; Bordi, L.; Taglietti, F.; Petrosillo, N. Cytomegalovirus Pneumonia in Immunocompetent Host: Case Report and Literature Review. J. Clin. Virol. 2012, 55, 356–359. [Google Scholar] [CrossRef]
- Carneiro, H.A.; Coleman, J.J.; Restrepo, A.; Mylonakis, E. Fusarium Infection in Lung Transplant Patients: Report Of 6 Cases and Review of the Literature. Medicine 2011, 90, 69–80. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 26 December 2022).
- Carpenter, R.E.; Sadia, A.; Brown, E.; Vine, T.; Sharma, R. COVIDSeqTM as Laboratory Developed Test (LDT) For Detection of SARS-Cov-2 Variants of Concern (VOC). Arch. Clin. Biomed. Res. 2022, 6, 954–970. [Google Scholar] [CrossRef]
- Mohammadi, M.R.; Omidi, A.H.; Sabati, H. Current Trends and New Methods of Detection of SARS-Cov-2 Infection. Cell. Mol. Biomed. Rep. 2022, 2, 138–150. [Google Scholar] [CrossRef]
- Yan, Y.; Chang, L.; Wang, L. Laboratory Testing of SARS-CoV, MERS-CoV, and SARS-CoV-2 (2019-nCoV): Current Status, Challenges, and Countermeasures. Rev. Med. Virol. 2020, 20, e2106. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Rothman, R.E. PCR-Based Diagnostics for Infectious Diseases: Uses, Limitations, and Future Applications in Acute-Care Settings. Lancet Infect. Dis. 2004, 4, 337–348. [Google Scholar] [CrossRef]
- Zhang, Z.; Bi, Q.; Fang, S.; Wei, L.; Wang, X.; He, J.; Zou, X. Insight into the Practical Performance of RT-PCR Testing for SARS-Cov-2 Using Serological Data: A Cohort Study. Lancet Microbe 2021, 2, e79–e87. [Google Scholar] [CrossRef]
- Mao, Q.; Wang, Y.; Yao, X.; Bian, L.; Wu, X.; Xu, M.; Liang, Z. Coxsackievirus A16: epidemiology, diagnosis, and vaccine. Hum. Vaccines Immunother. 2014, 10, 360–367. [Google Scholar] [CrossRef]
- Wang, C.Y.; Lu, F.L.; Wu, M.H.; Lee, C.Y.; Huang, L.M. Fatal Coxsackievirus A16 Infection. Pediatr. Infect. Dis. J. 2004, 23, 275–276. [Google Scholar] [CrossRef]
- Hung, H.M.; Yang, S.L.; Chen, C.J.; Chiu, C.H.; Kuo, C.Y.; Huang, K.Y.A.; Lin, T.Y.; Hsieh, Y.C.; Gong, Y.N.; Tsao, K.C.; et al. Molecular Epidemiology and Clinical Features of Rhinovirus Infections Among Hospitalized Patients in a Medical Center in Taiwan. J. Microbiol. Immunol. Infect. 2019, 52, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.; Yip, C.; Woo, P.; Yuen, K.Y. Human Rhinovirus C: A Newly Discovered Human Rhinovirus Species. Emerg. Health Threat. J. 2010, 3, 7106. [Google Scholar] [CrossRef]
- Hao, W.; Bernard, K.; Patel, N.; Ulbrandt, N.; Feng, H.; Svabek, C.; Wilson, S.; Stracener, C.; Wang, K.; Suzich, J.; et al. Infection and Propagation of Human Rhinovirus C in Human Airway Epithelial Cells. J. Virol. 2012, 86, 13524–13532. [Google Scholar] [CrossRef]
- Xiang, Z.; Gonzalez, R.; Xie, Z.; Xiao, Y.; Liu, J.; Chen, L.; Liu, C.; Zhang, J.; Ren, L.; Vernet, G.; et al. Human Rhinovirus C Infections Mirror Those of Human Rhinovirus A in Children with Community-Acquired Pneumonia. J. Clin. Virol. 2010, 49, 94–99. [Google Scholar] [CrossRef] [PubMed]
- Kumpitsch, C.; Koskinen, K.; Schöpf, V.; Moissl-Eichinger, C. The Microbiome of the Upper Respiratory Tract in Health and Disease. BMC Biol. 2019, 17, 87. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.W.; Choudhary, I.; Paudel, K.; Mao, Y.; Sharma, R.; Wang, Y.; Deshane, J.S.; Boucher, R.C.; Patial, S.; Saini, Y. The Innate Lymphoid System is a Critical Player in the Manifestation of Mucoinflammatory Airway Disease in Mice. J. Immun. 2020, 205, 1695–1708. [Google Scholar] [CrossRef]
- Janda, J.M.; Abbott, S.L. 16S rRNA Gene Sequencing for Bacterial Identification in the Diagnostic Laboratory: Pluses, Perils, and Pitfalls. J. Clin. Microbiol. 2007, 45, 2761–2764. [Google Scholar] [CrossRef]
- Muhamad Rizal, N.S.; Neoh, H.M.; Ramli, R.; Periyasamy, P.R.; Hanafiah, A.; Abdul Samat, M.N.; Tan, T.L.; Wong, K.K.; Nathan, S.; Chieng, S.; et al. Advantages and Limitations of 16S Rrna Next-Generation Sequencing for Pathogen Identification in the Diagnostic Microbiology Laboratory: Perspectives from a Middle-Income Country. Diagnostics 2020, 10, 816. [Google Scholar] [CrossRef]
- Li, n.; Ma, X.; Zhou, J.; Deng, J.; Gu, C.; Fei, C.; Tao, F. Clinical Application of Metagenomic Next-Generation Sequencing Technology in the Diagnosis and Treatment of Pulmonary Infection Pathogens: A Prospective Single-Center Study of 138 Patients. J. Clin. Lab. Anal. 2022, 36, e24498. [Google Scholar] [CrossRef]
- Takeuchi, S.; Kawada, J.I.; Horiba, K.; Okuno, Y.; Okumura, T.; Suzuki, T.; Torii, Y.; Kawabe, S.; Wada, S.; Ikeyama, T.; et al. Metagenomic Analysis Using Next-Generation Sequencing of Pathogens in Bronchoalveolar Lavage Fluid from Pediatric Patients with Respiratory Failure. Sci. Rep. 2019, 9, 12909. [Google Scholar] [CrossRef] [PubMed]
- Miao, Q.; Ma, Y.; Wang, Q.; Pan, J.; Zhang, Y.; Jin, W.; Yao, Y.; Su, Y.; Huang, Y.; Wang, M.; et al. Microbiological Diagnostic Performance of Metagenomic Next-Generation Sequencing When Applied to Clinical Practice. Clin. Infect. Dis. 2018, 67, S231–S240. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Fang, K.; Shi, X.; Yang, D.; Zhao, L.; Yu, W.; Zheng, Y.; Xu, Y.; Ma, X.; Chen, L.; et al. Enhanced DNA and RNA pathogen detection via metagenomic sequencing in patients with pneumonia. J. Transl. Med. 2022, 22, 195. [Google Scholar] [CrossRef]
- Huang, C.; Chen, H.; Ding, Y.; Ma, X.; Zhu, H.; Zhang, S.; Du, W.; Summah, H.D.; Shi, G.; Feng, Y. A Microbial World: Could Metagenomic Next-Generation Sequencing Be Involved in Acute Respiratory Failure? Front. Cell. Infect. Microbiol. 2021, 11, 738074. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.; Mun, S.; Oh, Y.; Cho, C.S.; Yun, K.; Ahn, Y.; Chung, W.H.; Lim, M.Y.; Lee, K.E.; Hwang, T.S.; et al. A qRT-PCR Method Capable of Quantifying Specific Microorganisms Compared to NGS-Based Metagenome Profiling Data. Microorganisms 2022, 10, 324. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Ye, J.; Yang, L.; Chen, X.; Fang, H.; Liu, Z.; Xia, G.; Zhang, Y.; Zhang, Z. Inconsistency Analysis Between Metagenomic Next-Generation Sequencing Results of Cerebrospinal Fluid and Clinical Diagnosis with Suspected Central Nervous System Infection. BMC Infect. Dis. 2022, 22, 764. [Google Scholar] [CrossRef] [PubMed]
- Ramchandar, n.; Burns, J.; Coufal, N.G.; Pennock, A.; Briggs, B.; Stinnett, R.; Bradley, J.; Arnold, J.; Liu, G.Y.; Pring, M.; et al. Use of Metagenomic Next-Generation Sequencing to Identify Pathogens in Pediatric Osteoarticular Infections. Open Forum Infect. Dis. 2021, 8, 346. [Google Scholar] [CrossRef]
- Scavizzi, F.; Bassi, C.; Lupini, L.; Guerriero, P.; Raspa, M.; Sabbioni, S. A comprehensive approach for microbiota and health monitoring in mouse colonies using metagenomic shotgun sequencing. Anim. Microbiome 2021, 3, 53. [Google Scholar] [CrossRef]
- Hirsch, H.H.; Martino, R.; Ward, K.N.; Boeckh, M.; Einsele, H.; Ljungman, P. Fourth European Conference on Infections in Leukaemia (ECIL-4): Guidelines for Diagnosis and Treatment of Human Respiratory Syncytial Virus, Parainfluenza Virus, Metapneumovirus, Rhinovirus, and Coronavirus. Clin. Infect. Dis. 2013, 56, 258–266. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, R.E. Learning as Cognition: A Developmental Process for Organizational Learning. Dev. Learn. Organ. Int. J. 2021, 35, 18–21. [Google Scholar] [CrossRef]
- Robbins, B.; Carpenter, R.E.; Long, M.; Perry, J. A Human Oral Fluid Assay for D-and L-Isomer Detection of Amphetamine and Methamphetamine Using Liquid-Liquid Extraction. J. Anal. Methods Chem. 2022, 2022, 4819599. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| FTD® PCR Panel | Microorganism Classification | PCR Positive |
|---|---|---|
| Moraxella catarrhalis | Bacteria | 13 (45%) |
| Haemophilus influenzae | Bacteria | 9 (31%) |
| Streptococcus pneumoniae | Bacteria | 9 (31%) |
| Staphylococcus aureus | Bacteria | 5 (17%) |
| Human rhinovirus | Virus | 4 (14%) |
| Influenza A virus (H3N2) | Virus | 4 (14%) |
| Human coronavirus OC43 | Virus | 3 (10%) |
| Human metapneumoviruses | Virus | 3 (10%) |
| Human respiratory syncytial viruses | Virus | 3 (10%) |
| Human parainfluenza 1 virus | Virus | 2 (7%) |
| Enterovirus | Virus | 1 (3%) |
| Human parainfluenza 2 virus | Virus | 1 (3%) |
| Human parainfluenza 3 virus | Virus | 1 (3%) |
| Human parainfluenza 4 virus | Virus | 1 (3%) |
| Illumina® RPIP mNGS Panel | Microorganism Classification | mNGS Positive |
|---|---|---|
| Moraxella catarrhalis | Bacteria | 12 (41%) |
| Dolosigranulum pigrum | Bacteria | 12 (41%) |
| Haemophilus influenzae | Bacteria | 9 (31%) |
| Streptococcus pneumoniae | Bacteria | 9 (31%) |
| Stenotrophomonas maltophilia | Bacteria | 7 (24%) |
| Pseudomonas aeruginosa | Bacteria | 6 (21%) |
| Staphylococcus aureus | Bacteria | 5 (17%) |
| Corynebacterium pseudodiphtheriticum | Bacteria | 4 (14%) |
| Human rhinovirus A | Virus | 4 (14%) |
| Influenza A virus (H3N2) | Virus | 4 (14%) |
| Corynebacterium propinquum | Bacteria | 3 (10%) |
| Human coronavirus OC43 | Virus | 3 (10%) |
| Human metapneumovirus | Virus | 3 (10%) |
| Ochrobactrum anthropi | Bacteria | 3 (10%) |
| Prevotella melaninogenica | Bacteria | 3 (10%) |
| Respiratory syncytial virus B | Virus | 3 (10%) |
| Rothia mucilaginosa | Bacteria | 3 (10%) |
| Streptococcus mitis | Bacteria | 3 (10%) |
| Actinomyces graevenitzii | Bacteria | 2 (7%) |
| Alternaria alternata | Fungus | 2 (7%) |
| Campylobacter concisus | Bacteria | 2 (7%) |
| Capnocytophaga leadbetteri | Bacteria | 2 (7%) |
| Cytomegalovirus (CMV) | Virus | 2 (7%) |
| Gemella haemolysans | Bacteria | 2 (7%) |
| Human parainfluenza virus 1 | Virus | 2 (7%) |
| SARS-CoV-2 (2019-nCoV) | Virus | 2 (7%) |
| Veillonella parvula | Bacteria | 2 (7%) |
| Achromobacter xylosoxidans | Fungus | 1 (3%) |
| Actinomyces naeslundii | Fungus | 1 (3%) |
| Coxsackievirus A | Virus | 1 (3%) |
| Enterovirus D68 | Virus | 1 (3%) |
| Fusarium proliferatum | Fungus | 1 (3%) |
| Fusobacterium necrophorum | Fungus | 1 (3%) |
| Haemophilus haemolyticus | Bacteria | 1 (3%) |
| Haemophilus parainfluenzae | Bacteria | 1 (3%) |
| Human parainfluenza 4 virus | Virus | 1 (3%) |
| Human parainfluenza virus 2 | Virus | 1 (3%) |
| Human parainfluenza virus 3 | Virus | 1 (3%) |
| Human rhinovirus C | Virus | 1 (3%) |
| Influenza A virus (H1N1) | Virus | 1 (3%) |
| Neisseria flavescens | Bacteria | 1 (3%) |
| Neisseria lactamica | Bacteria | 1 (3%) |
| Pseudomonas stutzeri | Bacteria | 1 (3%) |
| Streptococcus intermedius | Bacteria | 1 (3%) |
| Phenotypic Group 1 | ||
|---|---|---|
| No. | PCR | mNGS |
| 1 | Haemophilus haemolyticus | |
| 2 | Neisseria flavescens | |
| 3 | Neisseria lactamica | |
| 4 | Dolosigranulum pigrum | |
| 5 | Alternaria alternata | |
| 6 | Campylobacter concisus | |
| 7 | Capnocytophaga leadbetteri | |
| 8 | Gemella haemolysans | |
| 9 | Veillonella parvula | |
| 10 | Corynebacterium propinquum | |
| 11 | Ochrobactrum anthropi | |
| 12 | Prevotella melaninogenica | |
| 13 | Rothia mucilaginosa | |
| 14 | Corynebacterium pseudodiphtheriticum | |
| Phenotypic Group 2 | ||
| 1 | Moraxella catarrhalis | Moraxella catarrhalis |
| 2 | Haemophilus influenzae | Haemophilus influenzae |
| 3 | Streptococcus pneumoniae | Streptococcus pneumoniae |
| 4 | Achromobacter xylosoxidans | |
| 5 | Actinomyces naeslundii | |
| 6 | Fusobacterium necrophorum | |
| 7 | Haemophilus parainfluenzae | |
| 8 | Pseudomonas stutzeri | |
| 9 | Streptococcus intermedius | |
| 10 | Actinomyces graevenitzii | |
| 11 | Cytomegalovirus (CMV) | |
| 12 | Fusarium proliferatum | |
| 13 | Streptococcus mitis | |
| 14 | Pseudomonas aeruginosa | |
| 15 | Stenotrophomonas maltophilia | |
| Phenotypic Group 3 | ||
| 1 | Enterovirus | Enterovirus |
| 2 | Human parainfluenza virus 2 | Human parainfluenza virus 2 |
| 3 | Human parainfluenza virus 3 | Human parainfluenza virus 3 |
| 4 | Human parainfluenza virus 4 | Human parainfluenza virus 4 |
| 5 | Influenza A virus (H1N1) swl | Influenza A virus (H1N1) swl |
| 6 | Human parainfluenza virus 1 | Human parainfluenza virus 1 |
| 7 | Human coronavirus OC43 | Human coronavirus OC43 |
| 8 | Human metapneumovirus | Human metapneumovirus |
| 9 | Respiratory syncytial virus B | Respiratory syncytial virus B |
| 10 | Human rhinovirus A | Human rhinovirus A |
| 11 | Influenza A virus (H3N2) | Influenza A virus (H3N2) |
| 12 | Staphylococcus aureus | Staphylococcus aureus |
| 13 | Coxsackievirus A (CAV) | |
| 14 | Human rhinovirus C | |
| 15 | SARS-CoV-2 (2019-nCoV) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Almas, S.; Carpenter, R.E.; Singh, A.; Rowan, C.; Tamrakar, V.K.; Sharma, R. Deciphering Microbiota of Acute Upper Respiratory Infections: A Comparative Analysis of PCR and mNGS Methods for Lower Respiratory Trafficking Potential. Adv. Respir. Med. 2023, 91, 49-65. https://doi.org/10.3390/arm91010006
Almas S, Carpenter RE, Singh A, Rowan C, Tamrakar VK, Sharma R. Deciphering Microbiota of Acute Upper Respiratory Infections: A Comparative Analysis of PCR and mNGS Methods for Lower Respiratory Trafficking Potential. Advances in Respiratory Medicine. 2023; 91(1):49-65. https://doi.org/10.3390/arm91010006
Chicago/Turabian StyleAlmas, Sadia, Rob E. Carpenter, Anuradha Singh, Chase Rowan, Vaibhav K. Tamrakar, and Rahul Sharma. 2023. "Deciphering Microbiota of Acute Upper Respiratory Infections: A Comparative Analysis of PCR and mNGS Methods for Lower Respiratory Trafficking Potential" Advances in Respiratory Medicine 91, no. 1: 49-65. https://doi.org/10.3390/arm91010006
APA StyleAlmas, S., Carpenter, R. E., Singh, A., Rowan, C., Tamrakar, V. K., & Sharma, R. (2023). Deciphering Microbiota of Acute Upper Respiratory Infections: A Comparative Analysis of PCR and mNGS Methods for Lower Respiratory Trafficking Potential. Advances in Respiratory Medicine, 91(1), 49-65. https://doi.org/10.3390/arm91010006