Abstract
An intronic GAA repeat expansion in the FGF14 gene was recently identified as a common cause of autosomal dominant GAA-FGF14 ataxia (SCA27B). We aimed to characterize in detail the clinical and video-oculographic features in our cohort of SCA27B patients. We genotyped the FGF14 GAA repeat expansion in 52 patients with unsolved late-onset cerebellar ataxia. Brain MRI and nerve conduction study, as well as video-oculographic (VOG) assessment, were performed. Eight patients (15.4%) with pathogenic GAA repeat expansion in the FGF14 gene were found. The median age at onset was 51 years (range—23–63 years). Sensory axonal neuropathy was found in 5/8 patients. Cerebellar atrophy was observed in 5/8 patients, and in one case, pontocerebellar atrophy was found. All tested patients had impaired smooth pursuit, 5/6 patients had impaired vestibulo-ocular reflex suppression, nystagmus, and an increased number of square wave jerks, 4/6 patients had horizontal gaze-evoked nystagmus, 3/6 had spontaneous downbeat nystagmus, and 1/6 had an upbeat one. Video head impulse test gain was lower than 0.8 on both sides in 2/4 patients, along with the presence of overt saccades. Further studies in different cohorts are needed to complete the phenotype of the FGF14-related disorders.
1. Introduction
Late-onset cerebellar ataxias (LOCAs) is a highly heterogenous group of progressive neurological disorders with predominantly cerebellar involvement [1]. The most common LOCAs types are multiple system atrophy, cerebellar type (MSA-C), idiopathic cerebellar ataxia, alcohol-induced, and immune-mediated ataxias, as well as hereditary spinocerebellar ataxias (SCA) caused by microsatellite tandem repeat expansions (e.g., SCA types 1, 2, 3, 6, and cerebellar ataxia, neuropathy, and vestibular areflexia syndrome [CANVAS]) [2,3,4].po
It has been recently reported that GAA expansion of >250 repeats in the first intron of the fibroblast growth factor 14 gene FGF14 causes a new type of hereditary ataxia, SCA27B (GAA-FGF14), with SCA27A being associated with FGF14 point mutations [5,6]. Clinical, radiological, neurophysiological, and genetic features of SCA27B have already been studied in different countries and cohorts [5,7,8,9,10,11,12]. It is estimated that this type of ataxia may account for 15–30% of cases in European cohorts of patients with unverified LOCAs [13]. The main clinical features of SCA27B are slowly progressive cerebellar ataxia with a frequent combination of sensory and sensorimotor neuropathy, bilateral vestibulopathy, downbeat nystagmus, paroxysmal diplopia, vertigo or ataxia with cerebellar vermis, and hemispheres atrophy [8,9,14,15].
The aim of this study was to present our own first data on the frequency of SCA27B among Russian patients with unsolved adult-onset ataxia and to determine their clinical characteristics. We reveal some phenotypic features of this ataxia type in our cohort (frequency of sporadic cases, evident anticipation phenomenon in one family, and absence of episodic symptomatic fluctuations, upbeat nystagmus). We also attempt to estimate as many video-oculographic (VOG) parameters in SCA27B patients as possible to fully describe the oculomotor features in our cohort, while most of the published data have focused mainly on the video head impulse test (vHIT) gain and the nystagmus characteristics. While analyzing vHIT, we pay attention not only to the lowered gain but also to the presence of covert and/or overt saccades.
2. Materials and Methods
2.1. Study Population
Patients attending the specialized neurogenetic clinic at the Research Center of Neurology, Moscow, were enrolled from January 2017 to September 2023. This was a single-center retrospective study conducted using the collection of DNA samples and medical records data. Local Ethical Committee approval was received (protocol no. 3, 18 July 2019), and all participants signed an informed consent form.
We evaluated 52 unrelated patients with progressive ataxia (sporadic-44 patients, family cases-8 patients), age-56.8 ± 21.7 years, age at onset-48.3 ± 17.3 years (28 males). Also, all patients underwent preliminarily FXN, RFC1, ATXN1, ATXN2, ATXN3, CACNA1A, ATXN8/ATXN8OS, and TBP gene analysis to exclude pathogenic microsatellite tandem repeat expansions.
2.2. Clinical, Neurophysiology, and Neuroimaging Assessments
Apart from routine neurological examination, all patients were evaluated with the Scale for the Assessment and Rating of Ataxia (SARA). Mini-Mental State Examination (MMSE) performance was used as a screening tool for cognitive impairment. Brain magnetic resonance imaging (MRI) and nerve conduction study (NCS) with sensory nerve action potentials, muscle action potentials, and conduction velocity assessment [16] were performed in all patients.
VOG assessment was carried out using monocular EyeSeeCam Sci system (EyeSeeTec GmbH, Munich, Germany). Left eye movements were recorded. Test battery included reflexive and self-paced horizontal and vertical saccades, smooth pursuit, gaze holding during fixation and in darkness, and vestibulo-ocular reflex (VOR) suppression. Additional vHIT testing in the plane of horizontal canals was performed.
The reflexive saccade test had a pseudo-random step stimulation with an amplitude of 10 and 20° for vertical saccades and 15 and 30° for horizontal saccades. Self-paced saccades included stimulation at 20° for vertical and 30° for horizontal saccades. Smooth pursuit was performed with the target moving at 0.2 Hz, maximum velocity 20°/s. Gaze holding comprised fixation in primary gaze position, as well as towards 10° up, 10° down, 20° left, and 20° right stimuli. In the VOR suppression test, patients were asked to fixate on a head-fixed target while rotating in the darkness in the horizontal plane with a maximum head velocity of 50°/s.
Saccadic peak velocity, latency, and amplitude gain, as well as velocity gain of smooth pursuit, were calculated according to published data [17]. Saccadic parameters were then compared with data obtained from healthy subjects [18]. The presence of saccadic slowing was estimated based on saccadic velocity, and saccadic dysmetria was based on saccadic amplitude gain. Rate of square wave jerks was calculated manually; the cut-off value for a normal number of saccadic intrusions was set to less than 27 per minute. VOR suppression was regarded as impaired if the VOR gain obtained in this test exceeded 0.14. Smooth pursuit was considered to be impaired with a gain lower that 0.7 in at least two directions. The presence of nystagmus was established based on slow phase velocity parameter and confirmed upon visual analysis of recorded graphs.
2.3. Genetic Assessment
Genomic DNA was extracted from blood leucocytes using extraction kit Wizard Genomic DNA Purification Kit (Promega Corporation, Madison, WI, USA).
Screening of the FGF14 GAA repeat expansion was performed by flanking polymerase chain reaction (PCR) and repeat-primed polymerase chain reaction (RP-PCR).
The primers flank the trinucleotide repeat region and are predicted to amplify a 306 bp fragment based on hg38 reference sequence (which includes 50 GAA repeats) using standard PCR conditions. Products were visualized using agarose gel electrophoresis and automatic capillary electrophoresis (QSep100, BiOptic Inc., Taipei, Taiwan) fragment length analysis of FAM-labeled PCR products was performed on a Generic Analyzer Nanophore 05 (Institute for Analytical Instrumentation RAS, Moscow, Russia) using GeneMarker (SoftGenetics 3.0.1) (Figure 1).
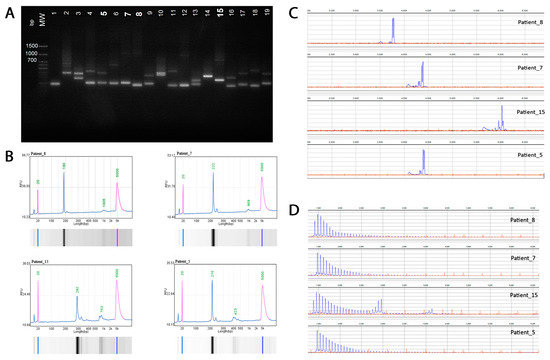
Figure 1.
Molecular analysis of the intronic FGF14 GAA repeat locus. (A) Agarose gel electrophoresis (1.5%): lanes 7, 8, 12, and 15, >250 repeat units. (B) Automatic capillary electrophoresis (QSep100) of four patients: Patient #8 (20/280 repeat units); Patient #7 (8/277 repeat units); Patient #15 (43/197 repeat units), and Patient #5 (18/87 repeat units). (C) Fragment length analysis of fluorescent PCR amplification products of four patients. (D) Results of RP-PCR for the expanded (GAA)n repeat of four patients demonstrated a characteristic ‘sawtooth’ pattern on the electropherogram.
The presence of an expanded (GAA)n repeat was confirmed by RP-PCR utilizing the following three primers: repeat specific forward primer (FGF14 RP F), anchor forward primer corresponding to the anchor tail of the forward primer (FGF14 RPa F), and locus-specific FAM labeled reverse primer (FGF14 R(Fam)). The presence of expansion was indicated by the appearance of a characteristic ‘sawtooth’ pattern on the electropherogram. Primer synthesis was carried out at the company “Syntol” (Moscow, Russia).
2.4. Statistical Analysis
We used standard descriptive statistics for values with non-normal distribution (median, range, min–max). To assess the normality of distribution, Shapiro–Wilk test was used. Spearman’s rank correlation coefficient was used to measure the association between length of GAA repeats and the age of disease onset. Statistical significance was indicated if p < 0.05.
3. Results
Eight patients (15.4%) with pathogenic GAA repeat expansion (median size-272.5 GAA repeats, range-256–290) in the FGF14 gene were found (median age-61 years, range-36–73 years, 5 males). Median age at onset was 51 years, range 23–63 years. All patients were of Slavic origin. Four patients had a positive family history corresponding to autosomal dominant inheritance. An anticipation phenomenon was discovered in only one family (patient #5), in which the age of onset was 23 years for the proband, 30 years for the mother, and 53 years for the grandfather (Table 1).

Table 1.
Clinical, neuroimaging and oculomotor characteristics of the patients with SCA27B. Missed values can be seen in the cells where the presence of the sign was not checked. NCS—nerve conduction study, SWJ—square-wave jerks, VOR—vestibulo-ocular reflex, vHIT—video head impulse test, NA—not available.
All SCA27B patients had cerebellar nystagmus and moderate gait ataxia (SARA 11–17.5 points) and walked without support. Other symptoms were cerebellar dysarthria (7/8), ataxia in the upper limbs (6/8), mild pyramidal signs (hyperreflexia, extensor plantar response, 2/8), overactive bladder (2/8), and mild cognitive impairment (MMSE < 27 points, 2/8).
In 5/8 patients, NCS showed a pronounced symmetrical sensory axonal polyneuropathy with absent or nearly absent sensory responses and almost no motor involvement.
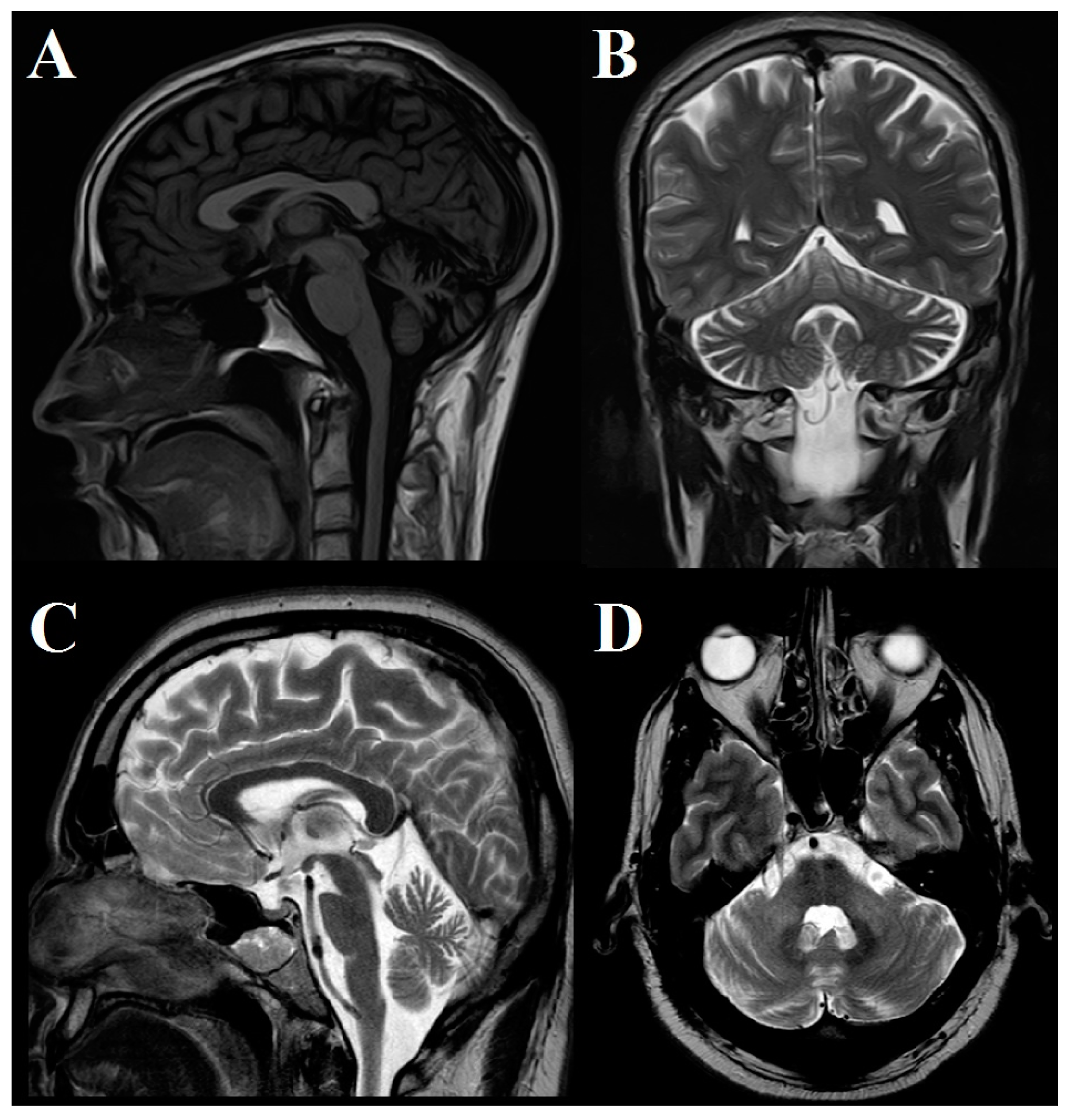
In two patients, brain MRI was normal; in 5 patients, mild to moderate atrophy of the cerebellar hemispheres and vermis was observed; and in 1 case, pontocerebellar atrophy was found (Figure 2).
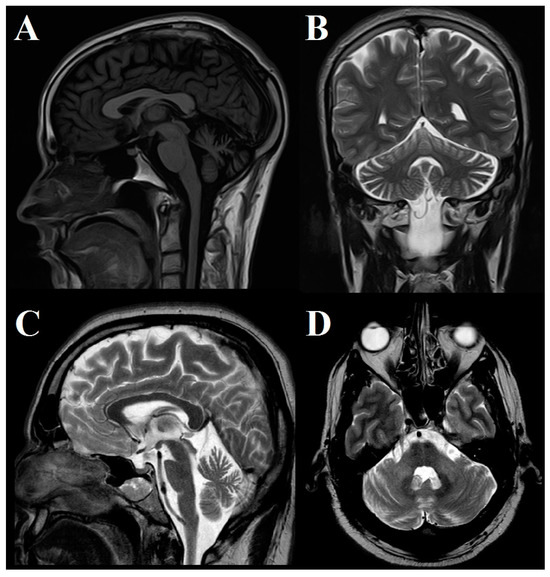
Figure 2.
Brain MRI study. (A) Saggital T2 FLAIR and (B) coronal T2 images in Patient #5 show moderate cerebellar atrophy involving both hemispheres and anterior and superior vermis. (C) Sagittal and (D) axial T2 images in Patient #7 show moderate cerebellar and brainstem atrophy.
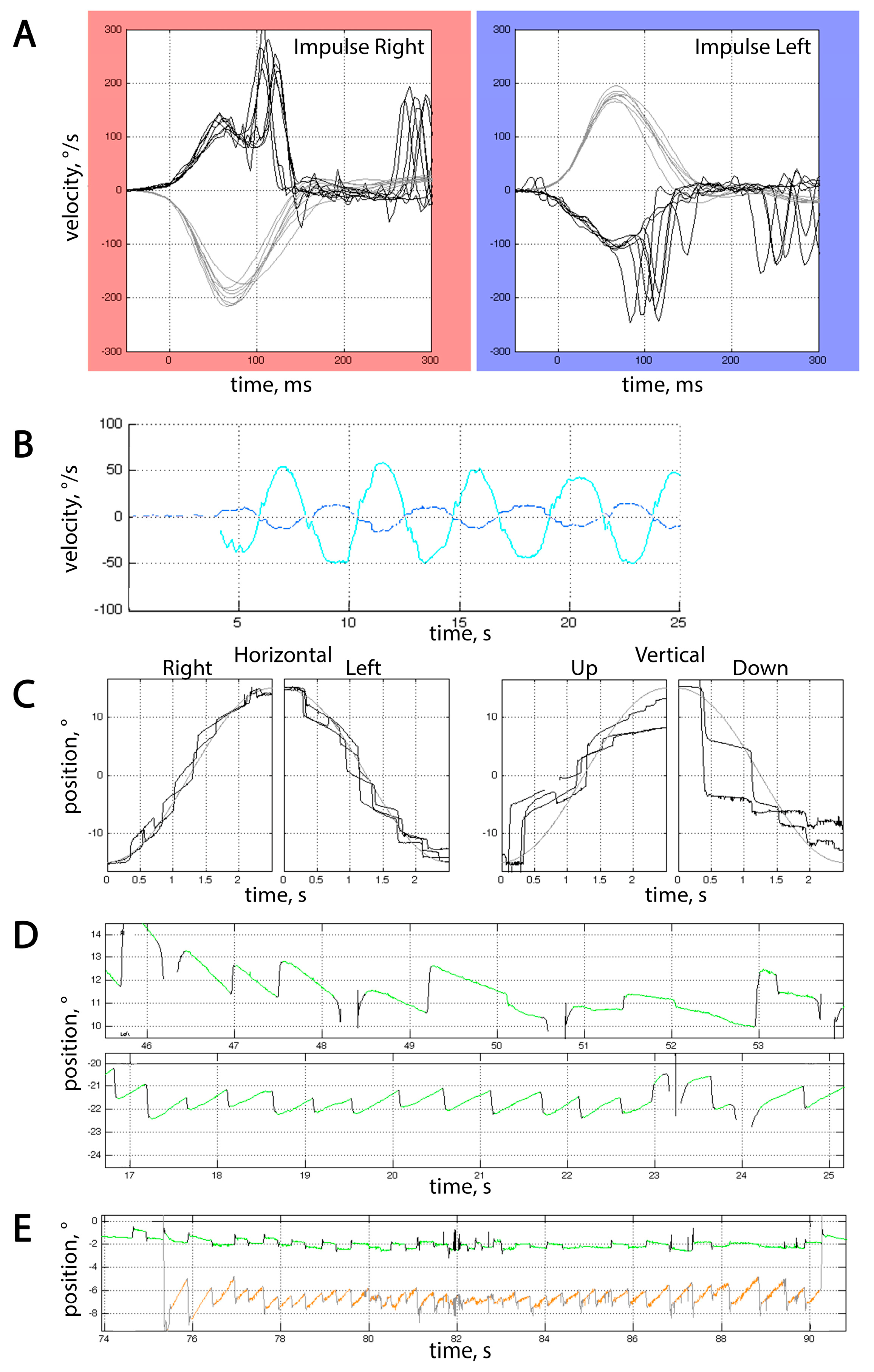
VOG results were available for 6 patients, and horizontal vHIT for 4 patients. All tested patients had impaired smooth pursuit with vertical positional gain of 0.31 [0.20; 0.45] and horizontal positional gain of 0.55 [0.43; 0.68] (Figure 3C). Besides that, all patients but 1 had impaired VOR suppression (gain 0.23 [0.2; 0.27]), nystagmus, and increased number of square wave jerks (SWJ) per minute (45.0 [40.0; 62.0]) in the gaze holding test, which was consistent with cerebellar dysfunction (Figure 3B,E). It is noteworthy that no saccadic dysmetria could be detected in our cohort of patients. No dysmetria of self-paced saccades could be detected in our cohort of patients, but reflexive saccades were either hypometric or hypermetric at least in one direction in 5/6 patients.
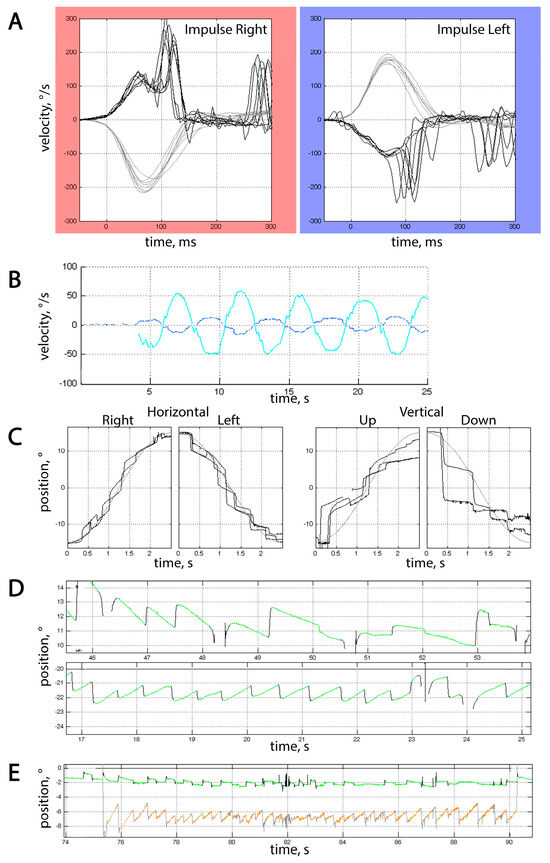
Figure 3.
Typical video-oculographic findings in SCA27B patients. (A) Horizontal vHIT in Patient #8. Grey curves represent the velocities of passive head rotation over time, and black curves represent eye velocity. Note the presence of both covert and overt saccades. Gain at 60th ms for rightward impulses was 0.65, and 0.57 for leftward ones. (B) Insufficient VOR suppression in Patient #8. The cy an curve represents horizontal head rotation over time, and the blue curve represents horizontal vestibular eye movements that the patient was unable to fully suppress, which resulted in a gain of 0.24. (C) Saccadic pursuit in Patient #1. Grey and black curves represent changes in stimulus and eye position over time, respectively. (D) Horizontal eye movements during rightward (upper part) and leftward (lower part) fixation in Patient #5. Horizontal gaze-evoked nystagmus can be seen, with a slow phase velocity (SPV) of 2°/s for leftward and 0.8°/s for rightward gaze holding. (E) Combination of spontaneous downbeat nystagmus and excessive SWJ in Patient #2. The green curve stands for horizontal eye position over time, and orange for vertical. SPV for downbeat nystagmus was 3.5°/s. In (B,D,E), upward deflections represent movement either up or to the right, and downward deflections represent movement either down or to the left, depending on the color of the curve.
Horizontal gaze-evoked nystagmus was the most prevalent type of nystagmus, seen in 4 of 6 patients (Figure 3D); in some cases, it was asymmetrical. In addition, spontaneous downbeat nystagmus could be seen in 3 of these patients, and upbeat in 1. One patient had right-beat fixation-off nystagmus while having normal gain in vHIT, which makes it less likely to be vestibular in origin.
Saccadic velocities were normal in all tested patients, corresponding to preserved brainstem function on neurological examination and absence of brainstem atrophy on MRI (the only patient with MRI brainstem atrophy did not undergo VOG study). vHIT gain was lower than 0.8 on both sides in two patients, along with the presence of overt saccades (Figure 3A), but it did not exceed the lower limit of 0.6; so, the obtained data were inconsistent with bilateral vestibulopathy [19]. Bilateral vHIT gain of <0.8 and >0.6 in one 73 y.o. patient led to the initial assumption of presbyvestibulopathy [20], but the presence of various types of vHIT abnormalities in all tested patients of different ages makes this hypothesis less probable. All tested patients, irrespective of vHIT gain, had overt saccades that could not be attributed to nystagmus or saccadic intrusions, and one patient with a gain of <0.8 also had covert saccades.
4. Discussion
In our study, we identified pathogenic FGF14 GAA repeat expansions in 8 out of 52 unrelated patients (15.4%) with progressive cerebellar ataxia. This study was conducted for the first time in Russia, and GAA-FGF14 repeat expansions have not been previously documented in Russian patients with unsolved LOCAs.
It was previously shown that SCA27B was found in different populations and ethnic groups among patients with adult-onset unverified ataxia, as follows: France-15/118 (12.7%) [8], Germany–26/79 (32.9%) [21], Spain-18/64 (28.1%) [22], Greece–19/540 (11.9%) [11], Brazil –8/93 (8.6%) [7], Japan-11/940 (1.2%) [12], China–17/1216 (1.3%) [23], and Serbia–9/167 (5.4%) [24]. The obtained data on the frequency of SCA27B in different countries need to be clarified. In some populations, a “founder effect” is possible (for example, in Western Europe compared to East Europe and Asian countries), or patient cohorts may vary across studies (e.g., all types of ataxia or predominantly familial cases, etc.). According to our preliminary data, SCA27B is one of the most common forms of autosomal dominant SCA in Russia; less common than SCA1 and SCA2, approximately equal in frequency to SCA3, and more common than SCA6, SCA7, SCA8, and SCA17 [25,26,27].
The median age at onset of our SCA27B patients was 51 years, which is younger than in most previous studies [8,10,11,12]. According to the available data, the GAA repeat length seems to only weakly correlate with age at onset [13]. Wirth T. et al. did not find a correlation between GAA expansion size and clinical features. including age of onset [8]. We also could not see a correlation between the length of GAA repeats and age of disease onset (rS = −0.10, p = 0.57).
SCA27B is characterized by a high frequency of sporadic cases, varying between 15 and 50% [22,28], and in our cohort, 50% of cases were sporadic. These findings are presumably associated with the significant instability of the FGF14 GAA repeat locus upon intergenerational transmission [13].
The main clinical characteristic of SCA27B is a slowly progressive pancerebellar syndrome, with gait ataxia being more severe than appendicular ataxia [8,15]. Sensory and sensorimotor axonal neuropathies are common manifestations of SCA27B, and demyelinating neuropathies are rare [8]. In our cohort, five patients had pure sensory axonal neuropathy without involvement of motor fibers. In such cases, ataxia is combined, sensory and cerebellar, resembling a CANVAS phenotype. Pyramidal syndrome is observed in rare patients with SCA27B and usually presents with hyperreflexia and Babinski sign without muscle weakness or severe spasticity [8,12]. Two patients had signs of neurogenic overactive bladder without post-void residue or orthostatic hypotension. Additionally, one of our patients had pontocerebellar atrophy according to the brain MRI study, which is also one of the signs of MSA-C. Cerebellar and brainstem atrophy have been previously described in rare patients with SCA27B [8], and cases with pathogenic GAA-FGF14 expansion have been also described in patients with clinically diagnosed MSA-C [23,29]. Summarizing these findings, we support the use of FGF14 GAA expansion screening in patients with suspected MSA-C in doubtful cases.
Cognitive functions in SCA27B have not been sufficiently studied yet. In summary, severe cognitive impairment and dementia are not typical for GAA-FGF14, even despite advanced age at examination [15]. In our cohort, mild cognitive impairment was detected in two patients and did not correlate with signs of small vessel disease according to brain MRI data (Fazekas grade 1).
It is known that point mutations in the FGF14 gene cause SCA27A and episodic ataxia type 9 [6,30]. Moreover, it has been shown that episodic ataxia and episodic symptomatic fluctuations are also characteristic of SCA27B [15]. Among our patients, there were no cases with paroxysmal ataxia or vertigo even at the beginning of the disease, and cerebellar ataxia was slowly progressive. In our cohort, there were also no cases of parkinsonism, dystonia, postural tremor, amyotrophia, or fasciculations, which were described in previous studies [7,8,15].
Brain MRI study in SCA27B often shows isolated cerebellar atrophy predominantly in the vermis, as observed in our cases [15]. Nevertheless, in some patients, cerebral MRI is normal, without signs of cerebellar hemispheric or vermis atrophy [9,13]. In sporadic cases with sudden onset of ataxia, vertigo or diplopia, and normal cerebral MRI, it is necessary to suspect immune-mediated ataxias, especially cerebellar ataxia with glutamic acid decarboxylase antibodies [31].
Smooth pursuit abnormalities found in all patients could be at least in part attributed to age-dependent changes in visual tracking. Other VOG findings suggestive of cerebellar abnormalities, such as nystagmus, SWJ, or VOR suppression impairment, are not a part of normal aging. In our cohort, horizontal gaze-evoked nystagmus was more frequent than downbeat nystagmus, similar to the data published in a Chinese cohort [23]. Upbeat gaze-evoked nystagmus in addition to horizontal gaze-evoked nystagmus was documented in our patient for the first time. Different results were obtained for positional accuracy of reflexive and self-paced saccades: the latter had no dysmetria, while reflexive was dysmetric to some extent in 83% of cases. According to published data, saccadic dysmetria was present in nearly one-third of SCA27B patients [32].
VOR pathology was previously described in SCA27B patients documented by either vHIT, rotatory chair, or caloric testing [13,32,33]. In examined patients, the prevalence of bilateral vestibulopathy (BV) varies between 11% and 67% according to the published data [13,24,32,33]. Such differences could depend on the size of the cohort, the severity of the disease course, and the vHIT gain value assumed to be characteristic for BV (<0.7 or <0.6 on both sides). None of our patients had BV, but some of them had VOR gain below normal values. In previously published data [32,33], only the vHIT gain was taken into account, according to the established BV criteria [19]. Paying attention to covert and overt saccades in addition to the gain allowed the observation that VOR was somewhat compromised in all of our SCA27B cohort. Mild VOR abnormalities in GAA-FGF14 could possibly have another pathophysiological mechanism than in the RFC1 spectrum, including cerebellar involvement [33]. The vHIT abnormalities present in some form in all of our tested patients, also being mild, show some overlap with other syndromes manifested by cerebellar ataxia, sensory axonal neuropathy, and vestibulopathy, especially CANVAS (given similar age of onset) and POLG-associated SANDO syndrome [34]. It remains unclear whether vHIT gain will gradually worsen as the disease progresses or whether it will remain stable. Both patients with decreased vHIT gains had axonal sensory polyneuropathy, but our cohort is too small for now to establish a clear relationship between vHIT gain and NCS parameters.
It is known that some drugs (e.g., benzodiazepines, tricyclic antidepressants, anticholinergic medications) can affect oculomotor functions. In our cohort, Patient #2 was taking imipramine. It has previously been shown that tricyclic antidepressants could cause oculomotor symptoms, including internuclear ophthalmoplegia, partial or total gaze palsy, and opsoclonus [35], but these are not the signs that were seen in this particular patient. Patient #6 was taking amantadine, and her oculomotor findings also could not be explained by medication intake. Other patients did not take any medications affecting oculomotor function.
The combination of VOG and vHIT abnormalities found in our patients (Figure 3) is not disease-specific, but it can help narrow down the spectrum of suspected diseases. A combination of impaired smooth pursuit, cerebellar nystagmus, impaired VOR with inconclusive vHIT changes, and no saccadic velocity/gain abnormalities should be critically revised as the number of cases examined increases.
Our study has some limitations. We did not include patients with MSA-C, so this could result in an underestimation of SCA27B patients with autonomic symptoms. As a retrospective study, the clinical, radiological, and VOG data were taken before the patients were proven to have SCA27B, so we did not have some important data (including segregation analysis in affected families). We also did not conduct reliability testing and training to ensure consistency among raters who tested patients with rating scales (SARA, MMSE).
5. Conclusions
Our study suggests that SCA27B ataxia is a common form among unverified late-onset cerebellar ataxia in a Russian cohort. According to our data, genetic testing (GAA expansion analysis in the FGF14 gene) should be performed in adult patients (sporadic and familial cases) with slowly progressive cerebellar and sensory ataxia with sensory axonal neuropathy, spontaneous vertical nystagmus, and vHIT abnormalities (suboptimal gain with covert saccades). Brain MRI study is non-specific and may present as normal or reveal isolated cerebellar atrophy or pontocerebellar atrophy. Further studies in different cohorts and ethnic groups are needed to complete the phenotype of the FGF14-related disorders.
Author Contributions
Conceptualization, E.N. and S.I.; investigation, E.N., A.P., A.B.-B. and E.L.; writing-original draft, E.N., N.A., A.P., A.B.-B. and E.L.; methodology, N.A. and A.B.-B.; writing-review and editing, E.F., S.K. and S.I.; conceptualization and supervision, S.I. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by a grant from the Russian Science Foundation (project no. 24-15-00209).
Institutional Review Board Statement
This study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Ethics Committee of the Research Center of Neurology (protocol no. 3, 18 July 2019).
Informed Consent Statement
All reported patient data are anonymized. Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data supporting the findings of this study are available within the article.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Giordano, I.; Harmuth, F.; Jacobi, H.; Paap, D.; Vielhaber, S.; Machts, J.; Schols, L.; Synofzik, M.; Sturm, M.; Tallaksen, C.; et al. Clinical and genetic characteristics of sporadic adult-onset degenerative ataxia. Neurology 2017, 89, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Hadjivassiliou, M.; Martindale, J.; Shanmugarajah, P.; Grünewald, R.A.; Sarrigiannis, P.G.; Beauchamp, N.; Garrard, K.; Warburton, R.; Sanders, D.S.; Friend, D.; et al. Causes of progressive cerebellar ataxia: Prospective evaluation of 1500 patients. J. Neurol. Neurosurg. Psychiatry 2017, 88, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Kuwabara, S.; Nakamura, K.; Abe, R.; Matsushima, A.; Beppu, M.; Yamanaka, Y.; Takahasi, Y.; Sasaki, H.; Mizusawa, H.; et al. Idiopathic cerebellar ataxia (IDCA): Diagnostic criteria and clinical analyses of 63 Japanese patients. J. Neurol. Sci. 2018, 384, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Cortese, A.; Simone, R.; Sullivan, R.; Vandrovcova, J.; Tariq, H.; Yau, W.Y.; Humphrey, J.; Jaunmuktane, Z.; Sivakumar, P.; Polke, J.; et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Genet. 2019, 51, 649–658. [Google Scholar] [CrossRef]
- Pellerin, D.; Danzi, M.C.; Wilke, C.; Renaud, M.; Fazal, S.; Dicaire, M.J.; Scriba, C.K.; Ashton, C.; Yanick, C.; Beijer, D.; et al. Deep Intronic FGF14 GAA Repeat Expansion in Late-Onset Cerebellar Ataxia. N. Engl. J. Med. 2023, 388, 128–141. [Google Scholar] [CrossRef]
- van Swieten, J.C.; Brusse, E.; de Graaf, B.M.; Krieger, E.; van de Graaf, R.; de Koning, I.; Maat-Kievit, A.; Leegwater, P.; Dooijes, D.; Oostra, B.A.; et al. A mutation in the fibroblast growth factor 14 gene is associated with autosomal dominant cerebellar ataxia. Am. J. Hum. Genet. 2003, 72, 191–199. [Google Scholar] [CrossRef]
- Novis, L.E.; Frezatti, R.S.; Pellerin, D.; Tomaselli, P.J.; Alavi, S.; Della Coleta, M.V.; Spitz, M.; Dicaire, M.-J.; Iruzubieta, P.; Pedroso, J.L.; et al. Frequency of GAA-FGF14 Ataxia in a Large Cohort of Brazilian Patients with Unsolved Adult-Onset Cerebellar Ataxia. Neurol. Genet. 2023, 9, e200094. [Google Scholar] [CrossRef]
- Wirth, T.; Clément, G.; Delvallée, C.; Bonnet, C.; Bogdan, T.; Iosif, A.; Schalk, A.; Chanson, J.-B.; Pellerin, D.; Brais, B.; et al. Natural History and Phenotypic Spectrum of GAA-FGF14 Sporadic Late-Onset Cerebellar Ataxia (SCA27B). Mov. Disord. 2023, 38, 1950–1956. [Google Scholar] [CrossRef]
- Rafehi, H.; Read, J.; Szmulewicz, D.J.; Davies, K.C.; Snell, P.; Fearnley, L.G.; Scott, L.; Thomsen, M.; Gilies, G.; Pope, K.; et al. An intronic GAA repeat expansion in FGF14 causes the autosomal-dominant adult-onset ataxia SCA50/ATX-FGF. Am. J. Hum. Genet. 2023, 110, 105–119. [Google Scholar] [CrossRef]
- Bonnet, C.; Pellerin, D.; Roth, V.; Clément, G.; Wandzel, M.; Lambert, L.; Frismand, S.; Douarinou, M.; Grosset, A.; Bekkour, I.; et al. Optimized testing strategy for the diagnosis of GAA-FGF14 ataxia/spinocerebellar ataxia 27B. Sci. Rep. 2023, 13, 9737. [Google Scholar] [CrossRef]
- Kartanou, C.; Mitrousias, A.; Pellerin, D.; Kontogeorgiou, Z.; Iruzubieta, P.; Dicaire, M.-J.; Danzi, M.C.; Koniari, C.; Athanassopoulos, K.; Panas, M.; et al. The FGF14 GAA repeat expansion in Greek patients with late-onset cerebellar ataxia and an overview of the SCA27B phenotype across populations. Clin. Genet. 2024, 105, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Ando, M.; Higuchi, Y.; Yuan, J.; Yoshimura, A.; Kojima, F.; Yamanishi, Y.; Aso, Y.; Izumi, K.; Imada, M.; Maki, Y.; et al. Clinical variability associated with intronic FGF14 GAA repeat expansion in Japan. Ann. Clin. Transl. Neurol. 2024, 11, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, D.; Danzi, M.C.; Renaud, M.; Houlden, H.; Synofzik, M.; Zuchner, S.; Zuchner, S.; Brais, B. Spinocerebellar ataxia 27B: A novel frequent and potentially treatable ataxia. Clin. Transl. Med. 2024, 14, e1504. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, D.; Wilke, C.; Traschütz, A.; Nagy, S.; Currò, R.; Dicaire, M.J.; Garcia-Moreno, H.; Anheim, M.; Wirth, T.; Faber, J.; et al. Intronic FGF14 GAA repeat expansions are a common cause of ataxia syndromes with neuropathy and bilateral vestibulopathy. J. Neurol. Neurosurg. Psychiatry. 2024, 95, 175–179. [Google Scholar] [CrossRef]
- Méreaux, J.L.; Davoine, C.S.; Pellerin, D.; Coarelli, G.; Coutelier, M.; Ewenczyk, C.; Monin, M.-L.; Anheim, M.; Le Ber, I.; Thobois, S.; et al. Clinical and genetic keys to cerebellar ataxia due to FGF14 GAA expansions. EbioMedicine 2024, 99, 104931. [Google Scholar] [CrossRef]
- Preston, D.C.; Shapiro, B.E. Electromyography and Neuromuscular Disorders, 3rd ed.; Elsevier Health Sciences: Philadelphia, PA, USA, 2012; 664p. [Google Scholar]
- Bremova-Ertl, T.; Abel, L.; Walterfang, M.; Salsano, E.; Ardissone, A.; Malinova, V.; Kolnikova, M.; Bayarri, J.G.; Tavasoli, A.R.; Ashrafi, M.R.; et al. A cross-sectional, prospective ocular motor study in 72 patients with Niemann-Pick disease type C. Eur. J. Neurol. 2021, 28, 3040–3050. [Google Scholar] [CrossRef]
- Hopf, S.; Liesenfeld, M.; Schmidtmann, I.; Ashayer, S.; Pitz, S. Age dependent normative data of vertical and horizontal reflexive saccades. PLoS ONE 2018, 13, e0204008. [Google Scholar] [CrossRef]
- Strupp, M.; Kim, J.S.; Murofushi, T.; Straumann, D.; Jen, J.C.; Rosengren, S.M.; della Santina, C.C.; Kingma, H. Bilateral vestibulopathy: Diagnostic criteria consensus document of the classification committee of the Barany society. J. Vestib. Res. Equilib. Orientat. 2017, 27, 177–189. [Google Scholar] [CrossRef]
- Agrawal, Y.; Van de Berg, R.; Wuyts, F.; Walther, L.; Magnusson, M.; Oh, E.; Sharpe, M.; Strupp, M. Presbyvestibulopathy: Diagnostic criteria Consensus document of the classification committee of the Bárány Society. J. Vestib. Res. 2019, 29, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Hengel, H.; Pellerin, D.; Wilke, C.; Fleszar, Z.; Brais, B.; Haack, T.; Traschütz, A.; Schöls, L.; Synofzik, M. As frequent as polyglutamine spinocerebellar ataxias: SCA27B in a large german autosomal dominant ataxia cohort. Mov. Disord. 2023, 38, 1557–1558. [Google Scholar] [CrossRef] [PubMed]
- Iruzubieta, P.; Pellerin, D.; Bergareche, A.; Albajar, I.; Mondragón, E.; Vinagre, A.; Fernandez-Torron, R.; Moreno, F.; Equiza, J.; Campo-Caballero, D.; et al. Frequency and phenotypic spectrum of spinocerebellar ataxia 27B and other genetic ataxias in a Spanish cohort of late-onset cerebellar ataxia. Eur. J. Neurol. 2023, 30, 3828–3833. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, R.; Wan, L.; Pellerin, D.; Long, Z.; Hu, J.; Jiang, Q.; Wang, C.; Peng, L.; Peng, H.; He, L.; et al. The genetic landscape and phenotypic spectrum of GAA-FGF14 ataxia in China: A large cohort study. EbioMedicine 2024, 102, 105077. [Google Scholar] [CrossRef] [PubMed]
- Milovanović, A.; Dragaševic-Mišković, N.; Thomsen, M.; Borsche, M.; Hinrichs, F.; Westenberger, A.; Klein, C.; Bruggemann, N.; Brankovic, M.; Marjanovic, A.; et al. RFC1 and FGF14 Repeat Expansions in Serbian Patients with Cerebellar Ataxia. Mov. Disord. Clin. Pract. 2024, 11, 626–633. [Google Scholar] [CrossRef] [PubMed]
- Shuvaev, A.N.; Belozor, O.S.; Smolnikova, M.V.; Yakovleva, D.A.; Shuvaev, A.N.; Kazantseva, O.M.; Pozhilenkova, E.A.; Mozhei, O.I.; Kasparov, S. Population genetics of spinocerebellar ataxias caused by polyglutamine expansions. Vavilov J. Genet. Breed. 2019, 23, 473–481. [Google Scholar] [CrossRef]
- Nuzhnyi, E.P.; Abramycheva, N.Y.; Chkhartishvili, I.A.; Protopopova, A.O.; Fedotova, E.Y.; Illarioshkin, S.N. Spinocerebellar ataxia type 8 in Russian patients. S.S. Korsakov J. Neurol. Psychiatry 2022, 122, 106–111. [Google Scholar] [CrossRef]
- Ivanova, E.; Nuzhnyi, E.; Abramycheva, N.; Klyushnikov, S.; Fedotova, E.; Illarioshkin, S. Mutation analysis of the TATA box-binding protein (TBP) gene in Russian patients with spinocerebellar ataxia and Huntington disease-like phenotype. Clin. Neurol. Neurosurg. 2022, 222, 107473. [Google Scholar] [CrossRef]
- Wilke, C.; Pellerin, D.; Mengel, D.; Traschütz, A.; Danzi, M.C.; Dicaire, M.J.; Neumann, M.; Lerche, H.; Bender, B.; Houlden, H.; et al. GAA-FGF14 ataxia (SCA27B): Phenotypic profile, natural history progression and 4-aminopyridine treatment response. Brain 2023, 146, 4144–4157. [Google Scholar] [CrossRef]
- Wirth, T.; Bonnet, C.; Delvallée, C.; Pellerin, D.; Bogdan, T.; Clément, G.; Schalk, A.; Chanson, J.-B.; Fleury, M.-C.; Piton, A.; et al. Does Spinocerebellar ataxia 27B mimic cerebellar multiple system atrophy? J. Neurol. 2024, 271, 2078–2085. [Google Scholar] [CrossRef]
- Piarroux, J.; Riant, F.; Humbertclaude, V.; Remerand, G.; Hadjadj, J.; Rejou, F.; Coubes, C.; Pinson, L.; Meyer, P.; Roubertie, A. FGF14-related episodic ataxia: Delineating the phenotype of Episodic Ataxia type. Ann. Clin. Transl. Neurol. 2020, 7, 565–572. [Google Scholar] [CrossRef]
- Matsumoto, S.; Kusuhara, T.; Nakajima, M.; Ouma, S.; Takahashi, M.; Yamada, T. Acute attacks nd brain stem signs in a patient with glutamic acid decarboxylase autoantibodies. J. Neurol. Neurosurg. Psychiatry 2002, 73, 345–346. [Google Scholar] [CrossRef]
- Pellerin, D.; Heindl, F.; Traschütz, A.; Rujescu, D.; Hartmann, A.; Brais, B.; Houlden, H.; Dufke, C.; Riess, O.; Haack, T.; et al. RFC1 repeat expansions in downbeat nystagmus syndromes: Frequency and phenotypic profile. J. Neurol. 2024, 271, 2886–2892. [Google Scholar] [CrossRef] [PubMed]
- Borsche, M.; Thomsen, M.; Szmulewicz, D.J.; Lübbers, B.; Hinrichs, F.; Lockhart, P.J.; Lohmann, K.; Helmnchen, C.; Bruggemann, N. Bilateral vestibulopathy in RFC1-positive CANVAS is distinctly different compared to FGF14-linked spinocerebellar ataxia 27B. J. Neurol. 2024, 271, 1023–1027. [Google Scholar] [CrossRef] [PubMed]
- Belyakova-Bodina, A.; Ratanov, M.; Schneider, E.; Seliverstov, Y.; Nuzhnyi, E.; Klyushnikov, S.; Broutian, A. Uncovering bilateral vestibulopathy in patients with SANDO syndrome caused by mutations in POLG gene: A case series. J. Neurol. 2021, 268, 3909–3912. [Google Scholar] [CrossRef] [PubMed]
- Leigh, R.J.; Zee, D.S. The Neurology of Eye Movements, 5th ed.; Contemporary Neurology Series; Oxford Academic: New York, NY, USA, 2015. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Published by MDPI on behalf of the Swiss Federation of Clinical Neuro-Societies. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).